

Abstract
The standard approach for documenting symptomatic adverse events (AEs) in cancer clinical trials involves investigator reporting using the National Cancer Institute’s (NCI’s) Common Terminology Criteria for Adverse Events (CTCAE). Because this approach underdetects symptomatic AEs, the NCI issued two contracts to create a patient-reported outcome (PRO) measurement system as a companion to the CTCAE, called the PRO-CTCAE. This Commentary describes development of the PRO-CTCAE by a group of multidisciplinary investigators and patient representatives and provides an overview of qualitative and quantitative studies of its measurement properties. A systematic evaluation of all 790 AEs listed in the CTCAE identified 78 appropriate for patient self-reporting. For each of these, a PRO-CTCAE plain language term in English and one to three items characterizing the frequency, severity, and/or activity interference of the AE were created, rendering a library of 124 PRO-CTCAE items. These items were refined in a cognitive interviewing study among patients on active cancer treatment with diverse educational, racial, and geographic backgrounds. Favorable measurement properties of the items, including construct validity, reliability, responsiveness, and between-mode equivalence, were determined prospectively in a demographically diverse population of patients receiving treatments for many different tumor types. A software platform was built to administer PRO-CTCAE items to clinical trial participants via the internet or telephone interactive voice response and was refined through usability testing. Work is ongoing to translate the PRO-CTCAE into multiple languages and to determine the optimal approach for integrating the PRO-CTCAE into clinical trial workflow and AE analyses. It is envisioned that the PRO-CTCAE will enhance the precision and patient-centeredness of adverse event reporting in cancer clinical research.
Adverse Event Reporting in Oncology
Adverse event (AE) reporting is mandatory in clinical research to assure patient safety and to understand the toxicity profiles of treatments. Therefore, methods for collecting this information must be accurate and reliable.
The standard approach to AE reporting in cancer trials is the Common Terminology Criteria for Adverse Events (CTCAE), which is maintained by the US National Cancer Institute (NCI). The current version of the CTCAE consists of 790 individual items, each representing a discrete event which is graded for severity on a five-point scale based on clinical criteria. Use of the CTCAE is required in NCI-sponsored trials, has become standard in industry-sponsored cancer trials and drug labels, and is widely used in oncology clinical practice to document the adverse effects of cancer treatment. The CTCAE was revised in 2009 to its current version (version 4) in order to harmonize its terminology with the Medical Dictionary for Medical Affairs (MedDRA), a standardized lexicon for reporting AEs in industry trials, which is required by some regulatory authorities.
There are three general categories of AEs in the CTCAE and MedDRA: 1) laboratory-based events (eg, neutropenia), 2) observable/measurable events (eg, retinal tear), and 3) symptomatic adverse events (eg, nausea). Current practice is for research staff to report all of these categories (1), but empirical evidence demonstrates that clinician reporting of symptomatic AEs lacks reliability (2), that clinicians under-report the incidence and severity of symptoms compared to patients’ direct reports (3,4,5), and that patient reports better reflect underlying health status than clinician reports (6,7). Moreover, direct patient reporting improves the characterization of baseline symptoms present at trial entry, and most patients are willing and able to self-report their own symptomatic AEs without substantial attrition, even among those with end-stage disease and poor performance status (3,6,8).
The current workflow for symptomatic AE reporting in cancer trials involves a cascade of data transfer between multiple professional staff members during which information may be lost or misinterpreted at each step (Figure 1A) (9). There is not a standard or required training of investigators or research staff or a standard workflow for the collection of adverse event information in clinical trials. Further, neither the NCI nor regulatory authorities have specified a methodology to elicit symptomatic AEs from study participants. The workflow for collecting and reporting information about these events can vary substantially between and within study sites. Direct patient reporting bypasses this workflow and preserves the fidelity of the patient perspective (Figure 1B).
Figure 1.
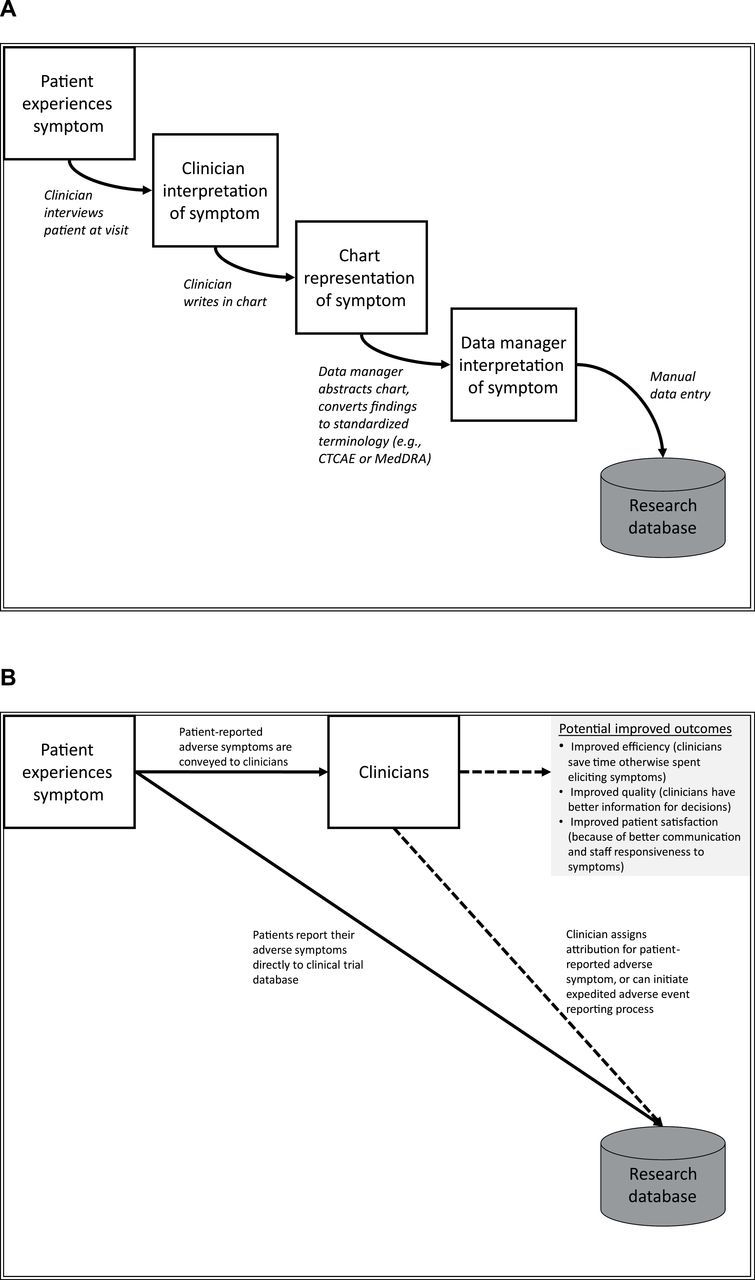
A) Current and B) proposed models for adverse symptom reporting in cancer clinical trials. Adapted with permission from: J Clin Oncol. 2005;23(15):3552–61 (9). A) The current model of adverse symptom reporting in clinical trials involves a cascade of information transfer and successive reinterpretations, with the end-result being systematic under-representation of patients’ actual symptoms in published results and drug labels. B) Integration of patient-reported adverse symptom reporting into clinical trials provides an unfiltered account of the patient experience with treatment, and can also inform clinicians’ treatment decisions. Clinicians can assign attribution of these symptoms to a study drug or other etiologies using current standard methods. CTCAE = Common Terminology Criteria for Adverse Events; MedDRA = Medical Dictionary for Regulatory Affairs.
