

Abstract
Background
Accelerated cell cycle progression is the common feature of most cancers. MiRNAs can act as oncogenes or tumor suppressors by directly modulating cell cycle machinery. It has been shown that miR-188 is upregulated in UVB-irradiated mouse skin and human nasopharyngeal carcinoma CNE cells under hypoxic stress. However, little is known about the function of miR-188 in cell proliferation and growth control.
Results
Overexpression of miR-188 inhibits cell proliferation, tumor colony formation and G1/S cell cycle transition in human nasopharyngeal carcinoma CNE cells. Using bioinformatics approach, we identify a series of genes regulating G1/S transition as putative miR-188 targets. MiR-188 inhibits both mRNA and protein expression of CCND1, CCND3, CCNE1, CCNA2, CDK4 and CDK2, suppresses Rb phosphorylation and downregulates E2F transcriptional activity. The expression level of miR-188 also inversely correlates with the expression of miR-188 targets in human nasopharyngeal carcinoma (NPC) tissues. Moreover, studies in xenograft mouse model reveal that miR-188 is capable of inhibiting tumor initiation and progression by suppressing target genes expression and Rb phosphorylation.
Conclusions
This study demonstrates that miR-188 exerts anticancer effects, via downregulation of multiple G1/S related cyclin/CDKs and Rb/E2F signaling pathway.
Electronic supplementary material
The online version of this article (doi:10.1186/s12964-014-0066-6) contains supplementary material, which is available to authorized users.
Keywords: MiR-188, Cell cycle, G1/S transition, CDK, Cyclin, Rb, E2F
Background
MicroRNAs (miRNAs) are a class of small non-coding RNAs that negatively regulate gene expression at the post-transcriptional level [1]. MiRNAs are important regulators of various biological and pathological processes, including cell proliferation, differentiation, apoptosis, metabolism and cancer [2-11]. As “micromanagers” of gene expression, miRNAs often have subtle influence on one single target. A variety of studies indicate that an individual miRNA may perform its function through targeting multiple components in one signaling pathway [9,12,13]. In addition, multiple miRNAs can act on the same signaling pathway or biological process [14-17].
Cell cycle progression is directly driven by a series of heterodimers formed by cyclins and cyclin-dependent kinases (CDKs) [18]. The decision for a cell to enter S phase is tightly controlled by cyclin D/CDK4/6 and cyclin E/CDK2 complexes, followed by cyclin A/CDK2 throughout S phase [18-20]. In mammalian cells, G1 to S phase progression is also regulated by the Retinoblastoma protein (Rb). Rb is a tumor suppressor protein that plays a pivotal role in the negative control of cell cycle and in tumor progression [21,22]. The Rb protein inhibits the expression of genes required for entry into S phase by sequestering the E2F family of transcription factors [23,24]. During G1 phase of the cell cycle, Rb binds to E2F-DP1 and inhibits downstream transcription. When it is time for a cell to enter S phase, the inactivating phosphorylation of Rb by the G1/S CDKs results in the release of Rb from E2F-DP1 and allows for the activation of E2F target genes that are responsible for facilitating G1/S transition and S phase progression.
Several lines of evidence indicate that miRNAs are important cell cycle regulators. Some miRNAs regulate cell cycle progression by directly suppressing the expression of cyclin/CDK complexes. For instance, miR-15/16 family induces cell cycle arrest by simultaneously targeting multiple cyclins that regulate G1/S transition; these include CCND3, CCNE1 and CDK6 [25,26]. CDK6 and CCND1 are also regulated by miR-34a, which induces a significant G1 cell cycle arrest in human lung carcinoma A549 cells [27]. CDK6 is targeted by miR-137 and miR-124a. The expression of both of these miRNAs is epigenetically silenced by hypermethylation in some human cancers [28-30]. Transfection of miR-137 or miR-124a causes G1 arrest in glioblastoma multiforme cells [31]. In addition, miR-206 suppresses CCND1 expression and plays a tumor suppressive function in skeletal muscle differentiation [32]. Besides cyclin/CDK complexes, miRNAs also modulate cell proliferation through interacting with critical cell cycle regulators that are involved in G1/S and G2/M transitions, such as Cip/Kip and INK4a/ARF family members, E2F family transcription factors, Cdc25, Wee1 and p53, etc. [33]. Therefore, better understanding the roles of miRNAs in cell proliferation and cell cycle checkpoints may shed light on cancer pathogenesis and lead to rational development of new types of cancer therapy.
