

Abstract
Ibrutinib, which irreversibly inhibits Bruton tyrosine kinase, was evaluated for antitumor activity in a panel of non–small cell lung cancer (NSCLC) cell lines and found to selectively inhibit growth of NSCLC cells carrying mutations in the epidermal growth factor receptor (EGFR) gene, including T790M mutant and erlotinib-resistant H1975 cells. Ibrutinib induced dose-dependent inhibition of phosphor-EGFR at both Y1068 and Y1173 sites, suggesting ibrutinib functions as an EGFR inhibitor. Survival was analyzed by Kaplan–Meier estimation and log-rank test. All statistical tests were two-sided. In vivo study showed that ibrutinib statistically significantly suppressed H1975 tumor growth and prolonged survival of the tumor bearing mice (n = 5 per group). The mean survival times for solvent- and erlotinib-treated mice were both 17.8 days (95% confidence interval [CI] = 14.3 to 21.3 days), while the mean survival time for ibrutinib-treated mice was 29.8 days (95% CI = 26.0 to 33.6 days, P = .008). Our results indicate that ibrutinib could be a candidate drug for treatment of EGFR-mutant NSCLC, including erlotinib-resistant tumors.
Ibrutinib has been reported to selectively and irreversibly inhibit Bruton tyrosine kinase (BTK) (1,2), which is specifically required for the B-cell antigen receptor signaling pathway (3). Previous studies revealed that ibrutinib specifically inhibited the proliferation of B-cell lymphoma with active B-cell antigen receptor signaling (3) and multiple myeloma cells expressing BTK (4). Oral administration of ibrutinib led to promising in vivo activity against spontaneous B-cell non-Hodgkin lymphoma in dogs and experimental rheumatoid arthritis in mice (1,2). Ibrutinib also inhibited growth of chronic lymphocytic leukemia and multiple myeloma cells inoculated into immune defective mice (4,5). Clinical trials have revealed that ibrutinib is well tolerated and elicits substantial activity in relapsed or refractory B-cell malignancies, with an objective response rate of 60–70% and a complete response of 16–20% (6–8). Ibrutinib was recently approved by the US Food and Drug Administration for treatment of mantle cell lymphoma.
We evaluated antitumor activity of ibrutinib in a panel of non–small cell lung cancer (NSCLC) cell lines and in six- to eight-week-old female nude mice with xenograft tumors derived from H1975 cells. Further details are available in the Supplementary Methods (available online). Animal experiments were carried out in accordance with Guidelines for the Care and Use of Laboratory Animals (NIH publication number 85-23) and the institutional guidelines of the M. D. Anderson Cancer Center. Statistical significance of the differences between treated samples was determined by the two-sided Student t test and one-way analysis of variance (ANOVA). Differences were considered statistically significant at P less than .05. The mean survival time and accumulative survival curve were determined by Kaplan–Meier estimation. The mean survival times were compared by log-rank test. All statistical tests were two-sided.
To test whether ibrutinib can be used for treatment of solid tumors, we evaluated its antitumor activities in a panel of lung cancer cell lines by using a cell viability assay (9) three days after treatment with 0.01 to 30 μM ibrutinib. For the 39 non–small cell lung cancer (NSCLC) cell lines tested, the 50% inhibitory concentration [IC50] of ibrutinib ranged from 0.002 to 30 μM. Among the 39 cell lines tested, 36 had IC50 values between 2 and 30 μM. For the remaining three cell lines, HCC827, H1975, and H292, the IC50 were between 0.002 and 0.195 μM (Figure 1A), all within the clinically achievable concentrations of ibrutinib in the doses used for treatment of lymphoma (6,7). HCC827 and H1975 cells are known to harbor epidermal growth factor receptor (EGFR) mutations, whereas the H292 cell line has wild-type EGFR. Our subsequent analysis showed that EGFR was constitutively active in H292 cells, and that H292 cells were also susceptible to the EGFR inhibitor erlotinib (10). These results suggest that ibrutinib is specific for EGFR-mutant or -constitutively active NSCLC cells.
Figure 1.
