

Abstract
Secondary cell death via gap junctions (GJs) plays a role in the propagation of neuronal loss under a number of degenerative disorders. Here, we examined the role of GJs in neuronal death in the retina, which has arguably the most diverse expression of GJs in the CNS. Initially, we induced apoptotic death by injecting single retinal ganglion cells and glia with cytochrome C and found that this resulted in the loss of neighboring cells to which they were coupled via GJs. We next found that pharmacological blockade of GJs eradicated nearly all amacrine cell loss and reduced retinal ganglion cell loss by ∼70% after induction of either excitotoxic or ischemic insult conditions. These data indicate that the GJ-mediated secondary cell death was responsible for the death of most cells. Whereas genetic deletion of the GJ subunit Cx36 increased cell survivability by ∼50% under excitotoxic condition, cell loss in Cx45 knock-out mouse retinas was similar to that seen in wild-type mice. In contrast, ablation of Cx45 reduced neuronal loss by ∼50% under ischemic insult, but ablation of Cx36 offered no protection. Immunolabeling of the connexins showed differential changes in protein expression consistent with their differing roles in propagating death signals under the two insults. These data indicate that secondary cell death is mediated by different cohorts of GJs dependent on the connexins they express and the type of initial insult. Our results suggest that targeting specific connexins offers a novel therapeutic strategy to reduce progressive cell loss under different neurodegenerative conditions.
Keywords: bystander effect, cell death, connexin, gap junctions, neuroprotection, retina
Introduction
In addition to the intrinsic mechanisms underlying primary cell death, intercellular communication appears to play a major, but presently unclear, role in so-called secondary cell death (Andrade-Rozental et al., 2000; Decrock et al., 2009). In this scheme, a primary insult leads to the death of a limited cohort of vulnerable cells, which, in turn, pass toxic molecules via gap junctions (GJs) to coupled neighbors. There is now substantial evidence that cells that are clustered and can thereby communicate via GJs tend to die en masse under a broad range of neurodegenerative conditions (Frantseva et al., 2002; Lei et al., 2009; Wang et al., 2010; Belousov and Fontes, 2013).
In contrast, some studies have reported that GJs may actually protect cells. Evidence for this “good Samaritan” role include the findings that GJ inhibitors can induce apoptosis (Lee et al., 2006; Hutnik et al., 2008) and that deletion of GJ connexins can increase neuronal loss (Naus et al., 2001; Striedinger et al., 2005). It has been posited that GJs are portals by which healthy cells provide dying neighbors with rescue signals or that toxic substances can be diluted within a coupled syncytium (Krysko et al., 2005). Apoptotic conditions induce various changes in the structure of GJs, including phosphorylation of connexins (Lin et al., 2007), suggesting that the connexin makeup of a GJ may be a critical factor in determining its contribution to cell death or survival.
The retina displays arguably the most diverse expression of GJs in the CNS, which are widely distributed among the five neuronal types and express a variety of connexin subunits (Bloomfield and Völgyi, 2009). GJ-mediated secondary cell death has been implicated in retinal neuron loss seen under a number of degenerative conditions, including retinitis pigmentosa, glaucoma, and ischemia (Ripps, 2002; Das et al., 2008). On the other hand, deletion of connexins has failed to increase the survivability of cone photoreceptors in a mouse model of retinitis pigmentosa (Kranz et al., 2013) and has been reported to increase cell loss after retinal trauma (Striedinger et al., 2005), suggesting that GJs can be neuroprotective. Thus, the role of retinal GJs in cell death/survivability remains unclear.
Here, we describe results of a comprehensive study of the role of GJs in secondary neuronal death in the retina initiated by excitotoxic or ischemic conditions. We found that both insults produce significant loss of retinal ganglion cells (RGCs), which leads to a subsequent loss of amacrine cells to which they are coupled. Moreover, pharmacological blockade of GJs or genetic deletion of connexins increased the survivability of neurons by up to 70%, indicating that GJ-mediated secondary cell death accounted for the loss of most retinal neurons. We also found that secondary cell death is mediated by different cohorts of GJs, based on the connexins they express, depending on the type of initial insult. Targeting specific connexins may thus offer a novel therapeutic approach to reduce progressive cell loss under different neurodegenerative conditions.
Materials and Methods
Retina-eyecup preparation.
All animal procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committees at State University of New York College of Optometry and New York University School of Medicine. Experiments were performed on retinas of wild-type (WT), connexin knock-out (KO) mice (Cx36−/−, Cx45−/−, and Cx36−/−/45−/− dKO), and their heterozygous (Het) littermates (WT: n = 97 of either sex; Cx36−/−: n = 7 male and 7 female; Cx45−/−: n = 11 female and 4 male; Cx36−/−/45−/− dKO: n = 1 female and 2 male; Cx36+/−: n = 4 female and 7 male; Cx45+/−: n = 4 female and 4 male; Cx36+/−/45+/−: n = 1 female and 2 male). The Cx36−/− mice and Het littermates were derived from F2 C57/B6–129SvEv mixed background litters (Deans et al., 2002) The Cx45−/− were generated by crossing Cx45fl/fl mice with mice expressing Cre recombinase under control of the neuron-directed Nestin promoter to yield Cx45fl/fl:Nestin-cre mice (Blankenship et al., 2011; Pang et al., 2013). The methods used to impale and label neurons have been described previously (Hu and Bloomfield, 2003; Völgyi et al., 2009). Briefly, flattened retina-eyecups were placed in a superfusion chamber, which was mounted on the stage of a BX51WI light microscope (Olympus) within a light-tight Faraday cage. An IR-sensitive CCD camera (Dage) captured the retinal image, which was displayed on a video monitor. The retina-eyecups were superfused with a oxygenated Ames medium (Sigma-Aldrich) supplemented with NaHCO3 and maintained at 35°C.
Intracellular injections.
For intracellular injections, neurons were visualized and impaled with standard, sharp glass microelectrodes filled with cytochrome C (CytC, 10 mg/ml, ∼1 mm) and Neurobiotin (4%) in 0.1 m Tris buffer. Substances were injected iontophoretically for 15–20 min using a sinusoidal current (3 Hz, 1–5 nA p-p).
Immunocytochemistry.
After experimental treatment, retinas were fixed with 4% PFA in a 0.1 m PBS, pH 7.4, for 30 min at room temperature, cryoprotected in 30% sucrose, embedded in Tissue-Tek OCT Compound (Andwin Scientific), and frozen. Cryosections (18–20 μm thick) were cut and mounted on microscope slides. For immunostaining, sections were blocked in 3% donkey serum in 0.1 m PBS supplemented with 0.5% Triton X-100, and 0.1% NaN3 for 1 h at room temperature. Primary antibodies were diluted in 0.1 m PBS with 0.5% Triton X-100, 0.1% NaN3, and 1% donkey or goat serum. Tissues were then incubated with primary antibodies for 3 h at room temperature or overnight at 4°C. The following primary antibodies were used: rabbit anti-calretinin 1:2000, rabbit anti-calbindin 1:1000, goat ant-ChAT (choline acetyltransferase) 1:100 (all from Millipore), rabbit anti-GFAP (glial fibrillary acidic protein) 1:1000 (Invitrogen), rabbit anti-active caspase 3 1:200 (Abcam); and mouse anti-Cx35/36 1:300 and mouse anti-Cx45 1:300 (both from Millipore). After extensive washing with 0.1 m PBS, tissues were incubated for 1 h in secondary anti-goat/rabbit/mouse antibodies conjugated to Alexa-488 or Alexa-594 (1:200, Invitrogen). Retinal sections were counterstained with the nuclear dye propidium iodide (Invitrogen). Neurobiotin was visualized with Alexa-488/594-conjugated streptavidin (Invitrogen, 1:200). Tissues were then flat-mounted in Vectashield media (Vector Labs), and fluorescent images were taken using a fluorescent microscope or a Zeiss LSM510 or Olympus FV1200 MPE confocal microscope.
Induction of cell death.
