

Abstract
Motoneuron death after transection of the axons (axotomy) in neonates is believed to share the same mechanistic bases as naturally occurring programmed cell death during development. The c-Jun N-terminal kinase pathway is activated in both forms of motoneuron death, but it remains unknown to what extent these two forms of motoneuron death depend on this pathway and which upstream kinases are involved. We found that numbers of facial motoneurons are doubled in neonatal mice deficient in either ZPK/DLK (zipper protein kinase, also known as dual leucine zipper kinase), a mitogen-activated protein kinase kinase kinase, or in MKK4/MAP2K4, a mitogen-activated protein kinase kinase directly downstream of ZPK/DLK, and that the facial motoneurons in those mutant mice are completely resistant to axotomy-induced death. Conditional deletion of MKK4/MAP2K4 in neurons further suggested that ZPK/DLK and MKK4/MAP2K4-dependent mechanisms underlying axotomy-induced death are motoneuron autonomous. Nevertheless, quantitative analysis of facial motoneurons during embryogenesis revealed that both ZPK/DLK and MKK4/MAP2K4-dependent and -independent mechanisms contribute to developmental elimination of excess motoneurons. In contrast to MKK4/MAP2K4, mice lacking MKK7/MAP2K7, another mitogen-activated protein kinase kinase directly downstream of ZPK/DLK, conditionally in neurons did not have excess facial motoneurons. However, some MKK7/MAP2K7-deficient facial motoneurons were resistant to axotomy-induced death, indicating a synergistic effect of MKK7/MAP2K7 on axotomy-induced death of these facial motoneurons. Together, our study provides compelling evidence for the pivotal roles of the ZPK/DLK and MKK4/MAP2K4-dependent mechanism in axotomy-induced motoneuron death in neonates and also demonstrates that axotomy-induced motoneuron death is not identical to developmental motoneuron death with respect to the involvement of ZPK/DLK, MKK4/MAP2K4 and MKK7/MAP2K7.
Keywords: injury response, mitogen-activated protein kinase, programmed cell death
Introduction
Injury responses of motoneurons (MNs) after transection of their axons (axotomy) vary depending on their maturational stages and MN subsets, and the site and type of axonal injury. In general, axotomized MNs in early neonates undergo acute degenerative changes, including substantial neuronal death (Snider et al., 1992), whereas a large proportion of them survive and regenerate axons in adults unless axons are transected in close proximity to the cell body (Koliatsos et al., 1994; Li et al., 1995). The mechanistic basis of this axotomy-induced MN death in neonates is believed to be analogous to that of naturally occurring programmed cell death (NPCD), a physiological process during development that establishes quantitative neuron–target matching by elimination of excess neurons (Buss et al., 2006). When developing MNs are susceptible to NPCD, the MNs that fail to establish axonal contact with target skeletal muscle fibers undergo apoptosis as a result of lack of muscle-derived trophic supports, and loss of axonal contact to muscles by axotomy in neonates thereby results in the same type of MN death as NPCD (Lowrie and Vrbová, 1992). In line with this, the c-Jun N-terminal kinase (JNK, also known as stress-activated protein kinase) pathway is activated in MNs during NPCD (Sun et al., 2005; Ribera et al., 2007), as well as after axotomy in neonates (Casanovas et al., 2001; Keramaris et al., 2005; Kanungo et al., 2008). Although these downstream molecular events leading to apoptosis of axotomized neonatal MNs and NPCD have been studied intensively, it remains to be elucidated which upstream kinases in the JNK pathway are involved in both types of MN death and, moreover, to what extent axotomy-induced death of neonatal MNs and NPCD do share the same upstream signaling molecules. In fact, some studies have suggested contributions of excitotoxic mechanisms to neuronal death after axonal injury (Lawson and Lowrie, 1998; Dekkers et al., 2004) and of death receptor-mediated mechanisms to NPCD (Haase et al., 2008).
Our recent finding that ZPK (also known as DLK, MUK, and MAP3K12; ZPK/DLK hereafter), one of the mitogen-activated protein kinase (MAPK) kinase kinase in the JNK pathway, is a key mediator of MN NPCD (Itoh et al., 2011) prompted us to test the hypothesis that ZPK/DLK-deficient MNs will remain viable even after axotomy if NPCD and axotomy-induced death of neonatal MNs indeed share ZPK/DLK in the cell-death pathway. Axotomy-induced death of MNs has been studied intensively particularly in the unilateral facial nerve axotomy model using neonatal rodents, primarily attributable to its advantages for quantitative analysis (Moran and Graeber, 2004). Here, using a unilateral facial nerve axotomy model, we demonstrate that, to our surprise, neonatal facial MNs deficient in either ZPK/DLK or MKK4/MAP2K4, a MAPK kinase (MAP2K) directly downstream of ZPK/DLK, are completely resistant against axotomy-induced death, indicating that ZPK/DLK and MKK4/MAP2K4 form the critical gateway to activation of the downstream molecules responsible for execution of the cell-death program in axotomized neonatal MNs. In contrast, developmental elimination of excess facial MNs is a rather complex process that includes both ZPK/DLK- and MKK4/MAP2K4-dependent and -independent mechanisms.
Materials and Methods
Animals.
ZPK/DLK-deficient mice were derived from the gene-trap embryonic stem cell clone RRN366 (B6;129P2-Map3k12Gt(RRN366)Byg/Mmucd, RRID:IMSR_MMRRC:030151) as reported in detail in a previous study (Itoh et al., 2009). MKK4/MAP2K4-flox mice (Map2k4tm1Ctr, Research Resource Identifier (RRID): MGI_MGI:3769121) and MKK7/MAP2K7-flox mice (Map2k7tm1.1Twad, RRID:MGI_MGI:4948959) were generated by C. Tournier's laboratory and J. M. Penninger's laboratory, respectively, as reported previously (Wang et al., 2007b; Schramek et al., 2011). Some of those mice were crossed onto the nestin promoter-driven and the synapsin-1 promoter-driven Cre recombinase transgenic mice from The Jackson Laboratory [Tg(nes::cre), RRID:IMSR_JAX:003771 and Tg(syn1::cre), RRID:IMSR_JAX:012687, respectively]. Animals were housed in standard laboratory cages with access to food and water ad libitum and maintained under 12 h light/dark cycles. To obtain embryos, timed pregnant females were used. All experiments using the animals were performed in conformity with protocols approved by the Institutional Animal Care and Use Committee of the University of California, Davis.
Facial nerve axotomy.
The main branch of the left facial nerve was transected at the site beneath the external acoustic meatus (refer to Fig. 6A) at postnatal day 2 (P2) under hypothermia anesthesia, and an ∼2-mm-long segment of the distal nerve end was resected to inhibit regeneration. The left retroauricular branch of the facial nerve and the right facial nerve were left intact. The incision was closed with DERMABOND, and the pups were allowed to recover on a heated pad before being returned to the dams. Mice were deeply anesthetized and perfused intracardially with PBS, immediately followed by 4% (w/v) paraformaldehyde in PBS for tissue collection at 24 h and 5 d after axotomy.
Figure 6.

Facial MNs are totally resistant against axotomy-induced death in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient neonates. A, Transection site of left facial nerve (scissors icons). Top is lateral view (M, external acoustic meatus; RA Br, retroauricular branch). Bottom is transverse sectional view. The path of facial motor axons from the facial nucleus (N VII) to the facial nerve (FN) is delineated in the coronal section of the brainstem (Genu, genu of the facial nerve; IV, fourth ventricle). The arrow in the top indicates the position of the distal nerve segment examined in Figure 11. B, C, Representative immunohistochemical images of the facial nuclei from ZPK/DLK-deficient mice (tp/tp) and wild-type littermates (+/+) (B) and from CNS MKK4/MAP2K4-deficient mice and littermate controls (C) at 5 d after axotomy at P2 (P7). Nuclei were counterstained with DAPI (blue). The MNs in the VM subnucleus remained viable on the transected side of wild-type mice (delineated by a dotted circle in B). D, Quantitative results of B and C. Three animals per genotype were examined. **p < 0.01 and not significant (NS) compared with the intact side (unpaired t test). Scale bars, 100 μm.