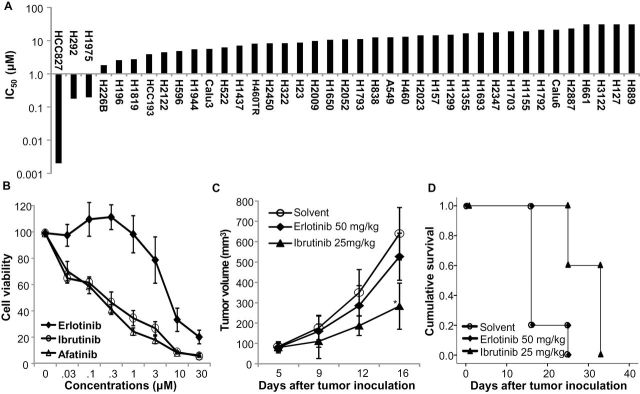
Antitumor activities of ibrutinib in non–small cell lung cancer (NSCLC) cell lines. A) Calculated 50% inhibitory concentration (IC50) on a logarithmic scale for 39 NSCLC cell lines. B) Dose–response curves of erlotinib, afatinib, and ibrutinib for the H1975 cell line, which has a T790M mutation. The data are means with standard deviations for two assays done in quadruplicate. The viability of control cells treated with dimethyl sulfoxide was assigned a value of 100. C) In vivo growth of H1795 tumors. The mice were treated as indicated. The values are means ±SD of data from five mice per group. * indicates P = .03 when compared with the control group, using a two-sided Student t test. D) Kaplan–Meier Survival Curve of the mice shown in (C). The mean survival times for solvent- and erlotinib-treated mice were both 17.8 days (95% confidence interval [CI] = 14.3 to 21.3 days). The mean survival time for ibrutinib-treated mice was 29.8 days (95% CI = 26.0 to 33.6 days; P = .008 when compared with solvent- or erlotinib-treated mice by log-rank test). All statistical tests were two-sided.
We next compared erlotinib’s and ibrutinib’s antitumor activities in nine NSCLC cell lines, six of which have mutations or deletions in the EGFR gene. Ibrutinib induced an antitumor spectrum similar to erlotinib in those cell lines, except for the H1975 cells, which harbor a T790M mutation in EGFR and were resistant to erlotinib but susceptible to ibrutinib (Supplementary Table 1, available online). The H1650 cells, which harbor EGFR mutation and PTEN loss (11), were resistant to erlotinib, ibrutinib, and afatinib (IC50 = 2.63 μM), a second generation of EGFR inhibitors that are approved for treatment of EGFR-mutant lung cancer (12). We also compared dose response of ibrutinib and afatinib in H1975 cells. The results showed that ibrutinib induced similar antitumor activity as afatinib in this cell line (Figure 1B).
To test whether ibrutinib can elicit in vivo antitumor activity in EGFR-mutant tumors, we established xenograft tumors from H1975 cells in nude mice and treated mice daily with ibrutinib (25mg/kg), erlotinib (50mg/kg), or solvent when tumors reached 4 to 5mm in diameters. The result showed that treatment with ibrutinib, but not erlotinib, statistically significantly suppressed H1975 tumor growth and prolonged survival of the tumor-bearing mice (Figure 1, C and D). While the mean survival times for solvent- and erlotinib-treated mice were both 17.8 days (95% confidence interval [CI] = 14.3 to 21.3 days), the mean survival time for ibrutinib-treated mice was 29.8 days (95% CI = 26.0 to 33.6 days, P = .008), demonstrating in vivo efficacy of ibrutinib in EGFR-mutant cancer.
We determined whether antitumor activity of ibrutinib in NSCLC cells was mediated by inhibition of BTK or by direct effect on EGFR. The expression of BTK was not detectable in any of the cell lines tested (Supplementary Figure 1, available online), indicating that ibrutinib-induced antitumor activity in these cells is not mediated by BTK. In contrast, treatment of H1975 and H3255 cells with erlotinib and ibrutinib led to a similar dose-dependent inhibition of phosphor-EGFR at the Y1068 site in H3255 cells. However, only ibrutinib inhibited pY1068 in H1975 cells (Figure 2A). The basal EGFR phosphorylation at Y1173 was only detectable in HCC827. Like erlotinib, ibrutinib-induced dose-dependent inhibition of EGFR Y1173 phosphorylation in HCC827 cells and constitutive Y1068 phosphorylation in H292 cells, although at relatively higher doses compared with those observed in EGFR-mutant cells. Similar results were observed for EGF-stimulated Y1068 phosphorylation in A549 cells (Figure 2B), suggesting that at a higher dose, ibrutinib was able to suppress wild-type EGFR activity, consistent with other studies on ibrutinib’s effect on EGFR (1,2). Ibrutinib-induced inhibition of EGFR phosphorylation occurred as early as 30min after the treatment (Figure 2C). We also tested whether ibrutinib-induced growth suppression or apoptosis in HCC827 cells. Flow cytometric analysis on apoptotic cells and western blot analyses of poly(ADP-ribose) polymerase (PARP1) and caspase-3 cleavage showed that ibrutinib induced dose-dependent increase of apoptotic cells (42% of apoptotic cells at 72 hours after treatment with 1 μM ibrutinib vs <10% of apoptotic cells in the control group) and cleavage of PARP1 and caspase-3 in HCC827 cells (Figure 2D), demonstrating that apoptosis is the major model of action in HCC827 cells.