Various methods were used to produce cell death. (1) Single-cell apoptosis: CytC was injected into individual RGCs or Müller cells for 15 min after which retinas were incubated for 4 h in oxygenated Ames medium before fixation. After streptavidin histology, apoptotic cells were detected with an anti-active caspase 3 antibody or with TUNEL staining. (2) Excitotoxicity: To assess the contribution of GJs to cell death within populations of RGCs and amacrine cells, we conducted parallel experiments in which retinas were preincubated for 20 min in normal Ames medium or in one containing the GJ blockers meclofenamic acid (MFA; 50 μm) or 18-β-glycyrrhetinic acid (18β-GA; 25 μm). Both control and drug-treated retinas were then exposed for 1 h to NMDA (100–300 μm) to induce excitotoxicity followed by 4 h in the control Ames solution. (3) Retinal ischemia: Transient in vivo retinal ischemia was induced by introducing into the anterior chamber a 33-gauge needle attached to a saline-filled reservoir (0.9% sodium chloride) that was raised above the animal so as to increase intraocular pressure to a level 120 mmHg above systolic blood pressure. MFA (2 μl, 500 μm) was administered intravitreally either 30 min before or 3 and 24 h after the ischemic insult. The opposite eye was cannulated but maintained at normal intraocular pressure to serve as a normotensive control. After 40–50 min, the needle was withdrawn and ischemia was evidenced by corneal whitening (Osborne et al., 2004). After 7 d of reperfusion, mice were killed and eyes were processed to assess retinal damage and neuronal death. We also used oxygen-glucose deprivation (OGD) to induce in vitro ischemia, in which retina-eyecups or isolated retinal whole mounts were exposed continuously to either a control, oxygenated Ringer solution or one that was glucose-free and deoxygenated by bubbling extensively with 95% N2/5% CO2 at 34°C. After 60 min in the OGD environment, retinas were transferred to a control, oxygenated Ames medium for 4 h before processing to evaluate cell death.
Retrograde labeling and visualization of coupled cells.
We used retrograde labeling of RGCs with Neurobiotin to visualize populations of amacrine cells in the inner nuclear layer (INL) to which they were coupled (Pang et al., 2011). Globes with attached optic nerves were submersed in oxygenated Ames medium, and a drop of Neurobiotin (4% in 0.1 m Tris buffer) was applied to the cut optic nerve for 40 min. For retrograde labeling limited to RGCs, Neurobiotin was replaced with Lucifer yellow (LY, 3%). In separate experiments, retrograde labeling was performed in both eyes before incubating one eye in Ames medium containing MFA (50 μm for 30 min) after which both eyes were exposed to NMDA (300 μm) for 1 h. Frozen retinal sections (20 μm thick) were cut on a cryostat and processed for assessment of cell death in the INL and ganglion cell layer (GCL).
Assessment of cell injury and cell death.
Apoptotic and necrotic cell death was assessed by cell counts following: (1) staining with Live/Death Viability Assay (calcein AM/ethidium homodimer (EthD) (Invitrogen); (2) TUNEL; or (3) labeling for activated caspase 3. For population studies, dead cells were counted manually within 500 × 500 μm areas (5 per retina) from micrographs of whole-mounts using images acquired by confocal microscope. Cell counts were made per unit length (500 μm) of frozen retinal cross-sections labeled with propidium iodide (2 μg/ml).
In some experiments, cells counts were limited to retrograde labeled RGCs in the GCL or certain subpopulation of amacrine cells, identified by specific markers, such as calretinin (CR), calbindin (CB), and ChAT in the INL. Fluorescence intensity measures were made of GFAP, which is overexpressed in Müller glial cells after retinal injury (Bringmann et al., 2006). Expression of GFAP and connexins was quantified by analysis of confocal images with Metavue software (Molecular Devices). Average pixel fluorescence intensities were measured by using uniform rectangular areas (3–5 per image) extending either from the GCL to the outer limiting membrane for GFAP or through the inner plexiform layer (IPL) for connexins. The intensity values were then averaged for at least 5 images from 3 to 5 independent experiments and data were normalized to controls.
Statistics.
Data are presented as mean ± standard mean error. The number of measurements performed for a given experiment (n) are given as x/y where x is the total number of samples in which the measures were made (e.g., flat mount areas or sectional lengths) and y is number of retinas. Statistical comparisons were assessed using Student's t test. Values of p ≤ 0.05 are considered statistically significant.
Results
GJs mediate secondary cell death in the retina
We initially examined whether GJ-mediated secondary cell death occurred in the retina. In these experiments, we performed intracellular injections of single RGCs with CytC to stimulate apoptosis and Neurobiotin to assess GJ coupling. To determine whether apoptosis in a single cell could spread to neighbors through a mechanism other than GJs, we randomly injected large somata in the GCL that were presumably RGCs. Following post hoc histological processing of Neurobiotin and anti-caspase3 labeling, we found that injection of CytC into uncoupled RGCs initiated cell death in the injected cell within the experimental timeframe of 3–4 h, but no other neighboring neurons were lost (Fig. 1A–C). In contrast, injection of CytC into RGCs that were coupled to other ganglion and/or amacrine cells resulted in the death of not only the injected cell, but also the coupled neighbors (Fig. 1D–F). These results confirmed that secondary cell death does occur in the retina and is dependent on functional GJs. It is important to note that CytC, at 12,000 Da, is far too large a molecule to pass across a GJ, indicating that other toxic molecules must be moving intercellularly to promote cell death in coupled neighbors. We also found that secondary cell death occurred in coupled Müller cells, indicating that this mechanism can result in progressive death of glia in addition to neurons (Fig. 1G–I).
Figure 1.
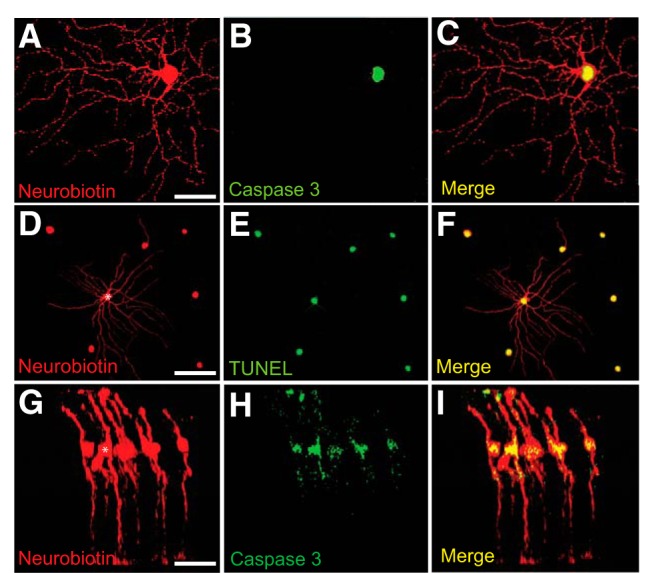
Injection of CytC into single cells results in GJ-mediated apoptosis of neighboring coupled cells. A–F, Representative photomicrographs of uncoupled (A–C) and coupled (D–F) RGCs in whole-mount retinas injected with the mixture of Neurobiotin and CytC to visualize GJ coupling and to induce cell apoptosis, respectively. C, Overlay of A and B represents apoptosis of the impaled RGC, but not any neighboring cells. F, Overlay of D and E represents that apoptosis was spread among neighboring cells coupled to impaled RGC (asterisk). G–I, Representative confocal image of coupled Müller cells in retinal vertical section, one of which (asterisk) was injected with Neurobiotin + CytC (G) and then labeled with anti-caspase 3 antibody (H) to detect apoptotic cells. I, Overlay of G and H represents spread of death in coupled neighboring Müller cells. Scale bars: A–C, G–I, 20 μm; D–F, 50 μm.