MiR-188, a miRNA located on the X chromosome in humans, was first identified in 2003 [34]. It has been reported that miR-188 is upregulated by the induction of long-term potentiation (LTP) and it serves to fine-tune synaptic plasticity by targeting neuropilin-2 (Nrp-2) expression in the nervous system [35]. Recent studies also show that miR-188 is upregulated in UVB-irradiated mouse skin and human nasopharyngeal carcinoma CNE cells under hypoxic stress [36,37]. However, little is known about the function of miR-188 on cell proliferation and cell cycle regulation. Here, we report that miR-188 is a potent tumor suppressor in human nasopharyngeal carcinoma cells. Overexpression of miR-188 inhibits cell proliferation and G1/S transition by directly targeting the expression of multiple cyclin/CDKs complexes, including CCND1, CCND3, CCNE1, CCNA2, CDK2 and CDK4. MiR-188 also suppresses Rb phosphorylation and E2F transcriptional activity. Further study from xenograft mouse model shows that miR-188 has anti-cancer potential through suppressing tumor initiation and progression. Taken together, our data highlight the anti-cancer potential of miR-188 and provide a novel mechanism whereby miR-188 controls cell cycle progression via downregulation of multiple cyclin/CDK complexes involved in G1/S transition.
Results
Overexpression of miR-188 suppresses cancer cell proliferation
To characterize the function of miR-188 on cancer cell proliferation, miR-188 mimics or negative control (miR-NC) were transiently transfected into human nasopharyngeal cancer CNE cells. Strikingly, we observed that overexpression of miR-188 attenuated cell proliferation in CNE cells (Figure 1A and B). The effect of miR-188 on cell proliferation was also evaluated using colony formation assay. MiR-188 transfected cells formed significantly less colonies than those transfected with the negative control (Figure 1C and D).
Figure 1.
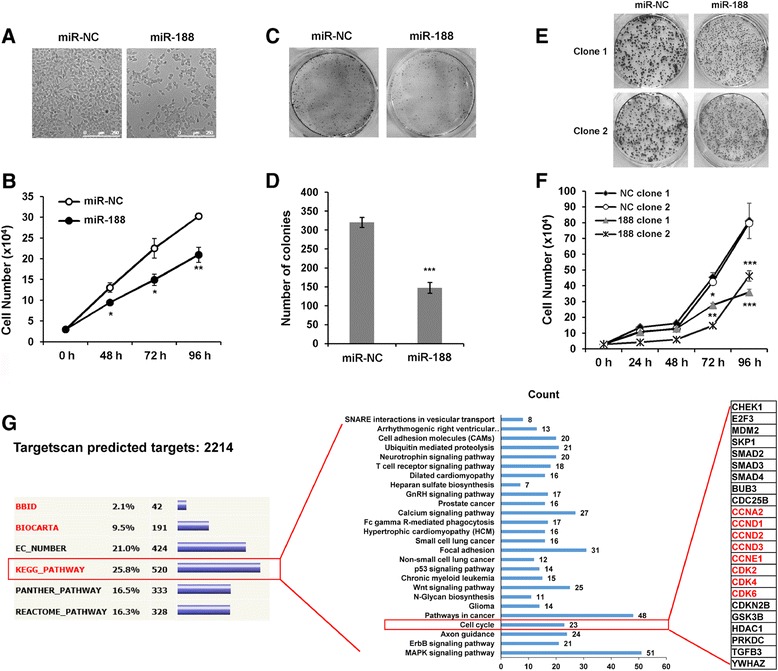
MiR-188 inhibits cell proliferation and tumor colony formation. (A) Overexpression of miR-188 inhibits cell growth in CNE cells. CNE cells were transfected with miR-188 or miR-NC for 72 h, the representative phase contrast images were shown. Scar bar: 250 μm. (B) Overexpression of miR-188 inhibits cell proliferation. Equal amounts of CNE cells were transfected with miR-188 or miR-NC, cell proliferation was monitored at indicated time points. Data presented are means ± SD from three independent experiments (Student t test, *p < 0.05, **p < 0.01). (C) Clonogenic assay of CNE cells transfected with miR-NC or miR-188. Cells were cultured for 10 days and stained with Crystal Violet. (D) The number of colonies were quantified and calculated as means ± SD of three independent experiments (Student t test, ***p < 0.001). (E) Clonogenic assay of CNE cells stably expressing miR-NC or miR-188. (F) Stable transfection of miR-188 inhibits cell proliferation. Equal amounts of miR-NC or miR-188 stable cell lines were cultured for 4 days, cell proliferation was monitored at indicated time points. Data shown are means ± SD from three independent experiments (Student t test, *p < 0.05, **p < 0.01, ***p < 0.001). (G) The functional association of miR-188 target genes. DAVID Functional Annotation tool was used for analysis of potential targets predicted from Targetscan and Findtar. A series of cell cycle related genes were potential targets of miR-188.