Immunohistochemistry.
CNS tissues were processed for immunohistochemistry as reported previously (Itoh et al., 2011). Six- and 12-μm-thick tissue sections were prepared by cryostat (CM1950; Leica), and 75-μm-thick sections were by vibratome (VT 100S; Leica). Six and 12 μm sections were blocked in the blocking solution [0.4% (w/w) Triton X-100, 10% (v/v) donkey serum, 15 mm HEPES, and 0.02% (w/v) sodium azide in 1× minimum essential medium without bicarbonate] and then incubated with primary antibodies in the blocking solution overnight at 4°C. The primary antibodies used for immunohistochemistry are listed in Table 1. The sections were washed with PBS, incubated for 1 h in appropriate secondary antibodies conjugated with rhodamine, DyLight 488, or DyLight 549 (1:500; Jackson ImmunoResearch) at room temperature, and mounted with ProLong Gold antifade reagent with 4,6-diamidino-2-phenylindole (DAPI; Invitrogen). Thick sections were processed as whole mounts in situ after fixation in 4% (w/v) paraformaldehyde in PBS at 4°C overnight. The sections were incubated in 0.1 m glycine in PBS, pH 7.3, for 30 min at room temperature, blocked in the blocking solution for 1 h, incubated in 100% methanol at −20°C for 7 min for permeabilization, rinsed three times in 0.5% Triton X-100 in PBS, and then incubated with the primary antibodies at 4°C overnight. The tissues were rinsed three times in 0.5% Triton X-100 in PBS, incubated with secondary antibodies at 4°C overnight, and mounted on slide glasses after rinsing in 0.5% Triton X-100 in PBS.
Table 1.
Primary antibodies used in this study
| Antibody name | Application | Citation data |
|---|---|---|
| Mouse anti-MKK4 monoclonal | IB | BD Biosciences catalog #554105, RRID:AB_395242 |
| Rabbit anti-MKK7 | IB | Cell Signaling Technology catalog #4172, RRID:AB_330914 |
| Rabbit anti-MAP3K12 | IB | Sigma-Aldrich catalog #HPA039936, RRID:AB_10795239 |
| Mouse anti-β-actin monoclonal | IB | Cell Signaling Technology catalog #3700S, RRID:AB_2242334 |
| Rabbit anti-β-actin monoclonal | IB | Cell Signaling Technology catalog #4970, RRID:AB_2223172 |
| Goat anti-ChAT | IHC | Millipore catalog #AB144, RRID:AB_90650 |
| Mouse anti-NeuN monoclonal | IHC | Millipore catalog #MAB377, RRID:AB_2298772 |
| Mouse anti-Islet-1,2 monoclonal | IHC | Developmental Studies Hybridoma Bank catalog #39.4D5, RRID:AB_528173 |
| Rabbit anti-cleaved Caspase-3 (Asp175) monoclonal | IHC | Cell Signaling Technology catalog #9664S, RRID:AB_331453 |
| Rabbit anti-phospho-c-Jun (Ser63) | IHC | Cell Signaling Technology catalog #9261S, RRID:AB_2130162 |
| Rabbit anti-Iba1 | IHC | Wako Chemicals catalog #019-19741, RRID:AB_839504 |
| Rabbit anti-neurofilament 200 | IHC | Sigma-Aldrich catalog #N4142, RRID:AB_477272 |
| Rat anti-myelin basic protein | IHC | Novus Biologicals catalog #NB600-717, RRID:AB_2139899 |
IB, Immunoblotting; IHC, immunohistochemistry.
Quantitative analysis of MNs and myelinated axons.
Consecutive 6-μm-thick coronal cryosections of the brainstem were prepared throughout the rostrocaudal extent of the facial motor nucleus, and every seventh section was immunostained for choline acetyltransferase (ChAT) and NeuN. Digitized fluorescent images were captured by a fluorescent microscope equipped with the Desk Scanning Unit (BX61; Olympus) and imaging software (Slidebox; Olympus), and ChAT and NeuN double-positive cells with a nucleus containing a distinct nucleolus were counted in each section. A total number of facial MNs in each facial motor nucleus was calculated according to the method by Clarke and Oppenheim (1995). Myelinated axons were counted in semithin sections as described previously (Itoh et al., 2011). At least three mice in each genotype were used for quantitative analysis.
Retrograde labeling with lysinated tetramethylrhodamine dextran.
Lysinated tetramethylrhodamine-dextran (fluoro-ruby) (4% in 0.9% sodium chloride; 0.8 μl total volume; Invitrogen), a retrograde tracer, was injected into the space adjacent to the proximal end of the axotomized nerve before closing the skin incision. At 24 h after axotomy, the animals were processed for immunohistochemistry as described above.
Immunoblotting.
Dissected tissues of the ventral brainstem containing the facial nucleus were homogenized in the lysis buffer, and immunoblotting was performed as reported previously (Horiuchi et al., 2006) except for the following. After incubation with the secondary antibodies, IRdye 800CW and IRDye 680RD (1:1000; LI-COR), immunoreactive signals were detected by Odyssey CLx (LI-COR). The primary antibodies used for immunoblotting are listed in Table 1.
Statistics.
Data are presented as means ± 1 SD of at least triplicate experiments. Welch's unpaired t test for unequal variances was used to determine p values between the two experimental groups. Otherwise, p values were calculated by ANOVA, followed by the Bonferroni's/Dunn's post hoc test. The significance level was set at p = 0.05.
Results
Numbers of facial MNs are doubled in ZPK/DLK-deficient and Nestin–Cre homozygous floxed MKK4/MAP2K4 mice
Facial MNs are a subset of branchial (pharyngeal) MNs in the brainstem (Chandrasekhar, 2004) and, unlike spinal cord MNs, are not dependent on homeodomain factor HB9 for their specification to MNs (Arber et al., 1999; Thaler et al., 1999). However, consistent with our previous finding of excess MNs in the spinal cord of ZPK/DLK-deficient (zpk/dlktp/tp) mice (Itoh et al., 2011), ZPK/DLK-deficient mice had twofold more ChAT-positive facial MNs than wild-type mice. The numbers of myelinated axons were also increased in the intracranial portion of the facial nerve (intracranial) and the extracranial main branch of the facial nerve immediately beneath the external acoustic meatus (extracranial, the site of axon injury; see below) in proportion to the increased numbers of facial MNs. The difference in axon numbers between the intracranial portion and the extracranial main branch corresponds to the axon numbers in the retroauricular branch, which bifurcates from the main branch at the exit of the stylomastoid foramen. MKK4/MAP2K4 and MKK7/MAP2K7 are known as substrates of ZPK/DLK at the MAP2K level in the JNK pathway (Hirai et al., 1997; Merritt et al., 1999; Xu et al., 2001). To explore MAP2Ks downstream of ZPK/DLK in quantitative control of MNs, we next examined the mice deficient in MKK4/MAP2K4 in the CNS [Tg(nes::cre)+/− mkk4/map2k4f/f, CNS MKK4/MAP2K4-deficient mice thereafter; Wang et al., 2007b], because constitutive deletion of either MKK4/MAP2K4 or MKK7/MAP2K7 results in early embryonic lethality (Ganiatsas et al., 1998; Wada et al., 2004). CNS MKK4/MAP2K4-deficient mice of 3 weeks of age demonstrated an almost identical increase in numbers of facial and spinal MNs and their axons to those observed in ZPK/DLK-deficient mice (Figs. 1, 2). As far as could be determined by immunoblots of the ventral part of the brainstem including the facial nuclei, ZPK/DLK protein was not decreased but rather slightly increased in CNS MKK4/MAP2K4-deficient mice, and MKK7/MAP2K7 was expressed at similar levels in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice (Fig. 1E). These results indicate that this phenotype is not a consequence of secondary reduction of ZPK/DLK in CNS MKK4/MAP2K4-deficient mice and vice versa and that MKK7/MAP2K7 cannot compensate for the functional roles of MKK4/MAP2K4 in this phenotype.