Pharmacological blockage of GJs reduces RGC death under excitotoxic or ischemic conditions
To determine the contribution of secondary cell death to the loss of RGCs produced under different neurodegenerative conditions, we examined the effect of GJ blockade on cell loss in the GCL. In initial experiments, we induced excitotoxic cell death by incubating retina-eyecups in 100–300 μm NMDA (Fig. 2A,C). Cell death counts were then performed in these retinas (n = 44/n = 5) and compared with retinas that were incubated in the nonselective GJ blockers 18β-GA (25 μm) or MFA (50 μm) before exposure to excitotoxic conditions (Fig. 2B,D). We found that prior blockade of GJs with either 18β-GA (n = 12/n = 3) or MFA (n = 42/n = 5) significantly reduced (p < 0.001 for both drugs) cell death in the GCL induced by excitotoxicity (Fig. 2E). Overall, NMDA-induced cell death was reduced dramatically in the GCL of mouse retinas pretreated with 18β-GA or MFA compared with those treated with NMDA alone. In control experiments, application of 18β-GA or MFA alone did not affect cell viability (p > 0.1), supporting the notion that the GJ blockers had no spurious toxic effects (Schulte et al., 2011). In addition, MFA, at the concentrations tested here, shows no inhibitory effect on NMDA receptors in neurons (Coyne et al., 2007), suggesting strongly that the attenuation of NMDA-induced cell death by GJ blockade was not due to reduced NMDA receptor activity. Overall, the significant protective effects of pharmacological GJ blockade suggest that secondary cell death is responsible for the progressive loss of the vast majority of RGCs under excitotoxic conditions.
Figure 2.
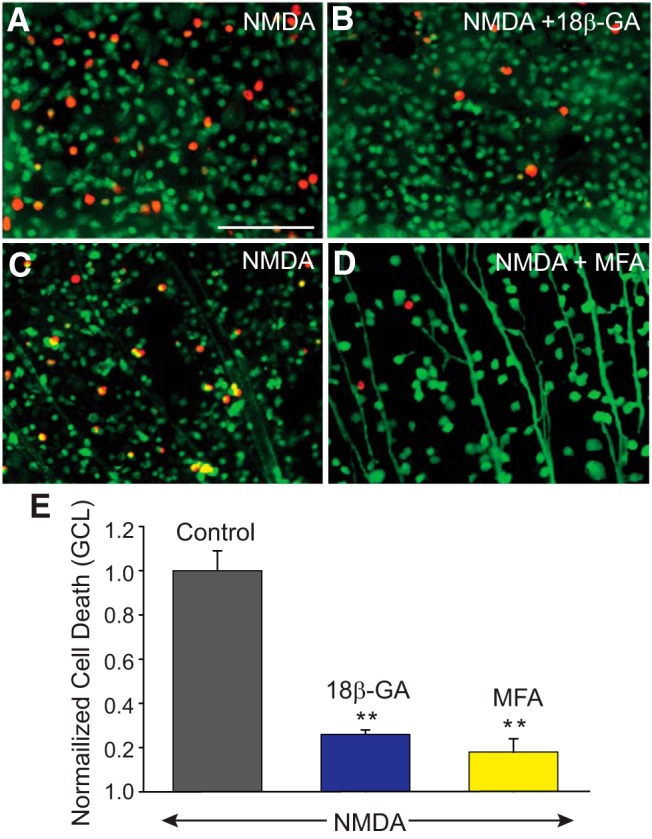
NMDA-induced excitotoxic cell death is significantly reduced by GJ blockers. A, C, NMDA-induced cell death in the GCL of control retinas, and those treated for 30 min with 25 μm of 18β-GA (B), or 50 μm of MFA (D) before exposing to NMDA (300 μm). Top, Experiments in which live and dead cells are marked with calcein-AM (green) and ethidium homodimer (EthD, red), respectively. Bottom, Experiments in which retrograde labeling through optic nerve cut with LY was used to label RGCs and then retinas processed with EthD to detect dead cells. E, Histogram represents the protective effect of GJ blockers on RGCs against NMDA-induced excitotoxicity. The number of dead cells was counted manually per unit area from 5 different visual fields in the GCL of whole-mount retinas pretreated with 18β-GA (n = 12/n = 3), or MFA (n = 42/n = 5), and numbers were normalized to one obtained in retinas exposed to NMDA alone (control, n = 44/n = 5). **p < 0.001 versus control. Scale bar, 100 μm.
In a second phase of experiments, we examined the role of secondary cell death in RGC loss associated with ischemic conditions of the retina. We assessed retinal damage subsequent to ischemia induced in vivo by raising intraocular pressure above normal systolic levels to compress the vasculature (Osborne et al., 2004). In control retinas (n = 19/n = 5), GFAP expression was confined exclusively to astrocytes in the GCL (Bringmann et al., 2006) (Fig. 3A,E). Expression of GFAP in ischemic retinas, however, was significantly upregulated as evidenced by the spread of immunolabeling to Müller cell processes at all retinal layers (p < 0.01) (Fig. 3B,E). Ischemia also produced a significant reduction in nuclear counts in the GCL (p < 0.01) accompanied by a marked thinning of the retina, particularly the inner layers (Fig. 3A,B,D).
Figure 3.

Blockade of GJs reduced retinal injury and cell death after ischemia-reperfusion. A, In control retinas, GFAP expression was confined exclusively to astrocytes in the GCL and nerve fiber layer (NFL). B, Seven days after reperfusion, the inner retinal thickness was reduced and the GFAP immunoreactivity appeared to traverse throughout the retinal layers in the Müller cell processes. C, Intravitreal injection of MFA (500 μm, 2 μl) prevented changes in the retinal morphology and maintained normal GFAP immunoreactivity. D, Histogram represents the nuclear counts in the GCL of vertical sections of control (n = 19/n = 5), and ischemic retinas in the presence (n = 16/n = 5) or absence (n = 16/n = 5) of MFA. E, Histogram represents the GFAP immunofluorescence intensity throughout the vertical section in control and ischemic retinas with or without MFA treatment. MFA was injected intravitreally either once 30 min before (blue, n = 16/n = 5), or twice at 3 and 24 h after (yellow, n = 12/n = 3) ischemic insult. Retinal sections were counterstained with propidium iodide (PI). *p < 0.01. Scale bar, 50 μm.
To assess the role of GJ-mediated secondary cell damage in ischemic injury of the retina, eyes were injected intravitreally with MFA either before or after insult. Treatment with MFA before ischemic induction preserved retinal thickness, cell counts in the GCL, and GFAP expression to levels seen in control retinas (Fig. 3C–E). Moreover, the protective effect of MFA administered 3 and 24 h after transient ischemia was evidenced by maintenance of normal levels of retinal thickness, GFAP expression, and cell counts in the GCL measured 1 week after insult; p < 0.01 for all measures (Fig. 3C–E).
GJs mediate amacrine cell loss after retinal injury
It has been reported that 15 of the 22 morphological subtypes of RGCs in the mouse retina are coupled to amacrine cells (Völgyi et al., 2009). This extensive coupling suggests that the GJ-mediated secondary cell death may also progress from RGCs to coupled amacrine cell neighbors. To test this idea, we examined the loss of amacrine cell populations immunolabeled with CR, CB, or ChAT.
Initial experiments were performed to determine whether CR-, CB-, and ChAT-immunopositive amacrine cells were indeed coupled to RGCs. Ganglion cell somata were retrogradely labeled with Neurobiotin through the cut optic nerve. Consistent with earlier reports, a large number of cells in the INL and GCL were immunoreactive to CR and ChAT with three distinct IPL bands of CR-labeled dendritic processes and two clear ChAT bands of starburst-a and starburst-b amacrine cells (Voigt, 1986; Haverkamp and Wässle, 2000) (Fig. 4A,C). In addition to CR-positive RGCs (or displaced amacrine cells) in the GCL, we found (n = 18/n = 4) that approximately one-half of the CR-positive, presumed amacrine cells in the INL showed Neurobiotin labeling (Fig. 4A,E,G), suggesting that they were coupled to RGCs. Indeed, blockade of GJs with MFA before retrograde labeling effectively eliminated all colocalized labeling of CR-positive amacrine cells in the INL with Neurobiotin (n = 8/n = 3, p < 0.01) (Fig. 4B,E). It has been shown that CB-immunoreactive labeling in the mouse retina mimics that described for CR, suggesting that they may label the same subpopulations of amacrine and RGCs (Haverkamp and Wässle, 2000). Consistent with this idea, our results from CB-immunolabeled retinas were similar to those described for CR (data not shown).