Figure 2.

Western blot analysis of epidermal growth factor receptor (EGFR) phosphorylation (p-EGFR) and cleavage of poly(ADP-ribose) polymerase (PARP) and caspase 3 (Casp-3). A) H1975 and H3255 cells were treated with erlotinib and ibrutinib with the dose as indicated. Phospho-Y1068 and total EGFR were determined at 24 hours after treatment. B) HCC827, H292, and A549 cells were treated with erlotinib or ibrutinib at the doses as indicated. Cell lysates were harvested for protein phosphorylation analyses at 24 hours. Note, for A549 cells, 10ng/ml EGF was added to the cells to activate EGFR 15min before harvesting cells. C) HCC827 cells were treated with 0.5 μM erlotinib or 0.5 μM ibrutinib and tested for EGFR phosphorylation at different time points as indicated. D) HCC827 cells were treated with ibrutinib and tested for EGFR phosphorylation and caspase-3 and PARP1 cleavages at 48 hours.
EGFR mutations are frequently detected in lung adenocarcinoma patients, especially those who have no smoking history (13,14). The high susceptibilities of EGFR-mutant lung cancer cells to gefitinib and erlotinib (13,15,16) have made these two agents the first choice for treatment of EGFR-mutant cancers. Unfortunately, despite dramatic responses of EGFR-mutant lung cancer patients to gefitinib or erlotinib, acquired resistance occurs at a median of 10 to 13 months after the treatment initiation (17,18). While a variety of mechanisms have been identified for the acquired resistance, including a second T790M mutation at exon 20 of the EGFR gene (19, 20), amplification of MET gene (21–23), mutations of the KRas gene (24), and activation of AXL or c-Src kinases (25–27), the most common cause of the resistance in clinics is the T790M mutation in the EGFR, which is found in about 50% of those patients (19,21,28). Effort has been made to develop EGFR kinase inhibitors that are effective for EGFR T790M mutants (29–31), including development and approval of afatinib for clinical application (12) and clinical trials on some novel anti-EGFR agents (32). Ibrutinib’s selective inhibition of EGFR-mutant NSCLC cells, including the T790M mutant cell line H1975 and its excellent safety profile in patients, indicate that this agent could be a good candidate for treatment of EGFR-mutant NSCLC.
Nevertheless, this study had some limitations, because ibrutinib’s inhibitory effect on EGFR was determined on cultured cell lines, not on recombinant EGFR proteins. The differential effects of ibrutinit on wild-type and mutant EGFRs remain to be determined. Moreover, the in vivo study was performed with subcutaneous tumors instead of tumors in orthotopic microenvironments. Because skin rash, a common dose-limiting side effect for EGFR inhibitors (12,33), was observed much less frequently in patients treated with ibrutinib (6–8), it also raises an intriguing question on whether ibrutinib can be used as an EGFR inhibitor to treat cancers in clinics.
Funding
This work was supported in part by the National Institutes of Health R01 grant CA124951 to BF, Specialized Program of Research Excellence (SPORE) grant CA070907 to JM and JAR, the University of Texas MD Anderson Cancer Center support grant CA-016672 (Lung Program, DNA Analysis and Flow Cytometry, and Cellular Imaging Core Facility), and by endowed funds to the University of Texas MD Anderson Cancer Center, including: Moon Shots Program, the Homer Flower Gene Therapy Research Fund, the Charles Rogers Gene Therapy Fund, the Flora & Stuart Mason Lung Cancer Research Fund, the Charles B. Swank Memorial Fund for Esophageal Cancer Research, the Phalan Thoracic Gene Therapy Fund, the M.W. Elkins Endowed Fund for Thoracic Surgical Oncology, Stading Lung Cancer Research Fund, the Chapman Foundation, the William Burchenal Memorial Research Fund, the Edward Crutchfield Fund, and the Marvin Kimmel Research Fund.