Figure 1.

ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice have approximately twofold more facial MNs and facial nerve axons than control littermates. A, Representative confocal scanning images of the facial nuclei of ZPK/DLK-deficient (tp/tp) mice and wild-type littermates (+/+) at P21 and those of heterozygous (+/flox) and homozygous (flox/flox) floxed MKK4/MAP2K4 Nestin–Cre [Tg(Nes::Cre)+/−] transgenic mice at P17. Facial MNs were identified as both ChAT- and NeuN-positive cells. B, Semithin cross-sections (500-nm-thick) of the extracranial portions of the facial nerves (the site of transection; see Fig. 6A) from the mice corresponding to A. C, Quantitative results of facial MNs in A. D, Myelinated axon numbers of the intracranial and extracranial portions of the facial nerves from the same groups of mice as those of B. The axons of visceral MNs in the intracranial portion of the facial nerve were not counted. E, Immunoblots for ZPK/DLK, MKK4/MAP2K4, and MKK7/MAP2K7 to demonstrate the protein levels of the ventral part of the brainstem containing the facial nuclei. One hundred micrograms of total proteins from a single P7 mouse of each genotype were applied to each lane. For some genotypes, two independent samples are shown to demonstrate the reproducibility. ACTB (beta-actin) was used as a loading control. **p < 0.01 compared with wild-type or control mice (unpaired t test), and at least three mice were used for quantitative analysis. Scale bars, 100 μm.
Figure 2.

Spinal MNs are doubled in CNS MKK4/MAP2K4-deficient mice. A, Representative confocal images of the fourth cervical (C4), fourth thoracic (T4), and fourth lumbar (L4) spinal segments from CNS MKK4/MAP2K4-deficient mice [Mkk4 flox/flox Tg(Nes::Cre)+/−] and control littermates [Mkk4 +/flox Tg(Nes::Cre)+/−] at P17. Cross-sections were immunolabeled for ChAT (red) and NeuN (green) to visualize MNs as a ChAT-positive neuronal subset (yellow to red). B, Quantitative analysis of MNs in the C4, T4, and L4 spinal segments from the mice of the genotypes as shown in A. MNs with visible nucleoli were counted in at least nine 6-μm-thick cross-sections per animal, and the average number of MNs per section of the anterior horn on one side was calculated. **p < 0.01 compared with littermate control (unpaired t test), and at least three mice were used for quantitative analysis. Scale bar, 100 μm.
ZPK/DLK- and MKK4/MAP2K4-dependent and -independent mechanisms contribute to the quantitative control of developing facial MNs
Taking advantage of facial MNs for quantitative analyses, we investigated the elimination of excess facial MNs in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice during development. In mice, cells committed to facial MNs appear within a column on either side of the floor plate, occupying the fourth rhombomere (r4) in the hindbrain and, by embryonic day 10.5 (E10.5), start migrating caudally into the rostral r6, forming the facial nucleus close to the pial surface and leaving their axons behind. These developing MNs forming the facial nuclei are identified as cells positive for Islet-1 (Ericson et al., 1992; Hafezparast et al., 2003). Aberrant migration of facial MNs was observed in homozygous embryonic mice carrying a point mutation in the cytoplasmic dynein heavy chain 1 (Dnchc1) gene (Loa) (Hafezparast et al., 2003). Moreover, ZPK/DLK and MKK4/MAP2K4 are essential for normal radial migration of cerebral cortical neurons (Hirai et al., 2002; Wang et al., 2007b). However, no ectopic facial nuclei were observed in consecutive sections of the brainstem from either ZPK/DLK-deficient or CNS MKK4/MAP2K4-deficient embryos. Most of developing Islet-1,2-positive facial MNs complete migration and settle down in the facial nucleus by approximately E14.5. We therefore quantified Islet-1,2-positive MNs and cleaved Caspase-3-positive apoptotic cells in the facial nuclei of ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient embryos from E14.5 to E18.5. Both mutant embryos already had significantly more Islet-1,2-positive MNs than control littermate embryos at E14.5 (Fig. 3). Two interpretations are possible for this result; more facial MNs are generated in these mutants than in controls and/or less facial MNs are eliminated before E14.5. The number of cleaved Caspase-3-positive cells was also greater in wild-type embryos than in ZPK/DLK-deficient embryos at E14.5, supporting the latter possibility, whereas this difference in apoptotic cells between CNS MKK4/MAP2K4-deficient embryos and littermate controls at E14.5 did not reach a statistical significance (Fig. 3B). We were unable to validate the former possibility, because cleaved Caspase-3-positive cells were already found even in the migratory flow of facial MNs at E13.5 before they reached the facial nucleus (Fig. 4). At E17.5, apoptotic cells further increased significantly in control embryos, which corresponds to NPCD of facial MNs (Ashwell and Watson, 1983) but not in both mutant embryos (Fig. 3B). This diminished NPCD as a result of loss of functional ZPK/DLK and MKK4/MAP2K4 was indeed associated with reduced activation of c-Jun in the nuclei of developing facial MNs (Fig. 5). Importantly, even in the absence of ZPK/DLK or MKK4/MAP2K4, cleaved Caspase-3-positive cells were constantly observed with a gradual but significant decrease in total numbers of facial MNs from E14.5 to E18.5. Although more facial MNs were eliminated in CNS MKK4/MAP2K4-deficient embryos than in ZPK/DLK-deficient embryos from E16.5 to E18.5, total numbers of facial MNs further decreased to similar values in both mutants by P7 (compare the numbers of facial MNs in E18.5 mutant embryos in Fig. 3B and those in P7 mutant pups on the intact side in Fig. 6D). We searched the facial nuclei of these P7 mutants for NeuN-positive but ChAT-negative neurons that might have escaped NPCD but failed to mature to become ChAT-positive neurons. However, as shown in Figure 6, B and C (the nuclei on the intact side), NeuN-positive, ChAT-negative neurons were only occasionally found in the facial nuclei of mutants and their littermate controls and never accumulated in mutants. Given that apoptotic cells and phosphorylated c-Jun-positive cells were still found in the facial nuclei of both ZPK/DLK-deficient and MKK4/MAP2K4-deficient embryos, it is quite likely that ∼2500 developing facial MNs are eliminated by apoptosis between E16.5 and P7 even in these mutants. Together, we found that there are two distinct mechanisms underlying quantitative control of developing facial MNs: ZPK/DLK- and MKK4/MAP2K4-dependent and ZPK/DLK- and MKK4/MAP2K4-independent mechanisms. It should be emphasized that, despite the reduced NPCD in the embryos lacking ZPK/DLK or MKK4/MAP2K4, the ZPK/DLK- and MKK4/MAP2K4-dependent mechanism contributes predominantly to quantitative control of developing facial MNs before E16.5, before the time window of their NPCD.
Figure 3.

ZPK/DLK- and MKK4/MAP2K4-dependent mechanism and ZPK/DLK- and MKK4/MAP2K4-independent mechanism contribute to elimination of excess facial MNs during development. A, ZPK/DLK-deficient (zpk/dlktp/tp) and CNS MKK4/MAP2K4-deficient [mkk4flox/flox Tg(nes::cre)+/−] embryos and their littermate controls were obtained from timed-pregnant mice, and serial cross-sections of the brainstem were double immunolabeled for Islet-1,2 and cleaved Caspase-3 (arrows and arrowheads). Facial nuclei are delineated by a dotted circle. Representative cleaved Caspase-3-positive cells (arrowheads in low-power micrographs) and their nuclei are magnified in color and inverted insets, respectively. Scale bars: 100 μm; insets, 10 μm. B, Quantitative results of facial MNs and cleaved Caspase-3-positive cells of ZPK/DLK-deficient embryos (tp/tp), wild-type littermates (+/+), and those of heterozygous (+/flox) and homozygous (flox/flox) floxed MKK4/MAP2K4 Nestin–Cre [Tg(Nes::Cre)+/−] embryos from E14.5 to E18.5. *p < 0.05, ***p < 0.001, and not significant (NS) compared with the indicated pair or with littermate controls (unpaired t test). At least four facial nuclei were counted in each dataset.