Figure 4.

Visualization of amacrine cell subpopulations with low and high degrees of coupling to retrograde-labeled RGCs. A, C, Retinal RGCs in control eyes were retrograde labeled for 40 min with Neurobiotin, and double labeled with antibodies against amacrine cell marker CR, or ChAT. Cells positive to both Neurobiotin and corresponding amacrine cell markers are shown by arrowheads. B, D, Blockade of GJs with MFA significantly reduced the number of Neurobiotin-labeled, CR-IR amacrine cells in the INL but had little effect on ChAT-IR cell numbers. E, Histogram represents that, whereas the total number of CR-IR cells (per 500 μm section) in the INL of MFA-treated retinas was comparable with that in control (n = 18/n = 4, p > 0.1), the number of those labeled with Neurobiotin was significantly reduced by MFA treatment (n = 8/n = 3). F, There was no significant difference in the number of Neurobiotin-positive ChAT-IR cells in control (n = 16/n = 3) and MFA-treated retinas (n = 6/n = 3, p > 0.1). G, Histogram quantifying the percentage of CR-IR and ChAT-IR amacrine cells in the INL that are coupled to RGCs under control conditions. *p < 0.01. Scale bars, 50 μm.
Although starburst amacrine cells in the rabbit retina are not coupled to RGCs (Xin and Bloomfield, 1997), we found a small number (<10%) of ChAT-positive cells in the INL of the mouse retina that were labeled with Neurobiotin (n = 16/n = 3) (Fig. 4C,F,G). However, application of MFA did not produce a significant reduction in the number of ChAT-positive amacrine cells labeled with Neurobiotin (n = 6/n = 3; p > 0.1) (Fig. 4D,F). We conclude that, at best, only a small number of starburst-a amacrine cells are coupled to RGCs.
In the next set of experiments, we examined how amacrine cell death due to excitotoxicity was affected by blockade of GJs with MFA. Application of NMDA produced a significant reduction of CR-immunopositive (n = 18/n = 3; p < 0.01) and CB-immunopositive (n = 22/n = 3; p < 0.01) subpopulations of amacrines in the INL (Fig. 5A–H). However, blockade of GJs with MFA prevented the loss of CR-immunolabeled (n = 10/n = 3) or CB-immunolabeled (n = 6/n = 3) amacrine cells (p < 0.01), preserving levels seen in control retinas (Fig. 5C,D,G,H). In contrast, neither NMDA-induced excitoxicity (n = 20/n = 4; p > 0.1) nor GJ blockade with MFA (n = 17/n = 4; p > 0.1) produced a significant reduction in the number of ChAT-positive cells, consistent with a lack of significant coupling to RGCs (Fig. 5L).
Figure 5.

Amacrine cells that are extensively coupled to RGCs show higher susceptibility to NMDA-induced excitotoxicity. A, CR labeled a large number of cells in the INL and GCL of control retina. B, In retinas exposed for 1 h to NMDA (300 μm), the number of CR-positive cells was significantly reduced. C, In vitro treatment of retinas with MFA (50 μm, 30 min) prevented the reduction in the number of CR-IR cells in the INL. D, Histogram summarizing NMDA-induced changes in the number of CR-positive amacrine cells in the INL of untreated (n = 18/n = 3) and MFA-treated retinas (n = 10/n = 3). E, CB-labeled horizontal cells in control retina. F, Exposure to NMDA reduced the number of CB-IR cells in the proximal INL. G, Blockade of GJs with MFA prevented such reduction. H, Histogram summarizing NMDA-induced changes in the number of CB-positive amacrine cells in the INL of untreated (n = 22/n = 3) and MFA-treated retinas (n = 6/n = 3). I–L, The number of ChAT-IR amacrine cells in control retinas. No significant change in the number of ChAT-IR amacrine cells was observed in NMDA-treated retinas compared with controls (n = 20/n = 4). Retinal sections were counterstained with propidium iodide (PI). *p < 0.01 versus control. Scale bars, 50 μm.
Amacrine cell death after transient ischemia paralleled the results seen after excitotoxic insult. Ischemia produced a significant loss of CR-immunoreactive (n = 48/n = 5; p < 0.001) and CB-immunoreactive (n = 17/n = 3; p < 0.001) subpopulations of amacrine cells in the INL and a disorganization of the dendritic bands in the IPL (Fig. 6A–H). Application of MFA prevented the loss of both the CR-immunoreactive (n = 26/n = 5; p < 0.001) and CB-immunoreactive (n = 21/n = 3; p < 0.001) amacrine cells, maintaining levels similar to those seen in control retinas (Fig. 6C,D,G,H). In contrast, induction of ischemia had no statistically significant impact on the number of ChAT-immunopositive amacrine cells in the INL (n = 25/n = 4; p > 0.1) (Fig. 6L). As expected, treatment of ischemic retinas with MFA had no effect on the number of ChAT-positive amacrine cells in the INL (n = 23/n = 4; p > 0.1) (Fig. 6K,L).
Figure 6.

Amacrine cells with a high degree of coupling to RGCs show higher susceptibility to ischemia-reperfusion injury. A–C, CR-IR cells in control retinas and in those subjected to ischemia-reperfusion injury in the absence and presence of MFA. D, Histogram summarizing alteration in the number of CR-IR amacrine cells in the INL of ischemic retinas with (n = 26/n = 5) or without (n = 48/n = 5) MFA treatment. E, CB immunoreactivity was localized to the horizontal cells and amacrine cells in the INL and sparse GCs. Three strata in the IPL were labeled as well. F, Changes in retinal morphology and the number of CB-IR cells after ischemia/reperfusion. G, Blockade of GJs by MFA largely prevented a reduction in the number of CB-IR cells. H, Histogram summarizing changes in CB-IR cells in the INL of control and ischemic retinas untreated (n = 17/n = 3) or treated (n = 21/n = 3) with MFA. I–L, No detectable change in the number of ChAT-IR amacrine cells was observed in ischemic retinas compared with levels in controls (n = 25/n = 4 each, p > 0.1). Retinal sections were counterstained with propidium iodide. **p < 0.001 versus control. Scale bars, 50 μm.
GJs mediate secondary cell death in a connexin-specific manner
The IPL of the retina contains an assortment of GJs formed between RGCs, amacrine cells, and bipolar cell axon terminals that express at least three different connexin subunits (Bloomfield and Völgyi, 2009). This diversity raises the notion that different cohorts of GJs, possibly based on their connexin profiles, may be responsible for secondary cell death in the inner retina arising from different primary insults. Our results using the nonspecific GJ blockers MFA and 18β-GA did not address this issue. To test this idea, we therefore examined the extent of excitotoxic and ischemic cell death in mice in which Cx36 and/or Cx45, the two most highly expressed subtypes in the IPL (Li et al., 2008; Bloomfield and Völgyi, 2009; Pan et al., 2010), were genetically deleted.
We induced excitotoxic cell death as described above by application of NMDA to retinas of Cx36−/−, Cx45−/−, and Cx36−/−/45−/− dKO mouse retinas and their heterozygous littermates. Detection of dead cells in the GCL showed that NMDA-induced excitotoxic cell death was markedly reduced in Cx36−/− mouse retinas (n = 35/n = 3) compared with levels found in Het littermates (n = 35/n = 3) or WT mice (n = 14/n = 3; p < 0.001 for both) (Fig. 7A,B,E). In contrast, the extent of cell death in the GCL of Cx45−/− mouse retinas after exposure to NMDA was not statistically different from control levels in Het or WT mice (n = 17/n = 3; p > 0.1) (Fig. 7A,C,E). We next induced excitotoxic cell death in Cx36−/−/45−/− dKO mouse retinas (n = 13/n = 3) and found that the level of cell death in the GCL was indistinguishable from those found in NMDA-treated retinas of Cx36−/− mice (p > 0.1) (Fig. 7B,D,E). Thus, the degree of cell death was not additive when both Cx36- and Cx45-expressing GJs were deleted. Overall, these results indicated that, whereas GJs expressing Cx36 played a role in secondary cell death associated with excitotoxicity, those expressing Cx45 made no significant contribution.