Patient-Reported Outcomes
Patient-reported outcomes (PROs) are already considered the gold standard for data collection in closely related research areas, including assessment of health-related quality of life (HRQL), treatment preferences, and satisfaction with care, and are of growing interest in comparative effectiveness research and quality assessment (10,11,12,13,14,15,16). In 2009, the US Food and Drug Administration (FDA) published a final guidance document for applicants seeking labeling claims (ie, claims of treatment benefit) recommending the incorporation of PRO instruments when seeking to measure concepts in clinical trials that are best measured from the patient perspective (17), with a similar position statement issued by the European Medicines Agency (18).
Yet there is no current standard measurement approach or instrument for collecting direct patient reports of AEs in clinical research. The need for such a system is particularly salient in oncology given that symptomatic AEs are common and many cancer therapies carry substantial toxicity burdens. Symptomatic AEs are increasingly recognized as a contributing factor to treatment noncompliance, discontinuation, or dose reduction (19,20,21,22). In a recent survey of over 700 clinical investigators and research staff, more than 90% felt patient reporting of symptomatic AEs would improve data completeness, accuracy, meaningfulness, and actionability (23).
There are several requirements specific to the measurement of symptomatic AEs using PROs, as distinct from the measurement of constructs such as symptom distress or HRQL more generally. This is because PRO-based AE reporting occurs within the general context of AE evaluation in clinical trials. Specifically, there must be a capacity to tailor the PRO measure to solicit the most salient AEs in any given trial, for example via a customizable case report form that draws select items from a larger library of available items. Available items must both include symptoms that are not typically represented in many symptom inventories or HRQL questionnaires (eg, blurred vision, injection site reaction, nail loss) as well as more commonly occurring symptoms (eg, nausea, pain, fatigue). The worst magnitude of AEs must be captured during any given reporting interval. AEs must be assessed at sufficiently frequent intervals such that the onset and resolution are captured without gaps in recall reference periods. Additionally, there must be an ability to capture unanticipated symptoms through verbatim reports that can be mapped to a structured terminology. Lastly, each AE must be scored independently, with analyses focused not on summative scores but on patterns of AEs towards an improved understanding of toxicity, tolerability, and safety. By comparison, many symptom inventories and HRQL measures focus on a discrete number of common domains and do not need to accommodate the routine collection of unsolicited symptoms; however, they offer the advantages of employing composite or summative scoring and comparisons with normative values (24). In the context of trials of cancer-directed therapy, measures of symptoms or HRQL are often included to inform determinations of treatment efficacy or overall clinical benefit, whereas PRO-based measures of symptomatic AEs provide data to help explicitly characterize toxicity.
A Patient-Centered Approach to Adverse Event Reporting
A patient-centered approach to AE reporting requires a standardized measurement system which accurately and reliably detects events from the patient perspective, as well as a model for implementation in trials that assures comprehensive surveillance without undue burden to patients.
To address this need, and to expand the scope of the CTCAE through direct integration of the patient perspective, the NCI issued two contracts (HHSN261201000043C and HHSN261201000063C) to develop and test the Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) Measurement System (Principal Investigator: EB). Project components included identifying AEs that are appropriate for patient self-reporting, creating PRO-CTCAE items to represent these AEs, refining and evaluating the measurement properties of PRO-CTCAE items through qualitative and quantitative studies, creating software for administering PRO-CTCAE items via web and interactive voice response (IVR) platforms, conducting usability testing to assure ease of software use for patients and research staff, and establishing the optimal implementation approach for integrating this tool into clinical trials. This Commentary describes PRO-CTCAE activities between 2008 and 2014 and provides a detailed description of how AEs were identified for patient-reporting and how the PRO-CTCAE items were created, as well as an overview of qualitative and quantitative assessments that have been reported elsewhere in detail.
A consortium was established including seven cancer centers (Memorial Sloan Kettering Cancer Center [coordinating center], Dana-Farber Cancer Institute, Duke Cancer Center, M.D. Anderson Cancer Center, Mayo Clinic, University of Pennsylvania, Emory University), five community oncology practice sites in the NCI Community Cancer Centers Program (NCCCP) network (in California, Connecticut, Delaware, Louisiana, and South Carolina) where clinical studies for instrument development would be conducted, a software development firm (SemanticBits), and a linguistic adaptation firm (FACITtrans). Multistakeholder committees were established for each component of the initiative and included clinical investigators, data administrators, health services researchers, psychometricians, health literacy experts, informaticians, biostatisticians, FDA observers, and representatives from the NCI’s Division of Cancer Control and Population Sciences (DCCPS), Division of Cancer Prevention (DCP), Cancer Therapy Evaluation Program (CTEP), and Center for Biomedical Informatics and Information Technology (CBIIT). Each committee included the Principal Investigator, NCI representatives, methodological and technical experts for that committee’s topic area, and one to three patient representatives. Two overall patient representatives provided input across committees. Broader feedback was elicited from patients through in-depth interviews and surveys at various points of PRO-CTCAE development.