Figure 4.
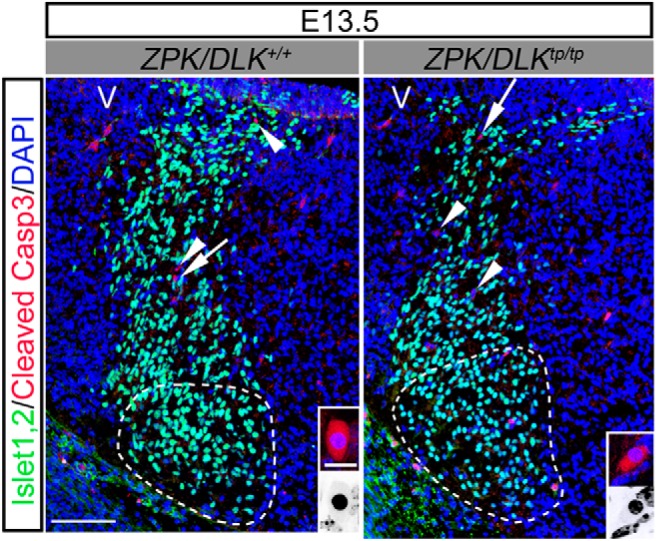
Apoptotic cells are observed in the migratory flow of Islet-1,2-positive developing MNs from their birthplace to the facial nucleus (delineated by a dotted circle) in E13.5 ZPK/DLK-deficient embryos (tp/tp) and littermate controls (+/+). Cross-sections of the brainstem were double immunolabeled for Islet-1,2 (green) and cleaved Caspase-3 (red, arrows, and arrowheads). Representative cleaved Caspase-3-positive cells (arrowheads in low-power micrographs) and their nuclei are magnified in color and inverted insets, respectively. V, Ventricular side. Scale bars: 100 μm; insets, 10 μm.
Figure 5.

c-Jun activation in the nuclei of developing facial MNs is significantly reduced in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient embryos at E17.5. A, Representative micrographs of the facial nuclei double immunolabeled for phospho-Ser63–c-JUN and Islet-1,2 in ZPK/DLK-deficient (zpk/dlktp/tp) and CNS MKK4/MAP2K4-deficient [mkk4flox/flox Tg(cre)+/−] embryos and their littermate controls at E17.5. Scale bars, 100 μm. B, Representative frequency histograms of the immunoreactive signals for nuclear phospho-Ser63–c-JUN in all MNs in a single cross-section of the facial nucleus from the embryos of indicated genotypes. Nuclei of facial MNs were gated by immunoreactivity for Islet-1,2 in the fluorescence micrographs, and averaged signal level of the fluorescence for phospho-c-Jun in each Islet-1,2-positive nucleus was calculated. Facial MNs positive for nuclear phospho-c-Jun were counted by the gating strategy indicated in the histograms. C, Percentages of nuclear phospho-c-Jun-positive MNs in ZPK/DLK-deficient (tp/tp) and CNS MKK4/MAP2K4-deficient (f/f) embryos compared with their littermate controls. Each dataset was from at least three embryos, and >1000 MNs were examined in each embryo. *p < 0.05, unpaired t test.
Facial MNs in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient neonatal mice are virtually resistant against axotomy-induced death
To determine contributions of ZPK/DLK and MKK4/MAP2K4 to axotomy-induced death of neonatal MNs, we performed unilateral facial nerve axotomy in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient P2 mice and quantified surviving facial MNs at 5 d after axotomy (P7). We transected the main branch of facial nerve immediately beneath the external acoustic meatus, thus preserving the retroauricular branch that consists of the axons originating from the ventromedial (VM) facial subnucleus (Fig. 6A), so that the intact MNs in the VM subnucleus serve as an additional internal control on the ipsilateral side (Olsson and Kristensson, 1979; Dubois-Dauphin et al., 1994). Five days was chosen as the longest interval after axotomy, because both ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice demonstrate severe growth retardation after birth even without any experimental insults and usually die before the time of weaning (Wang et al., 2007b; Itoh et al., 2009). At P7, ChAT-positive facial MNs were almost completely absent in the wild-type facial nuclei on the transected side save for the VM subnuclei. In clear contrast, almost all facial MNs of ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice were still viable without obvious degenerative changes and expressed ChAT at levels similar to those on the intact side even at 5 d after axotomy. Quantitative analysis confirmed that there was no statistical difference in numbers of surviving MNs between the transected and intact sides in these mutant mice (Fig. 6). In wild-type neonatal MNs, accumulation of phosphorylated c-Jun in the nucleus increased within 24 h after axotomy, and cleavage of Caspase-3 reached a peak at ∼24 h after axotomy. We confirmed that these events were virtually absent in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice after axotomy (Fig. 7). This experiment also revealed that cytosolic immunoreactivity for ChAT in facial MNs on the transected side was rapidly downregulated in control mice at 24 h after axotomy, whereas it remained unchanged in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice. Although the numbers of facial motor axons at the transection site correlated well with the increased numbers of facial MNs in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice, it remained to be verified that these excess facial MNs indeed sent their axons at least through the site of axonal injury. We used retrograde labeling with fluoro-ruby (Invitrogen) after axotomy to identify axotomized MNs and to assess neuronal ability to retrogradely transport fluoro-ruby from the transected axonal end to the cell body in the absence of ZPK/DLK or MKK4/MAP2K4. At 24 h after axotomy, almost all facial MNs, except for most MNs in the VM and dorsal accessory (DA) subnuclei, were labeled with fluoro-ruby on the transected side in both ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice (Fig. 8), indicating that the molecular mechanisms underlying retrograde transport of fluoro-ruby are not functionally defective in the absence of ZPK/DLK or MKK4/MAP2K4. In addition, accumulation of microglia in the facial nucleus on the transected side was not observed in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice (Fig. 9).
Figure 7.

Accumulation of activated c-Jun in the nucleus, cleavage of Caspase-3, and acute reduction in ChAT immunoreactivity did not occur after axotomy in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient facial MNs. A, Double immunohistochemistry for phospho-Ser63 of c-Jun (red) and ChAT (green) in the facial nuclei from ZPK/DLK-deficient mice (tp/tp) and wild-type littermates (+/+) and from CNS MKK4/MAP2K4-deficient mice (f/f) and littermate controls (+/f) at 24 h after axotomy at P2. All nuclei were visualized with DAPI (blue). Importantly, ChAT immunoreactivity was not reduced in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient MNs on the transected side compared with the corresponding controls. B, Quantitative analysis of nuclear phospho-Ser63–c-Jun-positive MNs in the facial nuclei of ZPK/DLK-deficient mice (tp/tp) and wild-type littermates (+/+) and those of CNS MKK4/MAP2K4-deficient mice (f/f) and littermate controls (+/f) on the transected and intact sides at 24 h after axotomy. C, Cleaved Caspase-3 (Cleaved Casp3, red)-positive MNs (arrows) were not increased in the ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient MNs at 24 h after axotomy. The insets represent high-power images of cleaved Caspase-3-positive MNs (left) and the fragmented and condensed nuclei (right, monochrome). D, Quantitative analysis of cleaved Caspase-3-positive MNs in the facial nuclei. Cleaved Caspase-3-positive MNs were counted in at least eight 6-μm-thick sections in each animal. Three animals were analyzed in each genotype. **p < 0.01 and not significant (NS) compared with the intact side (unpaired t test) or between the two groups indicated. Scale bars, 100 μm.
Figure 8.

Retrograde labeling of axotomized facial MNs with fluoro-ruby. Fluoro-ruby was applied to the proximal end of the transected nerve, and the facial nuclei were analyzed at 24 h after axotomy. Almost all facial MNs, except for most MNs in the VM (circumscribed by dots in the nuclei) and DA (arrows) subnuclei, were similarly labeled with fluoro-ruby regardless of the expression of either ZPK/DLK or MKK4/MAP2K4. Nevertheless, note that acute reduction in ChAT immunoreactivity in axotomized fluoro-ruby-positive MNs was not observed in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice at 24 h after axotomy. Scale bar, 100 μm.