Figure 7.
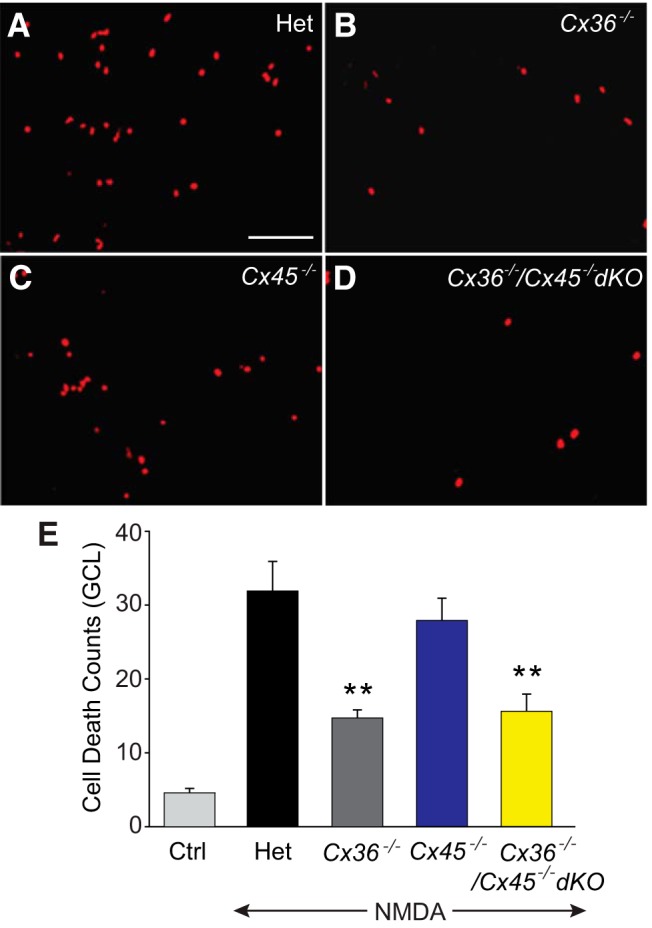
GJs formed by Cx36-mediated bystander cell death under excitotoxic conditions. A–D, Neuronal death was measured by EthD staining in whole-mount retinas of Het, Cx36−/−, Cx36−/−/45−/− dKO, and Cx45−/− mice. E, Histogram quantifying excitotoxic cell death in the GCL of retinas from Het and connexin KO mice. Compared with Hets (n = 35/n = 3), the reduction in NMDA-induced cell death was statistically significant in retinas of Cx36−/− (n = 35/n = 3), and Cx36−/−/Cx45−/− dKO (n = 13/n = 3) mice, but not in Cx45−/− (n = 17/n = 3; p > 0.1). **p < 0.001 versus Het. Scale bar, 50 μm.
In the next phase of experiments, we induced transient retinal ischemia in vivo in Cx36−/− and Cx45−/− mice and their Het littermates. After 7 d of survival, evaluation of retrograde labeling of GCs with LY in whole-mount retinas revealed a significant loss of axonal processes in Cx36−/− and Het mouse retinas compared with control levels (Fig. 8A–C). In contrast, ischemic retinas from Cx45−/− mice showed axonal labeling that was comparable with that seen in control retinas (Fig. 8A,D). Consistent with these findings, ischemia resulted in a marked reduction of cells in the GCL of Cx36−/− (n = 24/n = 4) and Het littermate mouse retinas (n = 28/n = 3; p < 0.001 for both) but no significant loss in the GCL of Cx45−/− mice (n = 74/n = 5), compared with control levels (n = 54/n = 5; p > 0.1) (Fig. 8I). The GFAP immunoreactivity was also markedly increased in Müller cell processes following ischemic insult of Cx36−/− (n = 24/n = 4) and Het (n = 24/n = 4) mouse retinas (p < 0.001) but showed levels in Cx45−/− mouse retinas (n = 22/n = 4) that were indistinguishable from that measured in control retinas (p > 0.1) (Fig. 8E–H,J).
Figure 8.

GJs formed by Cx45 mediate bystander cell death under ischemic conditions. A, LY retrogradely labeled RGC somas and their axons in control retina of Het mouse, ischemic Het retina (B), Cx36−/− retina (C), and Cx45−/− retina (D). E, Immunoreactivity of GFAP in vertical sections of control retina and ischemic retinas of Het (F), Cx36−/− (G), and Cx45−/− (H) mice. After ischemia-reperfusion, the GFAP immunoreactivity was upregulated throughout the retinal layers of Het and Cx36−/−, but not Cx45−/− mice. I, Histogram represents the nuclear cell count (per 500 μm length of vertical sections) in the GCL of control (n = 54/n = 5), and ischemic retinas of Het (n = 28/n = 3), Cx36−/− (n = 24/n = 4), and Cx45−/− (n = 74/n = 5) mice. J, Quantification of GFAP immunofluorescence intensity in the ischemic retinas of Het (n = 24/n = 4), Cx36−/− (n = 24/n = 4), and Cx45−/− (n = 22/n = 4, p > 0.1) mice. **p < 0.001 versus control. Scale bars: A–D, 100 μm; E–H, 50 μm.
A differential contribution of Cx36- and Cx45-expressing GJs to secondary neuronal death was also observed under condition of OGD, an in vitro model of ischemia. Exposure to OGD conditions produced a significant loss of neurons in the GCL of Cx36−/− (n = 12/n = 3) and Het mouse retinas (n = 12/n = 3; p < 0.001) but produced no significant loss of cells in the GCL of Cx45−/− mouse retinas (n = 10/n = 3; p > 0.1).
In the final phase of experiments, we investigated any changes in the distribution of Cx36- and Cx45-expressing GJs under excitotoxic and ischemic insult that could instruct their differential roles in secondary cell death (Fig. 9E,F,K,L). Induction of ischemia produced a dramatic reduction in the expression of Cx36 puncta in the IPL (n = 7/n = 3), compared with that in control retinas (n = 21/n = 3; p < 0.001) (Fig. 9A,B). Instead, we found intense Cx36 immunolabeling in RGC somata indicating an accumulation of the protein in the cytoplasm (Fig. 9B, inset). The expression of Cx45 puncta in the IPL was unaffected by ischemic insult (n = 26/n = 4; p > 0.1) (Fig. 9G,H,J). However, exposure of retinas to NMDA to induce excitotoxicity had no effect on the expression of Cx36 puncta in the IPL (n = 8/n = 3; p > 0.1) but dramatically reduced the expression of Cx45 (n = 8/n = 4), compared with control levels (n = 28/n = 4; p < 0.001) (Fig. 9I,J). Thus, ischemic and excitotoxic insult had opposite effects on the expression of Cx36- and Cx45-expressing GJs in the inner retina, consistent with their differential roles in mediating secondary cell death under these two pathological conditions (Fig. 9E,F,K,L).
Figure 9.