By quantifying the level of miR-188 in CNE cells, we found that the expression of miR-188 increased by nearly 1000 folds after transient transfection of miRNA mimics, which is far beyond the level of endogenous miR-188 (Additional file 1: Figure S1A). To create a scenario closer in resemblance to the normal physiological state, we generated CNE cells that stably expressed miR-188 to ascertain its effect on cell proliferation. We selected two stable clones in which the expression of miR-188 was nearly 4 folds higher than the negative control (Additional file 1: Figure S1B). Although, there was no significant difference among the colonies between each plate, the colony size of cells stably expressed miR-188 was much smaller than that of the control (Figure 1E). Similar to transiently transfected cells, cell proliferation rate was significantly reduced in the miR-188 stably expressed clones (Figure 1F). Our data indicate that miR-188 expressed cells have slower growth and cell proliferate rate.
MiR-188 targets multiple cyclin/CDKs involved in G1/S transition
To investigate the potential targets of miR-188, we searched the genome using miRNA target prediction algorithms, Targetscan and Findtar. We found that 2214 genes contain potential miR-188 target sites. We then employed DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/home.jsp) for functional annotation clustering and enrichment scoring [38,39]. According to pathway annotation summary, the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway was classified as containing most of the genes identified by DAVID as potential targets of miR-188 (Figure 1G left panel). The KEGG contains 26 signaling pathways, among these, MAPK signaling pathway (p value: 1.08 × 10−5), ErbB signaling pathway (p value: 3.57 × 10−4), axon guidance (p value: 5.16 × 10−3) and cell cycle (p value: 7.20 × 10−3) were the four pathways most significantly associated with miR-188 according to p values (Figure 1G middle panel, Additional file 2: Table S1). Since cell growth and cell proliferation are highly associated with cell cycle regulation, we analyzed 23 of the genes in the cell cycle pathway and identified a series of cyclins and CDKs involved in G1/S transition that could be potential miR-188 targets (Figure 1G right panel).
Using miRNA target prediction algorithms Findtar, we found that six G1/S related genes, including CCND1, CCND3, CDK4, CCNA2, CDK2 and CCNE1 contain miR-188 response elements in their 3′UTRs (Figure 2A). To validate, we overexpressed miR-188 in CNE cells, then harvested cells for mRNA and protein expression level measurements for these six genes. Results from qRT-PCR showed that overexpression of miR-188 resulted in 30-50% reduction in transcript abundance for CCND1, CCNA2, CCND3, CCNE1, CDK2 and CDK4 (Figure 2B). Similarly, western blot experiments demonstrated that the protein levels of these genes were significantly suppressed to various degrees by miR-188 overexpression in both transiently and stably transfected cells (Figure 2C, Additional file 3: Figure S2A, B).
Figure 2.