Figure 9.

Accumulation of IBA1/AIF1-positive microglia was not observed in the axotomized facial nuclei of ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice at 5 d after axotomy. Six-micrometer-thick cross-sections of the facial nuclei from ZPK/DLK deficient- and CNS MKK4/MAP2K4-deficient mice and their littermate controls of the indicated genotypes were immunolabeled for IBA1/AIF1 (red). Representative confocal images of the facial nuclei on the transected side were shown. Scale bars, 100 μm.
MKK4/MAP2K4-dependent MN-autonomous mechanisms mediate axotomy-induced death of neonatal facial MNs
Because nestin is expressed in early neuroepithelial cells giving rise to neurons, astroglia, and oligodendroglia, the phenotypes of CNS MKK4/MAP2K4-deficient mice might be attributable to MKK4/MAP2K4-deficient macroglia. To further examine whether MKK4/MAP2K4-mediated MN death is MN autonomous or not, we used another MKK4/MAP2K4-conditional knock-out mouse strain in which the mkk4/map2k4 gene was selectively inactivated in neurons [mkk4/map2k4f/f Tg(syn1::cre)+/−, neuronal MKK4/MAP2K4-deficient mice thereafter]. Unlike CNS MKK4/MAP2K4-deficient mice, neuronal MKK4/MAP2K4-deficient mice demonstrated neither gross anatomical anomaly nor growth retardation and survived into adulthood. Nevertheless, quantitative analysis of facial MNs revealed that neuronal MKK4/MAP2K4-deficient mice had ∼33% more facial MNs than littermate controls [mkk4/map2k4+/f Tg(syn1::cre)+/−]. Moreover, 70% of axotomized facial MNs survived in neuronal MKK4/MAP2K4-deficient mice in the same experiment as performed in Figure 6, whereas almost all facial MNs died except for those in the VM subnucleus, consisting of ∼20% of total facial MNs, in littermate controls (Fig. 10). These results clearly indicate that MKK4/MAP2K4-dependent MN-autonomous mechanisms are required for death of at least a significant proportion of axotomized neonatal facial MNs, as well as NPCD.
Figure 10.

Facial MNs are significantly resistant against axotomy-induced death in neuronal MKK4/MAP2K4-deficient neonates and, to a lesser extent, in neuronal MKK7/MAP2K7-deficient neonates. Left facial nerve axotomy was performed as described in Figure 6. A, Representative immunohistochemical images of the facial nuclei from neuronal MKK4/MAP2K4-deficient 7-d-old mice [mkk4/map2k4f/f Tg(syn1::cre)+/−] and their littermate controls [mkk4/map2k4+/f Tg(syn1::cre)+/−] and from neuronal MKK7/MAP2K7-deficient 7-d-old mice [mkk7/map2k7f/f Tg(syn1::cre)+/−] and their littermate controls [mkk7/map2k7+/f Tg(syn1::cre)+/−] at 5 d after axotomy. Sections were immunolabeled as in Figure 6. B, Quantitative results of A. Three animals per genotype were examined. In the right graphs, the data are calculated as percentage of total MNs on the transected side to those on the intact side. *p < 0.05, **p < 0.01, ***p < 0.001, and not significant (NS) compared with the intact side or with the indicated pairs (unpaired t test). Scale bars, 100 μm.
Activation of MKK7/MAP2K7-dependent MN-autonomous mechanisms contributes to axotomy-induced death of some neonatal facial MNs
Although CNS MKK4/MAP2K4-deficient mice demonstrated that MKK7/MAP2K7 cannot compensate for the functional roles of MKK4/MAP2K4 in NPCD, it remains to be clarified whether full activation of JNKs (and other downstream substrates) requires cooperative and synergistic phosphorylation by MKK4/MAP2K4 and MKK7/MAP2K7 in axotomy-induced death of neonatal facial MNs. Tg(nes::cre)+/− mkk7/map2k7f/f mice cannot be used, because they die soon after birth (Yamasaki et al., 2011). We thereby performed facial nerve axotomy using Tg(syn1::cre)+/− mkk7/map2k7f/f mice (neuronal MKK7/MAP2K7-deficient mice thereafter), which survived after birth without apparent abnormalities. In contrast to neuronal MKK4/MAP2K4-deficient mice, neuronal MKK7/MAP2K7-deficient mice had the same numbers of facial MNs as did littermate controls [Tg(syn1::cre)+/− mkk7/map2k7+/f]. However, at 5 d after axotomy, significantly more facial MNs survived in neuronal MKK7/MAP2K7-deficient mice than in littermate controls but to a lesser extent than those that survived in neuronal MKK4/MAP2K4-deficient mice (Fig. 10), supporting the notion that synergistic effects of MKK7/MAP2K7 are required for death of a fraction of axotomized facial MNs.
Degeneration of the distal motor axons after axotomy is distinct from axotomy-induced death of facial MNs in terms of contributions of ZPK/DLK and MKK4/MAP2K4
ZPK/DLK contributes to degeneration of the distal segment of a transected axon (Wallerian degeneration; Miller et al., 2009; Ghosh et al., 2011). We examined immunoreactivity for neurofilament 200 (NF200) in the distal portions of axotomized facial nerves in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice to evaluate axonal degeneration (for the anatomical position examined, refer to Fig. 6A). In contrast to almost complete resistance against axotomy-induced MN death, NF200-positive distal axons were no longer detected at 5 d after axotomy in both mutant mice. However, at 24 h after axotomy, more NF200-positive axons remained in ZPK/DLK-deficient mice compared with wild-type littermates, which is in agreement with the finding by Miller et al. (2009) that loss of ZPK/DLK delayed Wallerian degeneration of sensory axons. We also observed a similar delay in degeneration of distal axons at 24 h in CNS MKK4/MAP2K4-deficient mice (Fig. 11).
Figure 11.
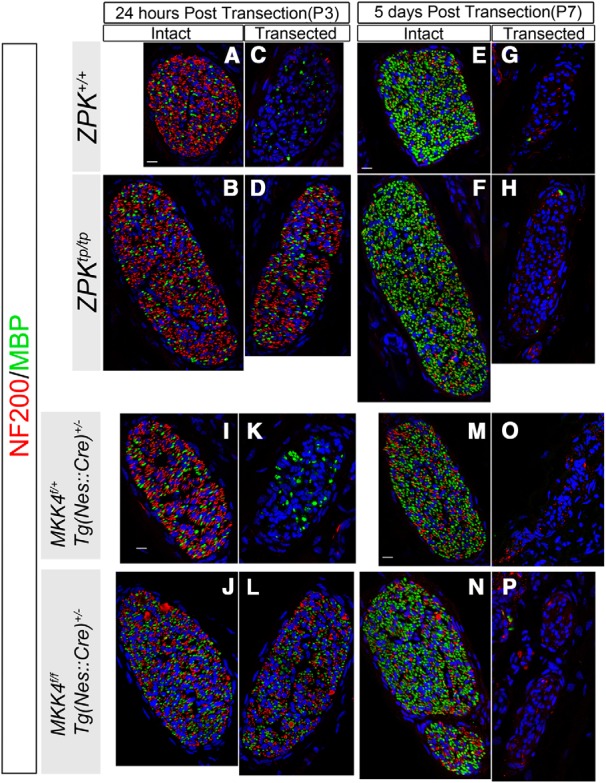
Degeneration of the distal facial nerves after axotomy in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient neonates. A–P, Representative confocal laser micrographs of the cross-sections of the main nasolabial branch at the same anatomical point. The facial nerves on the intact (A, B, E, F, I, J, M, N) and transected (C, D, G, H, K, L, O, P) sides from the mice of the indicated genotypes were double labeled for NF200 (red) and MBP (green) at 24 h and 5 d after axotomy at P2. Scale bars, 10 μm.