Identification of Adverse Events Amenable to Patient Reporting
The CTCAE was systematically analyzed to identify terms that represent subjective phenomena and that are amenable to patient self-reporting (ie, those which the patient is in the best position to identify). Five discrete categories were delineated based on the degree of subjective content in any given CTCAE term:
A. Laboratory/biomarker-based AEs (require equipment), eg, neutropenia
B. Observable/measurable AEs (require technical training, eg, retinal tear
C. Primarily subjective AEs, without observable components, eg, nausea
D. Primarily subjective AEs, with observable components, eg, vomiting
E. Primarily observable AEs, with subjective components, eg, nail discoloration
Categories A and B were considered inappropriate for patient self-reporting because of the requirement of technical equipment or clinical expertise for evaluation. Categories C and D were considered amenable to patient grading of magnitude. Category E was considered amenable to patient identification of presence/absence but necessitates a clinician with technical expertise to grade magnitude. Categorization was conducted independently by two subcommittees including experts in clinical trial design and patients with arbitration by the full committee in cases of disagreement. The full committee reviewed all selections prior to final NCI approval. Using this approach, out of 790 AEs in the CTCAE, 78 were identified as being amenable to self-reporting by adults with cancer in clinical trials evaluating systemic treatments, radiotherapy, and/or surgery.
Selection of Plain-Language Terminology for Adverse Events
Since CTCAE symptom terms are frequently technical (eg, “dysphagia”), it was necessary to identify plain-language descriptor terms for the PRO-CTCAE to represent analogous CTCAE terms. A structured literature review was conducted (by TM and TA) to identify existing symptom and HRQL measures (including both generic and disease-specific modules) and associated scientific publications for each CTCAE symptom term. Searches of PubMed, Psych INFO, and EMBASE were conducted, accompanied by hand searches of reference lists of relevant publications. Particular attention was given to publications that included the use of concept elicitation or cognitive interviews in supporting the content validity of symptom terms, and in demonstrating that specific terms correspond to symptom concepts of interest.
These data sources provided candidate terms, which were refined by the committee, including input from health literacy experts and patient representatives, then further refined based on a multicenter cognitive interviewing study (study chair: JH) involving 127 patients on active cancer treatment, with diverse racial and geographic backgrounds and including a substantial proportion (35%) with a high school education or less, as previously reported (25).
Design of PRO-CTCAE Items
A multistep process for developing the PRO-CTCAE items was developed, with patient input included at each step.
Identifying Appropriate Symptom Attributes
Grading of symptoms in the CTCAE is based on consideration of multiple attributes, including the frequency, severity, and/or interference with activities related to each AE. For example, CTCAE grade 3 diarrhea is defined as an “increase of ≥7 stools per day over baseline; incontinence… limiting self care activities of daily living.” Therefore, for each PRO-CTCAE symptom, it was considered essential to incorporate those attributes that are included in the corresponding CTCAE criteria. However, combining multiple attributes within each single patient question is undesirable, because it is cognitively demanding to ask respondents to simultaneously appraise several attributes of their experience (26). Thus, for each PRO-CTCAE symptom term, between one to three distinct items were developed to reflect the attributes of frequency, severity, and/or interference (as pertinent to the corresponding CTCAE criteria). Methodological work to determine the extent to which each of these attributes adds information when measuring a given AE is ongoing, and initial work demonstrates an independent distinct contribution from each attribute (27,28).
Item Structures
A generic structure for each symptom attribute was developed to reflect: 1) a plain language term for the symptom of interest, 2) the attribute of interest, specifically frequency, severity, or interference, and 3) the recall period for the item. Because the CTCAE requires reporting the “worst” magnitude of adverse events (29), the structure of the severity attribute items specified that the “worst” severity experienced should be recorded. Item structures used in existing validated questionnaires and principles of health literacy were employed to design simple and clear questions that would be amenable to adaptation in languages other than English, and feasible for administration via various electronic modes (26,30):
Frequency item structure: In the past [recall reference period], how OFTEN did you have [symptom]?
Severity item structure: In the past [recall reference period], what was the SEVERITY of your [symptom] at its worst?
Interference item structure: In the past [recall reference period], how much did [symptom] INTERFERE with your usual or daily activities?
Most symptoms could be characterized based on these three attributes. However, a structure was also needed for selected AEs where the patient is in a good position to report on presence/absence, but clinical expertise is necessary for assessing magnitude (eg, rash, bedsores, hives, photosensitivity reaction). Additionally, a structure was needed for AEs when “amount” is the most appropriate attribute (eg, alopecia or vaginal discharge). Cognitive interviews determined a high level of patient understanding of symptom questions using these various structures (25).
Response Option Format and Grading
To select response options for each type of attribute, various approaches used in instruments identified by the structured literature reviews were considered. These included: 0–10 numerical rating scale (NRS), visual analog scale (VAS), and verbal descriptor scale. VAS was not selected due to the difficulty of collecting VAS data via automated telephone IVR systems.