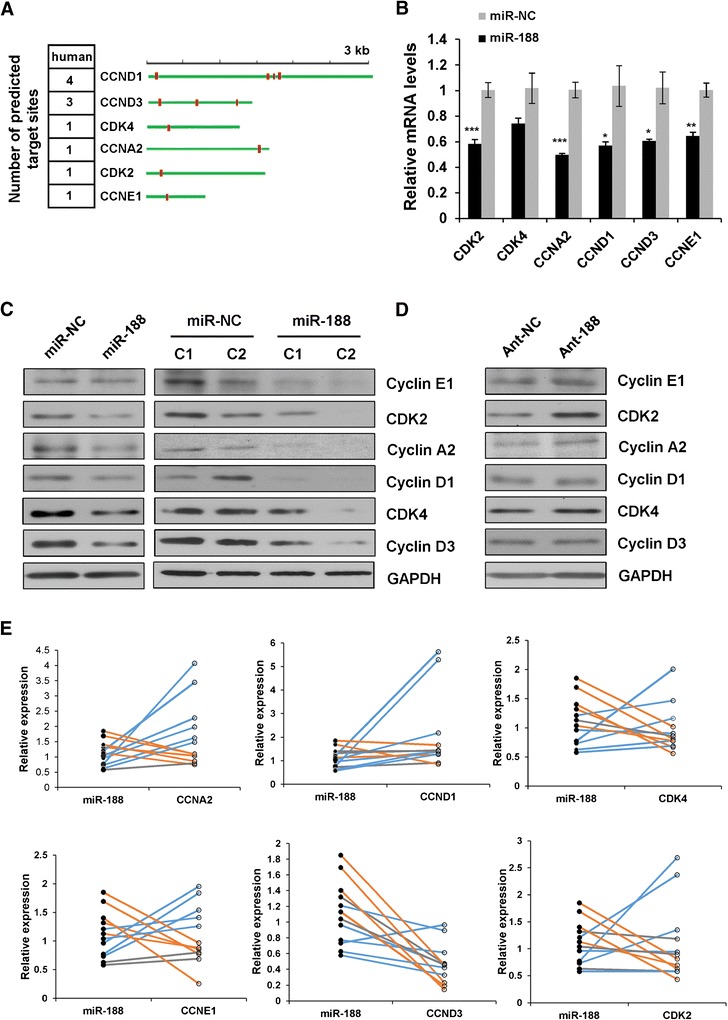
MiR-188 downregulates multiple genes related to G 1 /S transition. (A) CCND1, CCND3, CDK4, CCNA2, CDK2 and CCNE1 are predicted to be miR-188 target genes. (B) qRT-PCR analysis of mRNA expression levels of miR-188 targets in CNE cells transfected with miR-NC or miR-188. Data are presented as means ± SD of three independent experiments (Student t test, *p < 0.05, **p < 0.01, ***p < 0.001). (C) Overexpression of miR-188 downregulates the protein expression of cyclin E1, CDK2, cyclin A2, cyclin D1, cyclin D3 and CDK4. (C1, stable clone 1; C2, stable clone 2). All western blots were performed at least three times. (D) Western blot analysis of cyclin E1, CDK2, cyclin A2, cyclin D1, cyclin D3 and GAPDH in CNE cells transfected with Ant-188 or Ant-NC. All western blots performed at least three times. (E) Inverse correlation between miR-188 and its targets in NPC tissues. The relative levels of miR-188, CCND1, CCND3, CDK4, CCNA2, CDK2 and CCNE1 were quantified using qRT-PCR.
To test whether silencing endogenous miR-188 would affect target gene expression, we transfected cells with miR-188 antisense oligonucleotides (ant-188). Compared to control antisense oligonucleotides (ant-NC), the amounts of CCND1, CCNA2, CCND3, CCNE1, CDK2 and CDK4 proteins increased slightly when endogenous miR-188 expression was silenced (Figure 2D, Additional file 3: Figure S2C). Therefore, these results suggest that miR-188 inhibits cell proliferation, potentially via downregulation of multiple cyclin/CDK complexes involved in G1/S transition.
Inverse correlation of miR-188 and its target genes in NPC tissues
To define the clinical relevance of our findings that miR-188 suppressed the expression of G1/S related cyclin/CDKs, we showed that miR-188 expression was possibly associated with human nasopharyngeal carcinoma. Namely, we examined the expression of miR-188 and its target genes in NPC tissues using qRT-PCR, and an inverse correlation between miR-188 and CCND1, CCNA2, CCND3, CCNE1, CDK2 or CDK4 expression was identified in patient samples (Figure 2E). Thus, the in vitro and in vivo results further demonstrate that miR-188 targets the expression of multiple G1/S related cyclin/CDKs.
MiR-188 delays G1/S cell cycle progression
Having identified the potential targets of miR-188, we then wanted to determine the role of miR-188 on cell cycle progression, especially on G1/S transition. CNE cells transfected with miR-188 or miR-NC were synchronized at G1/S boundary by treatment with hydroxyurea. At 6 hours after release from hydroxyurea, the vast majority of control cells were in S and G2/M phase (Figure 3A). The G1 cell population was bigger in miR-188 transfected CNE cells (21.7 ± 1.38%) than that in control cells (14.6 ± 0.95%) (Figure 3A). Similar results were obtained from miR-188 stably overexpressed cells (Additional file 4: Figure S3A).
Figure 3.