Discussion
We here provide compelling evidence for the pivotal roles of ZPK/DLK and MKK4/MAP2K4 in axotomy-induced MN death in neonates. Remarkably, ablation of either ZPK/DLK or MKK4/MAP2K4 protected MNs almost completely from axotomy-induced death. Conversely, developmental death of facial MNs is rather complex in terms of dependence on ZPK/DLK and MKK4/MAP2K4. ZPK/DLK and MKK4/MAP2K4-mediated signals contribute not only to apoptosis during NPCD (a late MN death) but also to an early death of MN, before the time window of NPCD. This early neuronal death is likely to be independent of axonal contact to the target and equivalent to those observed in the germinal zones of various CNS regions (Buss et al., 2006; Vanderhaeghen and Cheng, 2010). ZPK/DLK and MKK4/MAP2K4-independent mechanisms also contribute to elimination of superfluous MNs particularly in the late phase of NPCD, which may involve non-apoptotic pathways. In fact, accumulation of activated c-Jun in the nucleus of facial MNs was still found in the E17.5 embryos lacking ZPK/DLK or MKK4/MAP2K4. Moreover, our results could not rule out the possibility that more facial MNs are generated in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient embryos than in control embryos. Therefore, the ZPK/DLK and MKK4/MAP2K4-dependent mechanism is a component of quantitative control of MNs during development but is solely responsible for axotomy-induced MN death in neonates.
A causative relationship of the JNK pathway with axotomy-induced MN death has been indicated by studies using mice deficient in JNKs. However, up to 50% of axotomized MNs still died in JNK2 and JNK3-double-deficient mice (Keramaris et al., 2005), suggesting redundancy in these downstream kinases in the pathway and/or involvement of other pathways in neonatal MN death after axotomy. Our results from ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient mice indicate that activation of ZPK/DLK and MKK4/MAP2K4 is absolutely required for execution of molecular events leading to MN death after axotomy. We confirmed that this ZPK/DLK- and MKK4/MAP2K4-dependent death of axotomized facial MNs is neuron autonomous, because ∼70% of facial MNs were still resistant to axotomy-induced death when MKK4/MAP2K4 was conditionally deleted only in neurons. This conclusion is consistent with other studies demonstrating that ZPK/DLK-mediated degeneration of other types of neurons after injury or trophic factor withdrawal is neuron autonomous (Ghosh et al., 2011; Watkins et al., 2013; Welsbie et al., 2013). However, our results do not exclude contributions of pathways other than the JNK pathway. Particularly, because MKK4/MAP2K4 is capable of activating p38 kinases (Brancho et al., 2003; Wang et al., 2007a), simultaneous activation of the p38 pathway might have synergistic effects on JNK-mediated MN death. Glutamatergic excitotoxic stress has also been proposed as an additional mechanism underlying axotomy-induced MN death (Iwasaki et al., 1995; Lawson and Lowrie, 1998; Dekkers et al., 2004), and ablation of ZPK/DLK protects neurons in adult mouse brain from kainate-induced excitotoxic degeneration (Pozniak et al., 2013). Our results predict that, although prolonged activation of glutamate receptors occurs after axotomy, simultaneous or subsequent activation of both ZPK/DLK and MKK4/MAP2K4 in the proximal axon or cell body is required for excitotoxic death of MNs. Recent studies of the Caenorhabditis elegans ZPK/DLK homolog DLK-1 have revealed a Ca2+-dependent mechanism that enables rapid and localized activation of the long isoform of DLK-1, DLK-1L, in response to axonal injury (Ghosh-Roy et al., 2010; Yan and Jin, 2012). If mammalian ZPK/DLK is proven to use a similar Ca2+-dependent mechanism for activation after axonal injury, this would provide a mechanistic link between Ca2+-mediated excitotoxic stress and ZPK/DLK and MKK4/MAP2K4-mediated MN death. However, Ca2+-dependent activation of C. elegans DLK-1L is critically regulated by a conserved C-terminus hexapeptide that is absent in mammalian ZPK/DLK but present in another mammalian MAP3K, LZK/MAP3K13 (Yan and Jin, 2012). Additional studies will be necessary to clarify this issue.
Although MKK7/MAP2K7 has been considered as the preferential substrate of ZPK/DLK (Merritt et al., 1999) at the MAP2K level, particularly in retinal ganglion neurons (Welsbie et al., 2013), our results indicate that MKK4/MAP2K4 is directly downstream of ZPK/DLK in the JNK pathway responsible for axotomy-induced death and NPCD and that MKK4/MAP2K4 cannot be replaced by MKK7/MAP2K7 in this functional role, although we also found that full activation of the cell-death pathway requires cooperative and synergistic phosphorylation by MKK4/MAP2K4 and MKK7/MAP2K7 (Kishimoto et al., 2003; Wang et al., 2007a) in death of some axotomized facial MNs. Non-redundant functional roles of MKK4/MAP2K4 and MKK7/MAP2K7 in MNs may be ensured by cell type-dependent and compartmentalized distribution of particular JNK phospho-relay modules, such as the three distinct JNK pools identified in cerebellar granular neurons (Coffey et al., 2000). The JIP (JNK-interacting protein) family of scaffold proteins is known to play a role in assembly and subcellular localization of distinct JNK signaling modules (Whitmarsh, 2006). ZPK/DLK has been shown to bind to JIP1 (Yasuda et al., 1999; Nihalani et al., 2001), JIP2 (Yasuda et al., 1999), and JIP3 (Ghosh et al., 2011), whereas MKK4/MAP2K4 interacts with JSAP1, an isoform of JIP3 (Ito et al., 1999), but with neither JIP1 nor JIP2 (Yasuda et al., 1999; Mooney and Whitmarsh, 2004). Therefore, it is conceivable that ZPK/DLK and MKK4/MAP2K4 interact directly with the aid of JIP3/JSAP1 in MNs. Our result also demonstrated that ZPK/DLK protein levels were increased in the brainstem of CNS MKK4/MAP2K4-deficient mice (Fig. 1E). Recent studies have proved that the protein level of ZPK/DLK is a critical determinant of downstream signaling in the JNK pathway, which is tightly regulated by the ubiquitin–proteasome system and by a JNK-dependent positive feedback mechanism (Bloom et al., 2007; Xiong et al., 2010; Huntwork-Rodriguez et al., 2013). However, the causative relationship between reduced MKK4/MAP2K4 protein level and increased ZPK/DLK protein level remains to be investigated.
Despite the total dependence of axotomy-induced death of MNs on ZPK/DLK and MKK4/MAP2K4, loss of either ZPK/DLK or MKK4/MAP2K4 only delayed Wallerian degeneration. This observation is consistent with the recent study by Osterloh et al. (2012) demonstrating that Wallerian degeneration is an active axon-autonomous self-destruction process that is molecularly distinct from apoptosis. Another critical difference between axotomy-induced MN death and Wallerian degeneration is that MN death is at least partly dependent on transcriptional changes after delivery of injury signals to the nucleus. Accumulation of activated c-Jun in the nucleus is a well studied transcriptional signature of MN responses to axonal injury, although it is not always proapoptotic but also important in axonal regeneration (Raivich et al., 2004; Itoh et al., 2009; Ruff et al., 2012; Shin et al., 2012; Watkins et al., 2013). Our results demonstrate that, as far as could be determined by nuclear accumulation of activated c-Jun, the nucleus fails to respond to axonal injury in the absence of either ZPK/DLK or MKK4/MAP2K4. This notion is also supported by lack of acute downregulation of ChAT immunoreactivity in axotomized ZPK/DLK-deficient and MKK4/MAP2K4-deficient MNs. It remains to be clarified how the nucleus senses axonal injury through activation of ZPK/DLK and MKK4/MAP2K4. One putative mechanism is referred to as “negative” injury signals (Abe and Cavalli, 2008). Insufficient supply of target-derived trophic signals to the cell body triggers nuclear responses to loss of axonal contact to the muscle. NPCD has been considered a typical case of this negative injury signal, because several exogenous neurotrophic molecules applied to the transected axons rescue MNs from axotomy-induced death (Sendtner et al., 2000; Gould and Oppenheim, 2011). The mechanistic similarity between axotomized neonatal MN death and an apoptotic component of NPCD in terms of the common dependence on ZPK/DLK and MKK4/MAP2K4 may support this mechanism and suggests that various trophic signals from exogenous trophic molecules commonly inhibit activation of ZPK/DLK and MKK4/MAP2K4 and thereby prevent MNs from apoptosis. Another putative mechanism, “positive” injury signals, depends on retrograde transport of injury-triggered signaling molecules from the injury site to the cell body. Activated ZPK/DLK and MKK4/MAP2K4 localized at the axonal injury site could themselves be signaling molecules or could play roles in generation or transport of signaling molecules. In support of the positive signals, a recent study by Shin et al. (2012) has demonstrated ZPK/DLK-dependent retrograde transport of signal molecules in the regenerative response of adult sensory nerves to axonal injury. However, as far as could be determined by a retrograde labeling method, we could not find defective retrograde transport in ZPK/DLK-deficient neonatal MNs. The mechanistic link between activated ZPK/DLK and MKK4/MAP2K4 and injury-induced degenerative or regenerative responses remain to be clarified in each neuronal subset at different maturational stages.