Changes in the immunolabeling of Cx36 and Cx45 in the IPL of retinas subjected to excitotoxic or ischemic insults. A, In control retina, Cx36 immunoreactivity appeared as punctate labeling predominantly in the inner half of the IPL. B, In ischemic retinas, the punctuate pattern of Cx36 labeling appeared as large clusters surrounding the cell nucleus. C, NMDA-induced excitotoxicity had little effect on the pattern of Cx36 immunolabeling. D, Histogram represents significant reduction in the intensity of punctate Cx36 immunoreactivity in the IPL of ischemic retinas (n = 7/n = 3) compared with that in controls (n = 21/n = 3). No detectable change in the Cx36 immunolabeling was observed under excitotoxic conditions (n = 8/n = 3). E, There was no significant difference in NMDA-induced cell death in the Het and Cx36−/− retinas (n = 21/n = 3; p > 0.1). F, NMDA-induced cell death was significantly less in Cx36−/− (n = 35/n = 3) compared with Het retina (n = 14/n = 3). G, Punctate immunolabeling for Cx45 was observed throughout the IPL of control retina. H, The punctuate pattern and the intensity Cx45 labeling were not altered by ischemia-reperfusion. I, NMDA markedly reduced Cx45 immunolabeling. Inset panels show high magnification details of IPL and GCL. J, Histogram quantifies Cx45 immunoreactivity in the IPL of retinas exposed to NMDA (n = 8/n = 4), compared with those in control (n = 28/n = 4), and ischemic retinas (n = 26/n = 4). K, Cell death was significantly less in ischemic retinas of Cx45−/− mice (n = 24/n = 5) compared with that in Het mice (n = 30/n = 5). L, There was no statistically significant difference in NMDA-induced cell death between Het and Cx45−/− retinas (n = 18/n = 3, p > 0.1). **p < 0.001. *p < 0.01. Scale bars, 20 μm.
Discussion
Together, our results provide clear evidence that GJ-mediated secondary cell death plays a significant role in the propagation of cell loss in the adult retina seen under a number of different primary insult conditions. First, we observed that injection of the apoptotic agent CytC into individual RGCs and glia led to the exclusive loss of neighboring neurons to which they were coupled via GJs. Second, pharmacological blockade of GJs under excitotoxic and ischemic insult increased the survival of RGCs by ∼70%, indicating that GJ-mediated secondary cell death plays a major role in cell loss. Further, under these same insult conditions, blockade of GJs prevented nearly all amacrine cell death presumably by eliminating the propagation of toxic signals from RGCs to which they were coupled. Finally, selective genetic ablation of the GJ subunits Cx36 or Cx45 found in the inner retina resulted in a significant reduction in the loss of RGCs normally seen after excitotoxic or ischemic insult. Our results thus support the growing evidence that damage in the CNS leads to primary cell death of the most vulnerable cells, induced by the initial insult, followed by a propagating secondary death of neighbors. In this scheme, GJs act as portals for the passage of apoptotic signals from injured cells to those to which they are coupled, which can ultimately be the cause of most cell loss (Kermer et al., 1999; Frantseva et al., 2002; Perez Velazquez et al., 2003; Decrock et al., 2009; Belousov and Fontes, 2013).
Our findings are consistent with evidence that GJs are involved in various neurodegenerative ocular disorders, including ischemic retinopathy, and glaucoma (Krysko et al., 2005; Malone et al., 2007; Das et al., 2008; Lei et al., 2009; Kerr et al., 2011; Danesh-Meyer et al., 2012). The topography of neuronal loss in the inner retina seen with these pathologies often includes both a diffuse, but clustered pattern, suggesting that dying RGCs influence neighboring cells resulting in secondary neuronal degeneration (Levkovitch-Verbin, 2001; Lei et al., 2009; Vander and Levkovitch-Verbin, 2012). The GJ-mediated bystander effect has also been implicated in the programmed cell death in the developing retina. As in the adult, dying cells in developing retina are spatially clustered into distinct networks (Cusato et al., 2003; de Rivero Vaccari et al., 2007). Dopamine, which is a modulator of GJ communication, as well as the GJ blockers octanol and carbenoxolone significantly reduce the rate of programmed and induced cell death in young retinas and the clustering of the remaining dying cells (Varella et al., 1999; Cusato et al., 2003).
Studies of glaucomatous human retinas have reported an apparent delayed or secondary degeneration of amacrine cells subsequent to RGC loss (Schwartz, 2003; Kielczewski et al., 2005; Moon et al., 2005). Consistent with this scenario is our present finding that CR- and CB-immunopositive amacrine cell loss resulting from excitoxicity or ischemia could be largely blocked by GJ blockade. These data suggest that, whereas RGCs were most vulnerable under our experimental insult conditions, the loss of amacrine cells was consequent to GJ-mediated bystander cell death. This hypothesis is supported by our finding that ChAT-immunopositive amacrine cells, which showed only minimal coupling to RGCs, were not significantly affected by excitotoxic or ischemic insult or by disruption of GJs. A downregulation of CR, CB, and ChAT has been reported in ischemic retinas (Dijk et al., 2004; Bernstein and Guo, 2011; Lee et al., 2011), suggesting that the loss of the amacrine cell immunoreactivity may be the result of reduced protein detection rather than cell death. However, we found that ChAT-immunoreactive cells were unaffected under our ischemic conditions and that MFA could maintain cell counts at control levels. These findings argue that the loss of CR and CB immunolabeling in ischemic retinas more likely reflected cell loss associated with GJ-mediated bystander cell death and not a downregulation of the protein markers.
In contrast to a role in secondary cell death, GJs have been reported to sometimes play a neuroprotective role (Andrade-Rozental et al., 2000; Decrock et al., 2009). Blockade of GJs or genetic ablation of certain connexins reduces neuronal loss in a rat model of brain trauma (Frantseva et al., 2002). Likewise, increased expression of Cx32 and Cx36 in the hippocampus after global brain ischemia has been suggested as an adapting mechanism to increase cell survivability (Oguro et al., 2001). In retina, focal lesions induced by laser coagulation produced a significantly higher loss of neurons in wild-type mice than in Cx36−/− mouse retinas (Striedinger et al., 2005). Although the results of this study are inconsistent with our present findings, it must be emphasized that the different insult conditions (trauma vs excitotoxicity and ischemia) were used. This raises the possibility that the role played by GJs in subserving cell death or neuroprotection may depend on the nature of the primary insult.
Our results revealed another important difference between GJs in terms of their role in secondary cell death promoted under different insult conditions. We found that, whereas genetic ablation of Cx36, but not Cx45, could significantly reduce cell loss under excitotoxic insult, ablation of Cx45, but not Cx36, protected cells from ischemic injury. These data indicate that different cohorts of GJs, dependent on their connexin makeup, subserve the bystander effect under different pathological conditions. Consistent with these findings, changes in the expression and function of certain connexin subtypes in CNS have been reported under a variety of pathological conditions (Rouach et al., 2002; Petrasch-Parwez et al., 2004; Eugenin et al., 2012; Kerr et al., 2012). However, to our knowledge, our results are the first to show that different cohorts of GJs, based on the connexin they express, subserve secondary cell death under different primary insult conditions. Our immunolabeling data suggest that this differential contribution of GJs under excitotoxic and ischemic conditions reflects changes in connexin protein expression and manifestation. Under ischemic insult, we found that Cx36 protein was accumulated mainly in a perinuclear region of RGCs, but the normal punctate immunolabeling in the IPL indicative of dendritic GJs was absent. A similar cytoplasmic internalization has been reported for Cx43 in ischemic cardiac tissue, resulting from a dysfunction in Cx43 trafficking linked to altered serine phosphorylation (Cone et al., 2014; Smyth et al., 2014). Thus, in our experiments, although Cx36 protein was still manufactured under ischemic conditions, it appears to have not been inserted in the membrane as functional GJs. In contrast, Cx45 punctate labeling in the IPL appeared normal. These findings can explain why ischemic cell loss was reduced in the Cx45−/− mouse retina, but not by ablation of Cx36, namely, that Cx36-expressing GJs were already disrupted by the insult. Oppositely, we found that Cx45 immunolabeling under excitotoxic insult was abnormal, with punctate labeling absent from the IPL, whereas Cx36 expression appeared normal. These data suggest a downregulation of Cx45 under excitotoxicity and a lack of functional GJs they constitute, which can explain why ablating Cx45 did not reduce cell loss after NMDA-induced excitotoxicity.