We would like to thank Arthur Gelmis for editorial review of this manuscript.
The study sponsors had no roles in the design of the study, the collection, analysis, or interpretation of the data, the writing of the manuscript, nor the decision to submit the manuscript for publication.
References
- 1. Honigberg LA, Smith AM, Sirisawad M, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA. 2010;107(29):13075–13080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan Z, Scheerens H, Li SJ, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2007;2(1):58–61 [DOI] [PubMed] [Google Scholar]
- 3. Davis RE, Ngo VN, Lenz G, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tai YT, Chang BY, Kong SY, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120(9):1877–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ponader S, Chen SS, Buggy JJ, et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood. 2012;119(5):1182–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu X, Guo W, Wu S, et al. Antitumor activity of a novel STAT3 inhibitor and redox modulator in non-small cell lung cancer cells. Biochem Pharmacol. 2012;83(10):1456–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shepherd FA, Rodrigues PJ, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353(2):123–132 [DOI] [PubMed] [Google Scholar]
- 11. Sos ML, Koker M, Weir BA, et al. PTEN loss contributes to erlotinib resistance in EGFR-mutant lung cancer by activation of Akt and EGFR. Cancer Res. 2009;69(8):3256–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31(27):3327–3334 [DOI] [PubMed] [Google Scholar]
- 13. Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci USA. 2004;101(36):13306–13311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ding L, Getz G, Wheeler DA, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350(21):2129–2139 [DOI] [PubMed] [Google Scholar]
- 16. Mukohara T, Engelman JA, Hanna NH, et al. Differential effects of gefitinib and cetuximab on non-small-cell lung cancers bearing epidermal growth factor receptor mutations. J Natl Cancer Inst. 2005;97(16):1185–1194 [DOI] [PubMed] [Google Scholar]
- 17. Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12(8):735–742 [DOI] [PubMed] [Google Scholar]
- 18. Lee CK, Brown C, Gralla RJ, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst. 2013;105(9):595–605 [DOI] [PubMed] [Google Scholar]
- 19. Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352(8):786–792 [DOI] [PubMed] [Google Scholar]
- 21. Bean J, Brennan C, Shih JY, et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc Natl Acad Sci USA. 2007;104(52):20932–20937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cappuzzo F, Janne PA, Skokan M, et al. MET increased gene copy number and primary resistance to gefitinib therapy in non-small-cell lung cancer patients. Ann Oncol. 2009;20(2):298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043 [DOI] [PubMed] [Google Scholar]
- 24. Linardou H, Dahabreh IJ, Kanaloupiti D, et al. Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer. Lancet Oncol. 2008;9(10):962–972 [DOI] [PubMed] [Google Scholar]
- 25. Byers LA, Diao L, Wang J, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res. 2013;19(1):279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang Z, Lee JC, Lin L, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nature Genet. 2012;44(8):852–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stabile LP, He G, Lui VW, et al. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin Cancer Res. 2013;19(2):380–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kosaka T, Yatabe Y, Endoh H, et al. Analysis of epidermal growth factor receptor gene mutation in patients with non-small cell lung cancer and acquired resistance to gefitinib. Clin Cancer Res. 2006;12(19):5764–5769 [DOI] [PubMed] [Google Scholar]
- 29. Zhou W, Ercan D, Chen L, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462(7276):1070–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kwak EL, Sordella R, Bell DW, et al. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc Natl Acad Sci USA. 2005;102(21):7665–7670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chang S, Zhang L, Xu S, et al. Design, synthesis, and biological evaluation of novel conformationally constrained inhibitors targeting epidermal growth factor receptor threonine790 -> methionine790 mutant. J Med Chem. 2012;55(6):2711–2723 [DOI] [PubMed] [Google Scholar]
- 32. Sequist LV, Soria JC, Gadgeel SM, et al. First-in-human evaluation of CO-1686, an irreversible, selective, and potent tryrosine kinase inhibitor of EGFR T790M. J Clin Oncol. 2013;31(Suppl):Abstract 2524. [Google Scholar]
- 33. Eilers RE, Jr, Gandhi M, Patel JD, et al. Dermatologic infections in cancer patients treated with epidermal growth factor receptor inhibitor therapy. J Natl Cancer Inst. 2010;102(1):47–53 [DOI] [PubMed] [Google Scholar]