A literature review demonstrated comparable psychometric performance and patient acceptance of the 0–10 NRS and five-point verbal descriptor scales. Based on expert consensus with NCI input, the five-point verbal descriptor approach was ultimately selected for the PRO-CTCAE in part because it corresponds to the five grade levels of the current CTCAE, and because the 0–10 numerical rating scale was seen as cumbersome to use in reporting symptom frequency (one of the selected PRO-CTCAE attributes). Specific verbal descriptor terms for each attribute’s response scale were selected based on those employed across commonly used questionnaires in oncology that have evidence for robust measurement properties, patient acceptance, and adaptability for linguistic translation (including the Memorial Symptom Assessment Scale [MSAS], Functional Assessment of Cancer Therapy [FACT] measure, Rotterdam Symptom Checklist, and European Organisation for Research and Treatment of Cancer [EORTC] QLQ-C30):
Frequency responses: Never / Rarely / Occasionally / Frequently / Almost constantly
Severity responses: None / Mild / Moderate / Severe / Very severe
Interference responses: Not at all / A little bit / Somewhat / Quite a bit / Very much
Recall Reference Period
The current recommended recall reference period for PRO-CTCAE items is specified as “the past 7 days” based on research indicating this as a reliable timeframe without substantial loss of information (31,32,33). The implication of this recall period is that for clinical trials in which the PRO-CTCAE is used to catalog cumulative AEs at the patient level, weekly self-reporting may be necessary so as not to miss any events. Multiple studies demonstrate that cancer trial participants are willing and able to complete surveys with this regularity without substantial attrition (8,34), and there are ongoing studies to test this assumption and compare alternative recall periods for the PRO-CTCAE (including two, three, and four-week recall).
Notably, when clinicians report AEs, they generally use a reference period “since the prior visit,” which can be variable between and within trials. Therefore, when both the PRO-CTCAE and CTCAE are employed in a trial, the frequency with which patients and clinicians are reporting AEs may differ. In trials that incorporate PRO-CTCAE, the time points of CTCAE and PRO-CTCAE reporting will need to be specified. However, this difference will not affect the standard analytic approach to adverse event reporting in oncology trials, which summarizes the cumulative incidence of the highest toxicity grade for each AE.
Item Library
The PRO-CTCAE item library version 1.0 is comprised of 78 symptomatic AEs (listed in Table 1). Because each AE is elicited using between one to three attribute questions (ie, representing the frequency, severity, and/or interference of the AE), there are 124 individual questions representing the 78 AEs in the library. For the most updated version of the PRO-CTCAE or information about translations, investigators should contact the Outcomes Research Branch of the National Cancer Institute (http://outcomes.cancer.gov/tools/pro-ctcae.html).
Table 1.
PRO-CTCAE version 1.0 items and corresponding CTCAE v4/MedDRA terms, organized by standard NCI “System Organ Class” categories*
| CTCAE v4 system organ class (SOC) | ||||||
|---|---|---|---|---|---|---|
| CTCAE v4 /MedDRA TERM | Corresponding PRO-CTCAE TERM | Attribute Items Included in PRO-CTCAE | ||||
| Frequency | Severity | Interference | Present/ absent | Amount | ||
| Cardiac disorders | ||||||
| Palpitations | Pounding or racing heartbeat (palpitations) | X | X | |||
| Ear and labyrinth disorders | ||||||
| Tinnitus | Ringing in your ears | X | ||||
| Eye disorders | ||||||
| Blurred vision | Blurry vision | X | X | |||
| Flashing lights | Flashing lights in front of your eyes | X | ||||
| Floaters | Spots or lines (floaters) that drift in front of your eyes | X | ||||
| Watering eyes | Watery eyes (tearing) | X | X | |||
| Gastrointestinal disorders | ||||||
| Abdominal pain | Pain in the abdomen (belly area) | X | X | X | ||
| Bloating | Bloating of the abdomen (belly) | X | X | |||
| Constipation | Constipation | X | ||||
| Diarrhea | Loose or watery stools (diarrhea) | X | ||||
| Dry mouth | Dry mouth | X | ||||
| Dyspepsia | Heartburn | X | X | |||
| Dysphagia | Difficulty swallowing | X | ||||
| Fecal incontinence | Loss of control of bowel movements | X | X | |||
| Flatulence | Increased passing of gas (flatulence) | X | ||||
| Mucositis oral | Mouth or throat sores | X | X | |||
| Nausea | Nausea | X | X | |||
| Vomiting | Vomiting | X | X | |||
| General disorders and administration site conditions | ||||||
| Chills | Shivering or shaking chills | X | X | |||
| Edema limbs | Arm or leg swelling | X | X | X | ||
| Fatigue | Fatigue, tiredness, or lack of energy | X | X | |||
| Injection site reaction | Pain, swelling, or redness at a site of drug injection or IV | X | ||||
| Pain | Pain | X | X | X | ||
| Injury, poisoning and procedural complications | ||||||
| Bruising | Bruise easily (black and blue marks) | X | ||||
| Dermatitis radiation | Skin burns from radiation | X | ||||
| Metabolism and nutrition disorders | ||||||
| Anorexia | Decreased appetite | X | X | |||
| Musculoskeletal and connective tissue disorders | ||||||
| Arthralgia | Aching joints (such as elbows, knees, shoulders) | X | X | X | ||
| Myalgia | Aching muscles | X | X | X | ||
| Nervous system disorders | ||||||
| Concentration impairment | Problems with concentration | X | X | |||
| Dizziness | Dizziness | X | X | |||
| Dysgeusia | Problems with tasting food or drink | X | ||||
| Headache | Headache | X | X | X | ||
| Memory impairment | Problems with memory | X | X | |||
| Peripheral sensory neuropathy | Numbness or tingling in your hands or feet | X | X | |||
| Psychiatric disorders | ||||||