In line with the failure in neuronal response to axonal injury, microglia around the axotomized facial nucleus also failed to become activated in ZPK/DLK-deficient and CNS MKK4/MAP2K4-deficient neonates (Fig. 9). Because microglia can detect even a subtle change in their microenvironment and respond to the alteration, this observation suggests that, after axotomy, the MNs lacking ZPK/DLK or MKK4/MAP2K4 remain “healthy” without any injury-induced changes that can be detectable by microglia.
In summary, our study demonstrates for the first time that activation of both ZPK/DLK and MKK4/MAP2K4 after axotomy is absolutely required for axotomy-induced MN death in neonates and may open a new avenue for therapeutic interventions directed toward ZPK/DLK and/or MKK4/MAP2K4 to inhibit MN degeneration after axonal injury in newborns.
Footnotes
This work was supported by Shriners Hospitals for Children Grants 85800 (A.I.) and 85400 (T.I.) and National Institutes of Health Grant NS025044 (D.P. and T.I.). We thank Teresa Ta for her technical assistance.
The authors declare no competing financial interests.
References
- Abe N, Cavalli V. Nerve injury signaling. Curr Opin Neurobiol. 2008;18:276–283. doi: 10.1016/j.conb.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/S0896-6273(01)80026-X. [DOI] [PubMed] [Google Scholar]
- Ashwell KW, Watson CR. The development of facial motoneurones in the mouse—neuronal death and the innervation of the facial muscles. J Embryol Exp Morphol. 1983;77:117–141. [PubMed] [Google Scholar]
- Bloom AJ, Miller BR, Sanes JR, DiAntonio A. The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev. 2007;21:2593–2606. doi: 10.1101/gad.1592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss RR, Sun W, Oppenheim RW. Adaptive roles of programmed cell death during nervous system development. Annu Rev Neurosci. 2006;29:1–35. doi: 10.1146/annurev.neuro.29.051605.112800. [DOI] [PubMed] [Google Scholar]
- Casanovas A, Ribera J, Hager G, Kreutzberg GW, Esquerda JE. c-Jun regulation in rat neonatal motoneurons postaxotomy. J Neurosci Res. 2001;63:469–479. doi: 10.1002/jnr.1041. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar A. Turning heads: development of vertebrate branchiomotor neurons. Dev Dyn. 2004;229:143–161. doi: 10.1002/dvdy.10444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PG, Oppenheim RW. Neuron death in vertebrate development: in vitro methods. Methods Cell Biol. 1995;46:277–321. doi: 10.1016/S0091-679X(08)61933-0. [DOI] [PubMed] [Google Scholar]
- Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J Neurosci. 2000;20:7602–7613. doi: 10.1523/JNEUROSCI.20-20-07602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers J, Bayley P, Dick JR, Schwaller B, Berchtold MW, Greensmith L. Over-expression of parvalbumin in transgenic mice rescues motoneurons from injury-induced cell death. Neuroscience. 2004;123:459–466. doi: 10.1016/j.neuroscience.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Dubois-Dauphin M, Frankowski H, Tsujimoto Y, Huarte J, Martinou JC. Neonatal motoneurons overexpressing the bcl-2 protooncogene in transgenic mice are protected from axotomy-induced cell death. Proc Natl Acad Sci U S A. 1994;91:3309–3313. doi: 10.1073/pnas.91.8.3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericson J, Thor S, Edlund T, Jessell TM, Yamada T. Early stages of motor neuron differentiation revealed by expression of homeobox gene Islet-1. Science. 1992;256:1555–1560. doi: 10.1126/science.1350865. [DOI] [PubMed] [Google Scholar]
- Ganiatsas S, Kwee L, Fujiwara Y, Perkins A, Ikeda T, Labow MA, Zon LI. SEK1 deficiency reveals mitogen-activated protein kinase cascade crossregulation and leads to abnormal hepatogenesis. Proc Natl Acad Sci U S A. 1998;95:6881–6886. doi: 10.1073/pnas.95.12.6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol. 2011;194:751–764. doi: 10.1083/jcb.201103153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh-Roy A, Wu Z, Goncharov A, Jin Y, Chisholm AD. Calcium and cyclic AMP promote axonal regeneration in Caenorhabditis elegans and require DLK-1 kinase. J Neurosci. 2010;30:3175–3183. doi: 10.1523/JNEUROSCI.5464-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould TW, Oppenheim RW. Motor neuron trophic factors: therapeutic use in ALS? Brain Res Rev. 2011;67:1–39. doi: 10.1016/j.brainresrev.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase G, Pettmann B, Raoul C, Henderson CE. Signaling by death receptors in the nervous system. Curr Opin Neurobiol. 2008;18:284–291. doi: 10.1016/j.conb.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hirai Si, Katoh M, Terada M, Kyriakis JM, Zon LI, Rana A, Avruch J, Ohno S. MST/MLK2, a member of the mixed lineage kinase family, directly phosphorylates and activates SEK1, an activator of c-Jun N-terminal kinase/stress-activated protein kinase. J Biol Chem. 1997;272:15167–15173. doi: 10.1074/jbc.272.24.15167. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S. MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development. 2002;129:4483–4495. doi: 10.1242/dev.129.19.4483. [DOI] [PubMed] [Google Scholar]
- Horiuchi M, Itoh A, Pleasure D, Itoh T. MEK-ERK signaling is involved in interferon-gamma-induced death of oligodendroglial progenitor cells. J Biol Chem. 2006;281:20095–20106. doi: 10.1074/jbc.M603179200. [DOI] [PubMed] [Google Scholar]
- Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW. JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol. 2013;202:747–763. doi: 10.1083/jcb.201303066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, Nakabeppu Y, Shiba T, Yamamoto KI. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a Scaffold factor in the JNK signaling pathway. Mol Cell Biol. 1999;19:7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T. Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun. 2009;383:258–262. doi: 10.1016/j.bbrc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Itoh A, Horiuchi M, Wakayama K, Xu J, Bannerman P, Pleasure D, Itoh T. ZPK/DLK, a mitogen-activated protein kinase kinase kinase, is a critical mediator of programmed cell death of motoneurons. J Neurosci. 2011;31:7223–7228. doi: 10.1523/JNEUROSCI.5947-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki Y, Ikeda K, Shiojima T, Kinoshita M. CNQX prevents spinal motor neuron death following sciatic nerve transection in newborn rats. J Neurol Sci. 1995;134:21–25. doi: 10.1016/0022-510X(95)00217-6. [DOI] [PubMed] [Google Scholar]
- Kanungo AK, Hao Z, Elia AJ, Mak TW, Henderson JT. Inhibition of apoptosome activation protects injured motor neurons from cell death. J Biol Chem. 2008;283:22105–22112. doi: 10.1074/jbc.M800988200. [DOI] [PubMed] [Google Scholar]
- Keramaris E, Vanderluit JL, Bahadori M, Mousavi K, Davis RJ, Flavell R, Slack RS, Park DS. c-Jun N-terminal kinase 3 deficiency protects neurons from axotomy-induced death in vivo through mechanisms independent of c-Jun phosphorylation. J Biol Chem. 2005;280:1132–1141. doi: 10.1074/jbc.M410127200. [DOI] [PubMed] [Google Scholar]