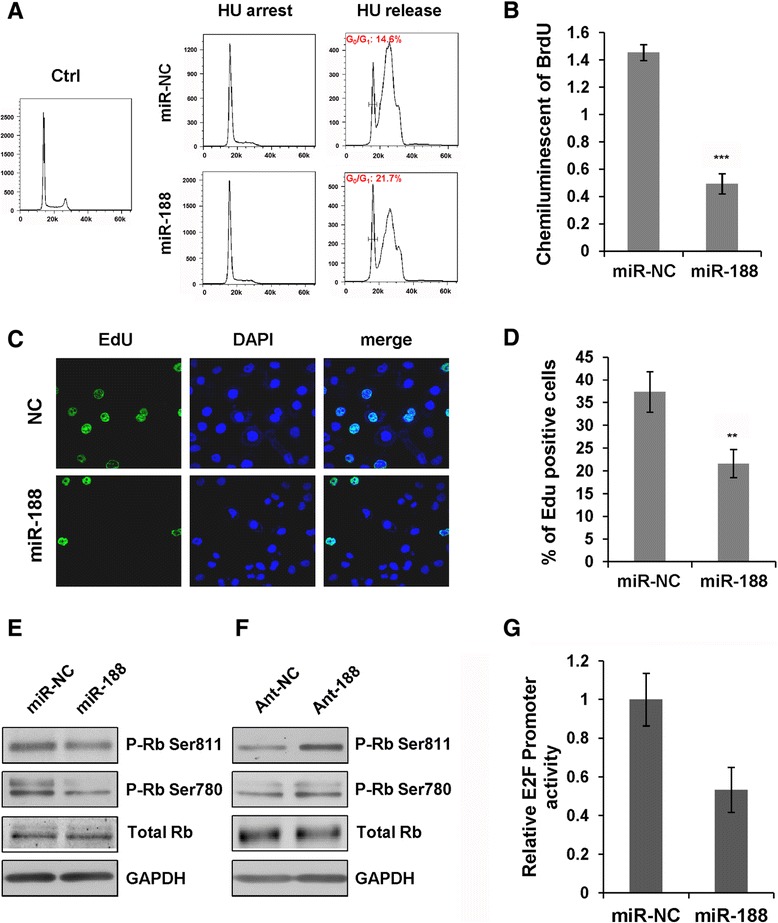
MiR-188 arrests cell cycle at G 1 /S transition through negative regulation of Rb-E2F axis. (A) CNE cells transfected with miR-188 or miR-NC were synchronized at G1/S boundary by hydroxyurea treatment. Cells were released from hydroxyurea block for 6 h, fixed and stained with propidium iodide (PI) for flow cytometry analysis. (B) miR-188 inhibits BrdU incorporation. BrdU Elisa Assay of CNE cells released from hydroxyurea block for 4 h. The BrdU incorporation was quantified by measuring the chemiluminescence. Data shown as means ± SD from three independent experiments (Student t test, ***p < 0.001). (C) Confocal immunofluorescence analysis of CNE cells stained with EdU (green) and DAPI (blue). Scar bar: 10 μm. (D) Quantification of EdU positive cells in CNE cells transfected with miR-188 or miR-NC released from hydroxyurea block for 2 h. Data are presented as means ± SD from three independent experiments (Student t test, **p < 0.01). (E) Immunoblot analysis of phosphorylated and total Rb expression in CNE cells transfected with miR-NC, miR-188, (F) Ant-NC or Ant-188 respectively. GAPDH were used as internal control. (G) E2F promoter activity was determined directly by use of a pE2F-TA-Luc luciferase reporter plasmid which contains four E2F binding sites upstream of TA promoter. Data shown as means ± SD of three independent experiments (Student t test, *p < 0.05).
One feature of G1/S transition is the beginning of genomic DNA synthesis. Chemicals, such as BrdU or EdU, can be inserted into newly synthesized DNA to allow for visualization of the chromosomes. A precise evaluation of cell proliferation can be performed by measuring BrdU or EdU incorporation in proliferating cells during DNA synthesis. First, we performed cell proliferation ELISA BrdU assay. Namely, CNE cells were synchronized at G1 phase by treatment with hydroxyurea. Subsequently, they were released and labeled with BrdU for 2 hours. The incorporation of BrdU was then determined by measuring chemiluminescence. There was a significant reduction of BrdU incorporation in miR-188 transfected cells (Figure 3B). This observation was confirmed by EdU image assay. The incorporation of EdU in miR-188 transfected cells was significantly less than that of control cells (Figure 3C and D). Together, these results demonstrate that miR-188 suppresses cell proliferation mainly by interrupting G1/S cell cycle progression.