| Anorgasmia | Unable to have orgasm or climax | X | ||||
| Anxiety | Anxiety | X | X | X | ||
| Delayed orgasm | Took too long to have an orgasm or climax | X | ||||
| Depression† | Feelings that nothing could cheer you up | X | X | X | ||
| Sad or unhappy feelings | X | X | X | |||
| Insomnia | Insomnia (including difficultly falling asleep, staying asleep, or waking up early) | X | X | |||
| Libido decreased | Decreased sexual interest | X | ||||
| Renal and urinary disorders | ||||||
| Urine discoloration | Urine color change | X | ||||
| Urinary frequency | Frequent urination | X | X | |||
| Urinary incontinence | Loss of control of urine (leakage) | X | X | |||
| Urinary tract pain | Pain or burning with urination | X | ||||
| Urinary urgency | Sudden urges to urinate | X | X | |||
| Reproductive system and breast disorders | ||||||
| Dyspareunia | Pain during vaginal sex | X | ||||
| Ejaculation disorder | Ejaculation problems | X | ||||
| Erectile dysfunction | Difficulty getting or keeping an erection | X | ||||
| Gynecomastia | Breast area enlargement or tenderness | X | ||||
| Irregular menstruation† | Irregular menstrual periods | X | ||||
| Miss an expected menstrual period | X | |||||
| Vaginal discharge | Unusual vaginal discharge | X | ||||
| Vaginal dryness | Vaginal dryness | X | ||||
| Respiratory, thoracic and mediastinal disorders | ||||||
| Cough | Cough | X | X | |||
| Dyspnea | Shortness of breath | X | X | |||
| Epistaxis | Nosebleeds | X | X | |||
| Hiccups | Hiccups | X | X | |||
| Hoarseness | Hoarse voice | X | ||||
| Voice alteration | Voice changes | X | ||||
| Wheezing | Wheezing (whistling noise in the chest with breathing) | X | ||||
| Skin and subcutaneous tissue disorders | ||||||
| Alopecia | Hair loss | X | ||||
| Body odor | Body odor | X | ||||
| Cheilitis | Skin cracking at the corners of your mouth | X | ||||
| Dry skin | Dry skin | X | ||||
| Hyperhidrosis | Unexpected or excessive sweating during the day or nighttime (not related to hot flashes) | X | X | |||
| Hypohidrosis | Unexpected decrease in sweating | X | ||||
| Nail discoloration | Change in the color of your fingernails or toenails | X | ||||
| Nail loss | Lose any fingernails or toenails | X | ||||
| Nail ridging | Ridges or bumps on your fingernails or toenails | X | ||||
| Palmar-plantar erythrodysesthesia syndrome | Hand-foot syndrome (a rash of the hands or feet that can cause cracking, peeling, redness, or pain) | X | ||||
| Photosensitivity | Increased skin sensitivity to sunlight | X | ||||
| Pruritus | Itchy skin | X | ||||
| Rash acneiform | Acne or pimples on the face or chest | X | ||||
| Rash maculo-papular | Rash | X | ||||
| Skin and subcutaneous tissue disorders - Other, specify (Striae) | Stretch marks | X | ||||
| Skin hyperpigmentation | Unusual darkening of the skin | X | ||||
| Skin ulceration | Bed sores | X | ||||
| Urticaria | Hives (itchy red bumps on the skin) | X | ||||
| Vascular disorders | ||||||
| Hot flashes | Hot flashes | X | X | |||
* CTCAE version 4 terms are identical to corresponding “Preferred Terms” from the Medical Dictionary for Regulatory Affairs (MedDRA), based on a harmonization process that occurred during the update from CTCAE version 3 to version 4. Each symptomatic adverse event (AE) may include up to three items that ask patients about the frequency, severity, activity interference, presence, and/or amount of that AE. For the most updated version of the PRO-CTCAE items, contact the Outcomes Research Branch at the National Cancer Institute (http://outcomes.cancer.gov/tools/pro-ctcae.html). Currently, use of the PRO-CTCAE items requires a no-cost Material Transfer Agreement with the National Cancer Institute. CTCAE = Common Terminology Criteria for Adverse Events; PRO = patient-reported outcome.
† For the CTCAE version 4 AEs depression and irregular menstruation, two PRO-CTCAE terms were mapped to each in order to capture a broader spectrum of patient experiences. Therefore, a total of 80 PRO-CTCAE terms are mapped to the 78 CTCAE version 4 AEs.
For any given clinical trial, it is envisioned that particular PRO-CTCAE items most salient to that study’s population and regimens would be preselected from the library to create a patient survey that would be administered at baseline and during the study, along with a structured mechanism for patients also to report unsolicited symptomatic AEs (35,36). Not all AEs are relevant to every disease or treatment context, and the large number of items in the PRO-CTCAE library make it impractical to administer all items to all patients. In a study of different modes of PRO-CTCAE administration (study chair: AB), it took between four and six minutes for patients to complete a 28-item questionnaire, administered by paper, web, or IVRS (37). It is generally recommended that the total time for PRO questionnaires at any particular time point in a clinical trial be limited to 20 minutes or less at baseline and 10–15 minutes (or less) at follow-up, to minimize respondent burden and to avoid missing data (36).
Software
A software platform was developed with the capacity for investigators to select items from the PRO-CTCAE library to generate tailored patient surveys and to schedule and electronically administer those surveys to study participants via web or automated telephone IVR. The software design was informed by prior research (38), incorporating features such as conditional branching and the ability for patients to report unsolicited symptoms. Automated alerts can be triggered for missed reports or symptoms that exceed prespecified thresholds. The software is hosted at the and is designed for compatibility with other data systems used in clinical research (39). The software was refined through a multicenter usability testing study in patients and clinical research staff (study chair: AA) (40). Selected screenshots are shown in Figure 2.
Figure 2.