- Kishimoto H, Nakagawa K, Watanabe T, Kitagawa D, Momose H, Seo J, Nishitai G, Shimizu N, Ohata S, Tanemura S, Asaka S, Goto T, Fukushi H, Yoshida H, Suzuki A, Sasaki T, Wada T, Penninger JM, Nishina H, Katada T. Different properties of SEK1 and MKK7 in dual phosphorylation of stress-induced activated protein kinase SAPK/JNK in embryonic stem cells. J Biol Chem. 2003;278:16595–16601. doi: 10.1074/jbc.M213182200. [DOI] [PubMed] [Google Scholar]
- Koliatsos VE, Price WL, Pardo CA, Price DL. Ventral root avulsion: an experimental model of death of adult motor neurons. J Comp Neurol. 1994;342:35–44. doi: 10.1002/cne.903420105. [DOI] [PubMed] [Google Scholar]
- Lawson SJ, Lowrie MB. The role of apoptosis and excitotoxicity in the death of spinal motoneurons and interneurons after neonatal nerve injury. Neuroscience. 1998;87:337–348. doi: 10.1016/S0306-4522(98)00120-1. [DOI] [PubMed] [Google Scholar]
- Li L, Wu W, Lin LF, Lei M, Oppenheim RW, Houenou LJ. Rescue of adult mouse motoneurons from injury-induced cell death by glial cell line-derived neurotrophic factor. Proc Natl Acad Sci U S A. 1995;92:9771–9775. doi: 10.1073/pnas.92.21.9771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrie MB, Vrbová G. Dependence of postnatal motoneurones on their targets: review and hypothesis. Trends Neurosci. 1992;15:80–84. doi: 10.1016/0166-2236(92)90014-Y. [DOI] [PubMed] [Google Scholar]
- Merritt SE, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. The mixed lineage kinase DLK utilizes MKK7 and not MKK4 as substrate. J Biol Chem. 1999;274:10195–10202. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, DiAntonio A. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney LM, Whitmarsh AJ. Docking interactions in the c-Jun N-terminal kinase pathway. J Biol Chem. 2004;279:11843–11852. doi: 10.1074/jbc.M311841200. [DOI] [PubMed] [Google Scholar]
- Moran LB, Graeber MB. The facial nerve axotomy model. Brain Res Brain Res Rev. 2004;44:154–178. doi: 10.1016/j.brainresrev.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Nihalani D, Meyer D, Pajni S, Holzman LB. Mixed lineage kinase-dependent JNK activation is governed by interactions of scaffold protein JIP with MAPK module components. EMBO J. 2001;20:3447–3458. doi: 10.1093/emboj/20.13.3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T, Kristensson K. Uptake and retrograde axonal transport of horseradish peroxidase in normal and axotomized motor neurons during postnatal development. Neuropathol Appl Neurobiol. 1979;5:377–387. doi: 10.1111/j.1365-2990.1979.tb00636.x. [DOI] [PubMed] [Google Scholar]
- Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, Powell EH, Sheehan AE, Avery MA, Hackett R, Logan MA, MacDonald JM, Ziegenfuss JS, Milde S, Hou YJ, Nathan C, Ding A, Brown RH, Jr, Conforti L, Coleman M, Tessier-Lavigne M, et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Sengupta Ghosh A, Gogineni A, Hanson JE, Lee SH, Larson JL, Solanoy H, Bustos D, Li H, Ngu H, Jubb AM, Ayalon G, Wu J, Scearce-Levie K, Zhou Q, Weimer RM, Kirkpatrick DS, Lewcock JW. Dual leucine zipper kinase is required for excitotoxicity-induced neuronal degeneration. J Exp Med. 2013;210:2553–2567. doi: 10.1084/jem.20122832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Da Costa C, Iwata O, Galiano M, Hristova M, Nateri AS, Makwana M, Riera-Sans L, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A. The AP-1 transcription factor c-Jun is required for efficient axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Ribera J, Ayala V, Casas C. Involvement of c-Jun-JNK pathways in the regulation of programmed cell death of developing chick embryo spinal cord motoneurons. Dev Neurosci. 2007;29:438–451. doi: 10.1159/000097318. [DOI] [PubMed] [Google Scholar]
- Ruff CA, Staak N, Patodia S, Kaswich M, Rocha-Ferreira E, Da Costa C, Brecht S, Makwana M, Fontana X, Hristova M, Rumajogee P, Galiano M, Bohatschek M, Herdegen T, Behrens A, Raivich G. Neuronal c-Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N-terminus are moderate. J Neurochem. 2012;121:607–618. doi: 10.1111/j.1471-4159.2012.07706.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramek D, Kotsinas A, Meixner A, Wada T, Elling U, Pospisilik JA, Neely GG, Zwick RH, Sigl V, Forni G, Serrano M, Gorgoulis VG, Penninger JM. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat Genet. 2011;43:212–219. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Pei G, Beck M, Schweizer U, Wiese S. Developmental motoneuron cell death and neurotrophic factors. Cell Tissue Res. 2000;301:71–84. doi: 10.1007/s004410000217. [DOI] [PubMed] [Google Scholar]
- Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, DiAntonio A. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, Elliott JL, Yan Q. Axotomy-induced neuronal death during development. J Neurobiol. 1992;23:1231–1246. doi: 10.1002/neu.480230913. [DOI] [PubMed] [Google Scholar]
- Sun W, Gould TW, Newbern J, Milligan C, Choi SY, Kim H, Oppenheim RW. Phosphorylation of c-Jun in avian and mammalian motoneurons in vivo during programmed cell death: an early reversible event in the apoptotic cascade. J Neurosci. 2005;25:5595–5603. doi: 10.1523/JNEUROSCI.4970-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–687. doi: 10.1016/S0896-6273(01)80027-1. [DOI] [PubMed] [Google Scholar]
- Vanderhaeghen P, Cheng HJ. Guidance molecules in axon pruning and cell death. Cold Spring Harb Perspect Biol. 2010;2:a001859. doi: 10.1101/cshperspect.a001859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Joza N, Cheng HY, Sasaki T, Kozieradzki I, Bachmaier K, Katada T, Schreiber M, Wagner EF, Nishina H, Penninger JM. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat Cell Biol. 2004;6:215–226. doi: 10.1038/ncb1098. [DOI] [PubMed] [Google Scholar]
- Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta. 2007a;1773:1349–1357. doi: 10.1016/j.bbamcr.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Wang X, Nadarajah B, Robinson AC, McColl BW, Jin JW, Dajas-Bailador F, Boot-Handford RP, Tournier C. Targeted deletion of the mitogen-activated protein kinase kinase 4 gene in the nervous system causes severe brain developmental defects and premature death. Mol Cell Biol. 2007b;27:7935–7946. doi: 10.1128/MCB.00226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, Berlinicke CA, Hackler L, Jr, Fuller J, Fu J, Cao LH, Han B, Auld D, Xue T, Hirai S, Germain L, Simard-Bisson C, Blouin R, Nguyen JV, Davis CH, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A. 2013;110:4045–4050. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh AJ. The JIP family of MAPK scaffold proteins. Biochem Soc Trans. 2006;34:828–832. doi: 10.1042/BST0340828. [DOI] [PubMed] [Google Scholar]
- Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, Collins CA. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Maroney AC, Dobrzanski P, Kukekov NV, Greene LA. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki T, Kawasaki H, Arakawa S, Shimizu K, Shimizu S, Reiner O, Okano H, Nishina S, Azuma N, Penninger JM, Katada T, Nishina H. Stress-activated protein kinase MKK7 regulates axon elongation in the developing cerebral cortex. J Neurosci. 2011;31:16872–16883. doi: 10.1523/JNEUROSCI.1111-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Jin Y. Regulation of DLK-1 kinase activity by calcium-mediated dissociation from an inhibitory isoform. Neuron. 2012;76:534–548. doi: 10.1016/j.neuron.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]