The finding that bystander cell death in retina is ultimately responsible for the loss of most retinal neurons suggests that GJs form a novel, important target for neuroprotection. GJs as a therapeutic target is lent support by our ability to significantly limit neuronal cell loss by blocking GJs with MFA administered after ischemic insult. Moreover, the finding that limited cohorts of GJs are responsible for bystander death is propitious, as it indicates that targeting of GJs can be selective and limited. Although available pharmacological blockers of GJs are largely nonspecific, the finding of blockers with greater connexin specificity (Srinivas et al., 2001; Cruikshank et al., 2004) holds promise for the creation of highly specific GJ blockers (Spray et al., 2002). Clearly, future work is called for to determine whether limited cohorts of GJs underlie secondary cell death associated with classic retinopathies, such as glaucoma, as well as similar degenerative disorders of the CNS.
Footnotes
This work was supported by National Institutes of Health Grant EY007360 to S.A.B. and Grant EY017832 to B.V. We thank Jacqueline A. Romero for assistance with some of the experiments.
The authors declare no competing financial interests.
References
- Andrade-Rozental AF, Rozental R, Hopperstad MG, Wu JK, Vrionis FD, Spray DC. Gap junctions: the “kiss of death” and the “kiss of life.”. Brain Res Rev. 2000;32:308–315. doi: 10.1016/S0165-0173(99)00099-5. [DOI] [PubMed] [Google Scholar]
- Belousov AB, Fontes JD. Neuronal gap junctions: making and breaking connections during development and injury. Trends Neurosci. 2013;36:227–236. doi: 10.1016/j.tins.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein SL, Guo Y. Changes in cholinergic amacrine cells after rodent anterior ischemic optic neuropathy (rAION) Invest Ophthalmol Vis Sci. 2011;52:904–910. doi: 10.1167/iovs.10-5247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship AG, Hamby AM, Firl A, Vyas S, Maxeiner S, Willecke K, Feller MB. The role of neuronal connexins 36 and 45 in shaping firing patterns in the developing retina. J Neurosci. 2011;31:9998–10008. doi: 10.1523/JNEUROSCI.5640-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomfield SA, Völgyi B. The diverse functional roles and regulation of neuronal gap junctions in the retina. Nat Rev Neurosci. 2009;10:495–506. doi: 10.1038/nrn2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Müller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Cone AC, Cavin G, Ambrosi C, Hakozaki H, Wu-Zhang AX, Kunkel MT, Newton AC, Sosinsky GE. Protein kinase co-mediated phosphorylation of connexin43 gap junction channels causes movement within gap junctions followed by vesicle internalization and protein degradation. J Biol Chem. 2014;289:8781–8798. doi: 10.1074/jbc.M113.533265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne L, Su J, Patten D, Halliwell RF. Characterization of the interaction between fenamates and hippocampal neuron GABA(A) receptors. Neurochem Int. 2007;51:440–446. doi: 10.1016/j.neuint.2007.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc Natl Acad Sci U S A. 2004;101:12364–12369. doi: 10.1073/pnas.0402044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusato K, Ripps H, Zakevicius J, Spray DC. Gap junctions remain open during cytochrome c-induced cell death: relationship of conductance to ‘bystander’ cell killing. Cell Death Differ. 2006;13:1707–1714. doi: 10.1038/sj.cdd.4401876. [DOI] [PubMed] [Google Scholar]
- Danesh-Meyer HV, Kerr NM, Zhang J, Eady EK, O'Carroll SJ, Nicholson LF, Johnson CS, Green CR. Connexin43 mimetic peptide reduces vascular leak and retinal ganglion cell death following retinal ischemia. Brain. 2012;135:506–520. doi: 10.1093/brain/awr338. [DOI] [PubMed] [Google Scholar]
- Das S, Lin D, Jena S, Shi A, Battina S, Hua DH, Allbaugh R, Takemoto DJ. Protection of retinal cells from ischemia by a novel gap junction inhibitor. Biochem Biophys Res Commun. 2008;373:504–508. doi: 10.1016/j.bbrc.2008.06.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deans MR, Volgyi B, Goodenough DA, Bloomfield SA, Paul DL. Connexin36 is essential for transmission of rod-mediated visual signals in the mammalian retina. Neuron. 2002;36:703–712. doi: 10.1016/S0896-6273(02)01046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JC, Corriveau RA, Belousov AB. Gap junctions are required for NMDA receptor dependent cell death in developing neurons. J Neurophysiol. 2007;98:2878–2886. doi: 10.1152/jn.00362.2007. [DOI] [PubMed] [Google Scholar]
- Decrock E, Vinken M, De Vuyst E, Krysko DV, D'Herde K, Vanhaecke T, Vandenabeele P, Rogiers V, Leybaert L. Connexin-related signaling in cell death: to live or let die? Cell Death Differ. 2009;16:524–536. doi: 10.1038/cdd.2008.196. [DOI] [PubMed] [Google Scholar]
- Dijk F, van Leeuwen S, Kamphuis W. Differential effects of ischemia/reperfusion on amacrine cell subtype-specific transcript levels in the rat retina. Brain Res. 2004;1026:194–204. doi: 10.1016/j.brainres.2004.08.034. [DOI] [PubMed] [Google Scholar]
- Eugenin EA, Basilio D, Sáez JC, Orellana JA, Raine CS, Bukauskas F, Bennett MV, Berman JW. The role of gap junction channels during physiologic and pathologic conditions of the human central nervous system. J Neuroimmune Pharmacol. 2012;7:499–518. doi: 10.1007/s11481-012-9352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantseva MV, Kokarovtseva L, Perez Velazquez JL. Ischemia-induced brain damage depends on specific gap-junctional coupling. J Cereb Blood Flow Metab. 2002;22:453–462. doi: 10.1097/00004647-200204000-00009. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Wassle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000;424:1–23. doi: 10.1002/1096-9861(20000814)424:1<1::AID-CNE1>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Hu EH, Bloomfield SA. Gap junctional coupling underlies the short latency spike synchrony of retinal alpha ganglion cells. J Neurosci. 2003;23:6768–6777. doi: 10.1523/JNEUROSCI.23-17-06768.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutnik CM, Pocrnich CE, Liu H, Laird DW, Shao Q. The protective effect of functional connexin43 channels on a human epithelial cell line exposed to oxidative stress. Invest Ophthalmol Vis Sci. 2008;49:800–806. doi: 10.1167/iovs.07-0717. [DOI] [PubMed] [Google Scholar]
- Kermer P, Klöcker N, Bähr M. Neuronal death after brain injury: models, mechanisms, and therapeutic strategies in vivo. Cell Tissue Res. 1999;298:383–395. doi: 10.1007/s004410050061. [DOI] [PubMed] [Google Scholar]
- Kerr NM, Johnson CS, Green CR, Danesh-Meyer HV. Gap junction protein connexin43 (GJA1) in the human glaucomatous optic nerve head and retina. J Clin Neurosci. 2011;18:102–108. doi: 10.1016/j.jocn.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Kerr NM, Johnson CS, Zhang J, Eady EK, Green CR, Danesh-Meyer HV. High pressure-induced retinal ischaemia reperfusion causes upregulation of gap junction protein connexin43 prior to retinal ganglion cell loss. Exp Neurol. 2012;234:144–152. doi: 10.1016/j.expneurol.2011.12.027. [DOI] [PubMed] [Google Scholar]