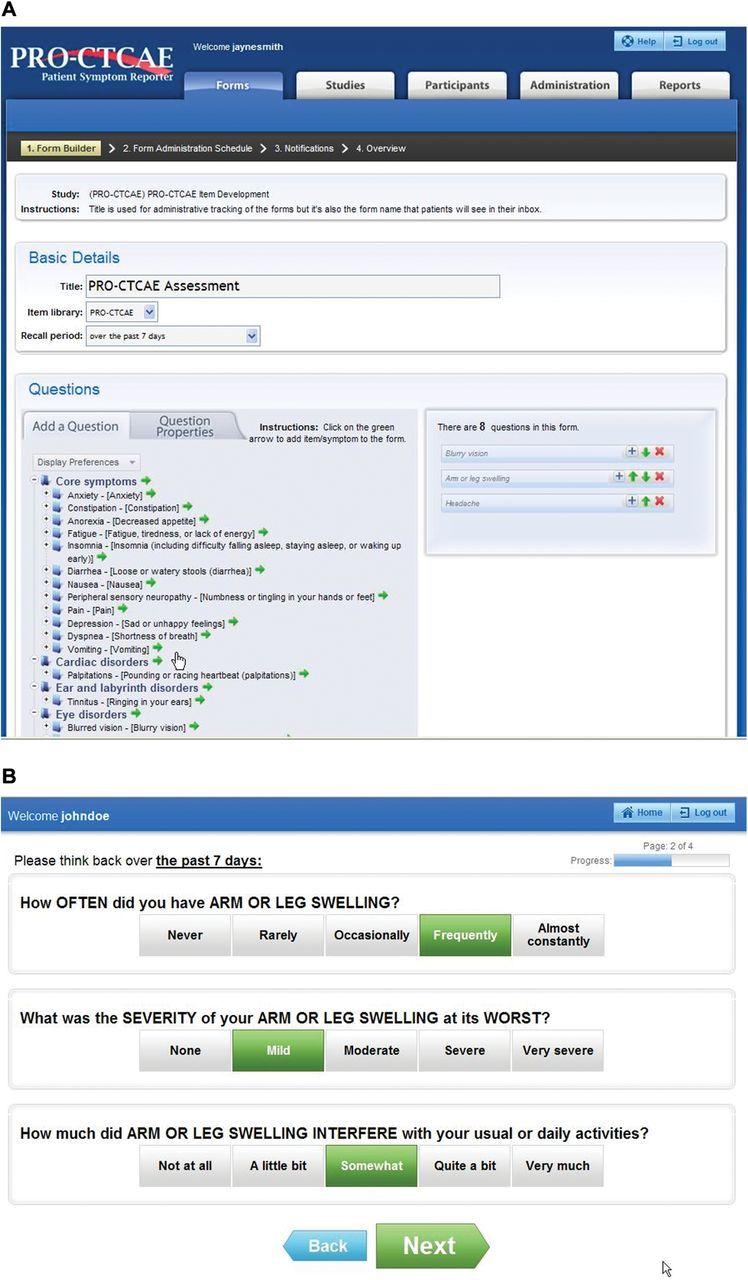
Example web screenshots from A) the investigator “form builder” web interface in which patient electronic forms can be created by selecting from the Patient-Reported Outcome version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) item library, and B) the patient PRO-CTCAE web data entry interface via which study participants can complete forms.
Measurement Properties
In addition to content validity evaluated through a cognitive interviewing study (25), construct validity, reliability, and responsiveness were examined in a US multicenter study including 940 patients with diverse cancer types (including breast, gastrointestinal, genitourinary, gynecologic, head and neck, lymphoma, leukemia, melanoma, myeloma, neurologic, and thoracic malignancies), US geographic locations, educational backgrounds, racial/ethnic backgrounds, and performance status (study chair: ACD). Most patients (N=522) had received systemic chemotherapy in the prior two weeks; 424 received radiotherapy within the prior two weeks, and 35 had undergone surgery within the prior two weeks. Results have been presented previously (28). In brief, validity was assessed based on associations between PRO-CTCAE items with HRQL scales from the EORTC QLQ-C30 instrument; associations with clinician-assigned ECOG performance status scores; and known-group differences between cancer types and treatments. Pearson correlations in the expected direction were observed for all PRO-CTCAE items with related QLQ-C30 scales. Statistically significant correlations for one or more validity criteria were seen for 120 items (97%). Scores for 94/124 (76%) PRO-CTCAE items were higher in the ECOG performance status 2–4 group vs the 0–1 group. Ongoing analyses are further evaluating the four outlier items, which represent the three symptomatic AEs: “nosebleeds,” “pain during vaginal sex,” and “pain, swelling or redness at site of drug injection or intravenous therapy.” These items represent events that were rare (ie, infrequently endorsed) in the study population, which limited their analysis.
Test-retest reliability was observed across 48 preselected items. Items were sensitive to change over time in relationship to functional status variations. In a comparison of web, IVR, and paper administration of PRO-CTCAE items, high correlations were observed between paper, web, and automated telephone IVR administration of items, demonstrating acceptable equivalence between these modes (37).
Envisioned Uses of the PRO-CTCAE in Clinical Research and Decision-Making
Several specific uses of the PRO-CTCAE are envisioned in cancer clinical research:
In early-phase trials, to collect initial information about tolerability from the patient perspective, and to assist in selecting appropriate dose levels and schedules,
In phase 3 trials, to better characterize symptoms at baseline, to assess adverse reactions generally, to support dose modification decisions, and to provide data for comparing tolerability between regimens,
In postmarketing studies, comparative effectiveness research (observational studies/registries) and safety surveillance systems to detect treatment impacts in targeted or broad populations and/or with long-term treatment.
Ongoing PRO-CTCAE Work
The PRO-CTCAE has been integrated into several multicenter clinical trials, both in the NCI-supported cooperative groups and through industry partnerships. These studies are addressing key methodological and implementation issues at each of these phases of research, including: how best to integrate PRO-CTCAE into existing clinical trial procedures and workflow, the incremental effort and cost associated with using the PRO-CTCAE in a trial, the role that PRO-CTCAE may play in the regulatory context, strategies to minimize missing data, and the ability of PRO-CTCAE to distinguish toxicities between study arms (including at baseline) and between low-grade and high-grade toxicities compared with investigator-reported CTCAE. A mapping algorithm to convert PRO-CTCAE verbal responses into numerical grades analogous to CTCAE grades is underway. An evaluation of associations between PRO-CTCAE grades and clinician CTCAE grades is planned. Linguistic adaptation and validation is in progress in multiple languages (with Spanish, German, and Japanese versions now completed), as is development of a pediatric version of the PRO-CTCAE (41).