- Kielczewski JL, Pease ME, Quigley HA. The effect of experimental glaucoma and optic nerve transection on amacrine cells in the rat retina. Invest Ophthalmol Vis Sci. 2005;46:3188–3196. doi: 10.1167/iovs.05-0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranz K, Paquet-Durand F, Weiler R, Janssen-Bienhold U, Dedek K. Testing for a gap junction-mediated bystander effect in retinitis pigmentosa: secondary cone death is not altered by deletion of connexin36 from cones. PLoS One. 2013;8:e57163. doi: 10.1371/journal.pone.0057163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krysko DV, Leybaert L, Vandenabeele P, D'Herde K. Gap junctions and the propagation of cell survival and cell death signals. Apoptosis. 2005;10:459–469. doi: 10.1007/s10495-005-1875-2. [DOI] [PubMed] [Google Scholar]
- Lee JH, Shin JM, Shin YJ, Chun MH, Oh SJ. Immunochemical changes of calbindin, calretinin and SMI32 in ischemic retinas induced by increase of intraocular pressure and by middle cerebral artery occlusion. Anat Cell Biol. 2011;44:25–34. doi: 10.5115/acb.2011.44.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NP, Leung KW, Wo JY, Tam PC, Yeung WS, Luk JM. Blockage of testicular connexins induced apoptosis in rat seminiferous epithelium. Apoptosis. 2006;11:1215–1229. doi: 10.1007/s10495-006-6981-2. [DOI] [PubMed] [Google Scholar]
- Lei Y, Garrahan N, Hermann B, Fautsch MP, Johnson DH, Hernandez MR, Boulton M, Morgan JE. Topography of neuron loss in the retinal ganglion cell layer in human glaucoma. Br J Ophthalmol. 2009;93:1676–1679. doi: 10.1136/bjo.2009.159210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkovitch-Verbin H, Quigley HA, Kerrigan-Baumrind LA, D'Anna SA, Kerrigan D, Pease ME. Optic nerve transection in monkeys may result in secondary degeneration of retinal ganglion cells. Invest Ophthalmol Vis Sci. 2001;42:975–982. [PubMed] [Google Scholar]
- Li X, Kamasawa N, Ciolofan C, Olson CO, Lu S, Davidson KG, Yasumura T, Shigemoto R, Rash JE, Nagy JI. Connexin45-containing neuronal gap junctions in rodent retina also contain connexin36 in both apposing hemiplaques, forming bihomotypic gap junctions, with scaffolding contributed by zonula occludens-1. J Neurosci. 2008;28:9769–9789. doi: 10.1523/JNEUROSCI.2137-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D, Shanks D, Prakash O, Takemoto DJ. Protein kinase C gamma mutations in the C1B domain cause caspase3-linked apoptosis in lens epithelial cells through gap junctions. Exp Eye Res. 2007;85:113–122. doi: 10.1016/j.exer.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone P, Miao H, Parker A, Juarez S, Hernandez MR. Pressure induces loss of gap junction communication and redistribution of connexin 43 in astrocytes. Glia. 2007;55:1085–1098. doi: 10.1002/glia.20527. [DOI] [PubMed] [Google Scholar]
- Moon JI, Kim IB, Gwon JS, Park MH, Kang TH, Lim EJ, Choi KR, Chun MH. Changes in retinal neuronal populations in the DBA/2J mouse. Cell Tissue Res. 2005;320:51–59. doi: 10.1007/s00441-004-1062-8. [DOI] [PubMed] [Google Scholar]
- Naus CC, Ozog MA, Bechberger JF, Nakase T. A neuroprotective role for gap junctions. Cell Commun Adhes. 2001;8:325–328. doi: 10.3109/15419060109080747. [DOI] [PubMed] [Google Scholar]
- Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, Grooms SY, Bennett MV, Zukin RS. Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci. 2001;21:7534–7542. doi: 10.1523/JNEUROSCI.21-19-07534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne NN, Casson RJ, Wood JP, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Prog Retin Eye Res. 2004;23:91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Pan F, Paul DL, Bloomfield SA, Völgyi B. Connexin36 is required for gap junctional coupling of most ganglion cell subtypes in the mouse retina. J Comp Neurol. 2010;518:911–927. doi: 10.1002/cne.22254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Paul DL, Wu SM. Survey on amacrine cells coupling to retrograde-identified ganglion cells in the mouse retina. Invest Ophthalmol Vis Sci. 2013;54:5151–5162. doi: 10.1167/iovs.13-11774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez Velazquez JL, Frantseva MV, Naus CC. Gap junctions and neuronal injury: protectants or executioners? Neuroscientist. 2003;9:5–9. doi: 10.1177/1073858402239586. [DOI] [PubMed] [Google Scholar]
- Petrasch-Parwez E, Habbes HW, Weickert S, Löbbecke-Schumacher M, Striedinger K, Wieczorek S, Dermietzel R, Epplen JT. Fine-structural analysis and connexin expression in the retina of a transgenic model of Huntington's disease. J Comp Neurol. 2004;479:181–197. doi: 10.1002/cne.20327. [DOI] [PubMed] [Google Scholar]
- Ripps H. Cell death in retinitis pigmentosa: gap junctions and the ‘bystander’ effect. Exp Eye Res. 2002;74:327–336. doi: 10.1006/exer.2002.1155. [DOI] [PubMed] [Google Scholar]
- Rouach N, Avignone E, Même W, Koulakoff A, Venance L, Blomstrand F, Giaume C. Gap junctions and connexin expression in the normal and pathological central nervous system. Biol Cell. 2002;94:457–475. doi: 10.1016/S0248-4900(02)00016-3. [DOI] [PubMed] [Google Scholar]
- Schwartz M. Neuroprotection as a treatment for glaucoma: pharmacological and immunological approaches. Eur J Ophthalmol Suppl. 2003;3:S27–S31. doi: 10.1177/112067210301303s05. [DOI] [PubMed] [Google Scholar]
- Smyth JW, Zhang SS, Sanchez JM, Lamouille S, Vogan JM, Hesketh GG, Hong T, Tomaselli GF, Shaw RM. A 14-3-3 mode-1 binding motif initiates gap junction internalization during acute cardiac ischemia. Traffic. 2014;15:684–699. doi: 10.1111/tra.12169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spray DC, Rozental R, Srinivas M. Prospects for rational development of pharmacological gap junction channel blockers. Curr Drug Targets. 2002;3:455–464. doi: 10.2174/1389450023347353. [DOI] [PubMed] [Google Scholar]
- Srinivas M, Hopperstad MG, Spray DC. Quinine blocks specific gap junction channel subtypes. Proc Natl Acad Sci U S A. 2001;98:10942–10947. doi: 10.1073/pnas.191206198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striedinger K, Petrasch-Parwez E, Zoidl G, Napirei M, Meier C, Eysel UT, Dermietzel R. Loss of connexin36 increases retinal cell vulnerability to secondary cell loss. Eur J Neurosci. 2005;22:605–616. doi: 10.1111/j.1460-9568.2005.04228.x. [DOI] [PubMed] [Google Scholar]
- Vander S, Levkovitch-Verbin H. Regulation of cell death and survival pathways in secondary degeneration of the optic nerve: a long-term study. Curr Eye Res. 2012;37:740–748. doi: 10.3109/02713683.2012.673679. [DOI] [PubMed] [Google Scholar]
- Varella MH, de Mello FG, Linden R. Evidence for an antiapoptotic role of dopamine in developing retinal tissue. J Neurochem. 1999;73:485–492. doi: 10.1046/j.1471-4159.1999.0730485.x. [DOI] [PubMed] [Google Scholar]
- Voigt T. Cholinergic amacrine cells in the rat retina. J Comp Neurol. 1986;248:19–35. doi: 10.1002/cne.902480103. [DOI] [PubMed] [Google Scholar]
- Völgyi B, Chheda S, Bloomfield SA. Tracer coupling pattern of the ganglion cells subtypes in the mouse retina. J Comp Neurol. 2009;512:664–687. doi: 10.1002/cne.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Denisova JV, Kang KS, Fontes JD, Zhu BT, Belousov AB. Neuronal gap junctions are required for NMDA receptor mediated excitotoxicity: implications in ischemic stroke. J Neurophysiol. 2010;104:3551–3556. doi: 10.1152/jn.00656.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin D, Bloomfield SA. Tracer coupling pattern of amacrine and ganglion cells in the rabbit retina. J Comp Neurol. 1997;383:512–528. doi: 10.1002/(SICI)1096-9861(19970714)383:4<512::AID-CNE8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]