Of particular interest is the relationship of patient-to-clinician symptomatic AE reporting in clinical trials. The underlying premise of the PRO-CTCAE program is that patients are generally in the best position to report their own experiences. Nonetheless, clinicians bear the ultimate responsibility for adverse event reporting and patient safety in clinical trials. Therefore, both patients and clinicians will likely continue to play a role in symptomatic AE detection and documentation. Based on evidence that patient self-reports can enhance the predictive accuracy of clinician ratings (7), a model in which self-reports are reviewed by investigators to inform their own AE documentation (ie, patient-informed investigator AE reporting) has been piloted, finding that providers agree with patients’ self-reported AE grades in most cases (42). A model in which the patient’s report serves as a stand-alone representation of symptomatic AEs has also been proposed (3). There is ongoing evaluation of both of these models within cooperative group clinical trials.
To date, PRO-CTCAE item development research has focused on symptomatic toxicities associated with chemotherapy, targeted therapy, and/or radiation across diverse tumor types. Although many patient participants in PRO-CTCAE studies have undergone prior surgical treatment, items have not been administered at a consistent postoperative time point, and there was limited variation in surgical procedures. Although the PRO-CTCAE system is readily adaptable for assessing PROs following surgery, a limitation of development to date is that it included limited and nonsystematic representation of surgical patients to permit assessment of the items in surgical contexts. Future research to evaluate PRO-CTCAE validity and sensitivity to change in populations that have recently undergone surgical cancer treatment is warranted.
Conclusion
A patient-centered approach to adverse event assessment in clinical research has been developed for the National Cancer Institute under contract by a group of collaborating multidisciplinary investigators and patients and is designed to complement the CTCAE. It is envisioned that the PRO-CTCAE will ultimately provide a more representative account of patients’ treatment experiences, thereby enabling more informed decisions by patients/clinicians facing treatment choices, investigators/regulators seeking to better understand toxicity and tolerability, and guideline developers/payers assessing the risks and value of alternative treatment strategies.
Funding
Work described in this report was supported by contracts from the US National Cancer Institute (HHSN261201000043C and HHSN261201000063C), Principal Investigator Dr. Ethan Basch.
The authors report no financial conflicts of interest
References
- 1. Trotti A, Colevas AD, Setser A, Basch E. Patient-Reported Outcomes and the Evolution of Adverse Event Reporting in Oncology. J Clin Oncol. 2007;25(32):5121–5127 [DOI] [PubMed] [Google Scholar]
- 2. Atkinson TM, Li Y, Coffey CW, et al. Reliability of adverse symptom event reporting by clinicians. Qual Life Res. 2012;21:1159–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Basch E. The Missing Voice of Patients in Drug-Safety Reporting. N Engl J Med. 2010;362(10):865–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fromme EK, Eilers KM, Mori M, Hsieh YC, Beer TM. How accurate is clinician reporting of chemotherapy adverse effects? A comparison with patient-reported symptoms from the Quality-of-Life Questionnaire C30. J Clin Oncol. 2004;22(17):3485–3490 [DOI] [PubMed] [Google Scholar]
- 5. Pakhomov SV, Jacobsen SJ, Chute CG, Roger VL. Agreement between patient-reported symptoms and their documentation in the medical record. Am J Manag Care. 2008;14(8):530–539 [PMC free article] [PubMed] [Google Scholar]
- 6. Basch E, Jia X, Heller G, et al. Adverse symptom reporting by patients versus clinicians: relationships with clinical outcomes. J Natl Cancer Inst. 2009;101(23):1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quinten C, Maringwa J, Gotay CC, et al. Patient self-reports of symptoms and clinician ratings as predictors of overall cancer survival. J Natl Cancer Inst. 2011;103(24):1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Meacham R, McEngart D, O’Gorman H, Wenzel K. Use and compliance with electronic patient reported outcomes within clinical drug trials. International Society for Pharmacoeconomics and Outcomes Research (ISPOR) Annual International Meeting, 2008; Toronto, Canada [Google Scholar]
- 9. Basch E, Artz D, Dulko D, et al. Patient online self-reporting of toxicity symptoms during chemotherapy. J Clin Oncol. 2005;23(15):3552–3561 [DOI] [PubMed] [Google Scholar]
- 10. Bruner DW, Bryan CJ, Aaronson N, et al. Issues and challenges with integrating patient-reported outcomes in clinical trials supported by the National Cancer institute-sponsored clinical trials network. J Clin Oncol. 2007;25(32):5051–5057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Minasian L, O’Mara A. Introduction to special JNCI monograph on patient-reported outcomes. J Natl Cancer Inst Monogr. 2007:1–4 [Google Scholar]
- 12. Ganz PA, Gotay CC. Use of patient-reported outcomes in phase III cancer treatment trials: lessons learned and future directions. J Clin Oncol. 2007;25(32):5063–5069 [DOI] [PubMed] [Google Scholar]
- 13. Lipscomb J, Reeve BB, Clauser SB, et al. Patient-reported outcomes assessment in cancer trials: taking stock, moving forward. J Clin Oncol. 2007;25(32):5133–5140 [DOI] [PubMed] [Google Scholar]
- 14. Rock EP, Kennedy DL, Furness MH, Pierce WF, Pazdur R, Burke LB. Patient-reported outcomes supporting anticancer product approvals. J Clin Oncol. 2007;25(32):5094–5099 [DOI] [PubMed] [Google Scholar]
- 15. Wu AW, Snyder C, Clancy CM, Steinwachs DM. Adding the patient perspective to comparative effectiveness research. Health Aff (Millwood). 2010;29(10):1863–1871 [DOI] [PubMed] [Google Scholar]
- 16. Cella D, Riley W, Stone A, et al. The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005–2008. J Clin Epidemiol. 2010;63(11):1179–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.U.S. Department of Health and Human Services, Food and Drug Administration. Guidance for industry: Patient-reported outcomes measures: Use in medical product development to support labeling claims. December 2009. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM193282.pdf. Accessed March 24, 2014
- 18. European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Pre-authorisation evaluation of medicines for human use: Reflection paper on the regulatory guidance for the use of health-related quality of life (HRQL) measures in the evaluation of medicinal products. January 2006. Available at: http://www.ispor.org/workpaper/emea-hrql-guidance.pdf. Accessed March 24, 2014).
- 19. Wagner LI, Zhao F, Chapman J-AW, et al. Patient-Reported Predictors of Early Treatment Discontinuation: NCIC JMA.27/E1Z03 Quality of Life Study of Postmenopausal Women with Primary Breast Cancer Randomized to Exemestane or Anastrozole. Presented at San Antonio Breast Conference; December 2011; San Antonio, TX [Google Scholar]
- 20. Bhattacharya D, Easthall C, Willoughby KA, Small M, Watson S. Capecitabine non-adherence: Exploration of magnitude, nature and contributing factors. J Oncol Pharm Pract. 2012;18(3):333–342 [DOI] [PubMed] [Google Scholar]
- 21. Eliasson L, Clifford S, Barber N, Marin D. Exploring chronic myeloid leukemia patients’ reasons for not adhering to the oral anticancer drug imatinib as prescribed. Leuk Res. 2011;35(5):626–630 [DOI] [PubMed] [Google Scholar]
- 22. Timmers L, Boons CC, Mangnus D, et al. The use of erlotinib in daily practice: a study on adherence and patients’ experiences. BMC Cancer. 2011;11:284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bruner DW, Hanisch LJ, Trotti A, et al. Stakeholder perspectives on implementing the National Cancer Institute’s Patient-Reported Outcomes Version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). Trans Behav Med. 2011;1(1):110–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Reilly CM, Bruner DW, Mitchell SA, et al. A literature synthesis of symptom prevalence and severity in persons receiving active cancer treatment. Support Care Cancer. 2013;21(6):1525–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hay JL, Atkinson TM, Reeve BB, et al. ; NCI PRO-CTCAE Study Group. Cognitive interviewing of the U.S. National Cancer Institute’s patient-reported outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). Qual Life Res. 2014;23(1):257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Willis GB. Cognitive Interviewing: A Tool for Improving Questionnaire Design. Thousand Oaks, CA: Sage Publications; 2005 [Google Scholar]
- 27. Mitchell SA, Lang K, Nichols C, et al. Validation of the NCI Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE) in women receiving treatment for metastatic breast cancer. Annual Meeting of the American Society of Clinical Oncology; 2012; Chicago, IL [Google Scholar]
- 28. Dueck AC, Mendoza TR, Mitchell SA, et al. Validity and reliability of the patient-reported outcomes version of the common terminology criteria for adverse events (PRO-CTCAE). Presented at 2012 ASCO Annual Meeting; Chicago, IL: Abstract 9047. [Google Scholar]
- 29. Trotti A, Colevas AD, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol. 2003;13(3):176–181 [DOI] [PubMed] [Google Scholar]
- 30. Converse JM, Presser S. Survey Question: Handcrafting the Standardized Survey Questionnaire. Newbury Park, CA: Sage; 1986:1–80 [Google Scholar]
- 31. Bennett AV, Amtmann D, Diehr P, Patrick DL. Comparison of seven day recall and daily diary reports of COPD symptoms and impacts. Value in Health. 2012;15(3):466–474 [DOI] [PubMed] [Google Scholar]
- 32. Stull DE, Leidy NK, Parasuraman B, et al. Optimal recall periods for patient-reported outcomes: challenges and potential solutions. Curr Med Res Opin. 2009;25(4):929–942 [DOI] [PubMed] [Google Scholar]
- 33. Broderick JE, Schwartz JE, Vikingstad G, Pribbernow M, Grossman S, Stone AA. The accuracy of pain and fatigue items across different reporting periods. Pain. 2008;139(1):146–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Basch E, Iasonos A, Barz A, et al. Long-term toxicity monitoring via electronic patient-reported outcomes in patients receiving chemotherapy. J Clin Oncol. 2007;25(34):5374–5380 [DOI] [PubMed] [Google Scholar]
- 35. National Cancer Institute, Symptom Management and Quality of Life Steering Committee. Clinical Trials Planning Meeting - Building Bridges: The Identification of Core Symptoms and Health-Related Quality of Life Domains for use in Cancer Research. September 22–23, 2011. Available at: http://transformingtrials.cancer.gov/files/SxQOLSCPROCTPMExecutive%20Summary_FINAL.pdf. Accessed March 24, 2014
- 36. Basch E, Abernethy AP, Mullins CD, et al. Recommendations for Incorpor ating Patient-Reported Outcomes Into Clinical Comparative-Effectiveness Research in Adult Oncology. J Clin Oncol. 2012;30(34):4249–4255 [DOI] [PubMed] [Google Scholar]
- 37. Bennett AB, Dueck AC, Mitchell SA, et al. Mode Equivalence and Acceptability of Web-, Interactive Voice Response System-, and Paper-based Administration of US National Cancer Institute’s Patient-Reported Outcomes version of the Common Terminology Criteria for Adverse Events (PRO-CTCAE). International Society for Quality of Life Research (ISOQOL) Annual Meeting; October 2013; Miami, FL. [DOI] [PMC free article] [PubMed]
- 38. Basch E, Artz D, Speakman J, et al. Design, implementation, and evaluation of a web portal for patient symptom monitoring. J Am Med Inform Assoc. 2007;14(3):264–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agarwal H, Gulati M, Baumgartner P, et al. Web interface for the patient-reported version of the CTCAE. NCI caBIG Annual Meeting; July 2009; Washington, DC [Google Scholar]
- 40. Fawzy MR, Abernethy AP, Schoen MW, et al. Usability Testing of the PRO-CTCAE Measurement System in Patients with Cancer. Presented at 2013 ASCO Annual Meeting, Chicago, IL. J Clin Oncol. 2013;e17560 [Google Scholar]
- 41. Reeve BB, Withycombe JS, Baker JN, et al. The first step to integrating the child’s voice in adverse event reporting in oncology trials: a content validation study among pediatric oncology clinicians. Pediatr Blood Cancer. 2013;60(7):1231–1236 [DOI] [PubMed] [Google Scholar]
- 42. Basch E, Bennett A, Pietanza MC. Use of patient-reported outcomes to improve the predictive accuracy of clinician-reported adverse events. J Natl Cancer Inst. 2011;103(24):1808–1810 [DOI] [PubMed] [Google Scholar]