MiR-188 suppresses Rb phosphorylation and E2F transcriptional activity
E2F activity is crucial for G1/S transition and DNA replication in mammalian cells. The tumor suppressor Rb is the primary negative regulator of E2F. Disruption of Rb/E2F interaction is achieved through CDK-mediated phosphorylation of Rb. The initial phosphorylation is performed by Cyclin D/CDK4/CDK6 and followed by additional phosphorylation by Cyclin E/CDK2. Since we have shown that miR-188 downregulates the expression of Cyclin D/CDK4 and Cyclin E/CDK2 complexes, we wanted to ask whether overexpression of miR-188 would have an impact on Rb phosphorylation. The phosphorylation status of Rb at ser780 and ser811 was detected using western blot. We found that miR-188 suppressed CDK-mediated Rb phosphorylation since silencing endogenous miR-188 with ant-188 increased the amount of Rb phosophorylation while miR-188 transfected cells showed less Rb phosphorylation (Figure 3E and F, Additional file 4: Figure S3B). Similarly, Rb phosphorylation at S811 and S780 residues were significantly reduced in CNE cells stably expressing miR-188 (Additional file 4: Figure S3C, D). These results indicate that miR-188 also plays an important role on Rb phosphorylation.
E2F acts as a transcription factor in the nucleus and activates down-stream gene expression to drive cell cycle progression. A reduction of Rb phosphorylation would affect its dissociation from E2F and therefore, affect E2F activation. Thus, we speculated that overexpression of miR-188 would lead to a downregulation of E2F transcriptional activity. Using a pE2F-TA-Luc plasmid that contains four copies of E2F binding elements, we showed that enforced expression of miR-188 remarkably decreased E2F transcriptional activity, as determined by reduced luciferase activity (Figure 3G). These results suggest that miR-188 downregulates CDK-mediated Rb phosphorylation and subsequent activation of E2F.
MiR-188 directly binds to the 3′UTRs of target genes
To determine whether miR-188 indeed target CCND1, CCND3, CCNA2, CCNE1, CDK2 and CDK4, we employed luciferase reporter assay. The potential miR-188 binding sequences within the 3′UTRs of target genes were predicted by FindTar (Figure 4). The 3′UTR fragments of target genes containing wild-type (WT) or seed region mutated (Mu) of miR-188 binding sites were inserted into a dual-luciferase reporter vector. When introduced into cells, the wild-type 3′UTR reporters of CCND1, CCND3, CCNA2, CCNE1, CDK2 and CDK4 exhibited a significant reduction of luciferase activity in the miR-188 transfected cells as compared to control. In contrast, the reporter vectors carrying seed mutated 3′UTR fragments abrogated the inhibitory effect of miR-188 on luciferase activity (Figure 4). Therefore, we concluded that miR-188 inhibits target-gene expression through direct interaction with their 3′UTRs.
Figure 4.
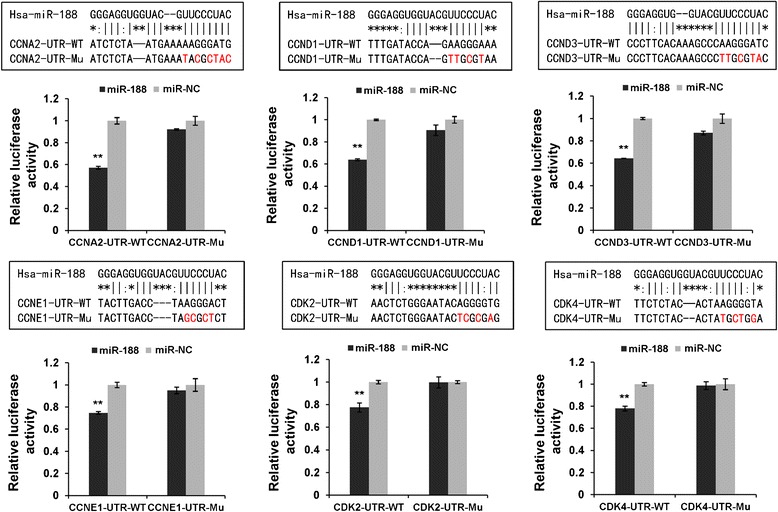
MiR-188 regulates target genes expression through direct binding to their 3′UTRs. Sequences of miR-188 and predicted miR-188-binding sites at CCNA2, CCND1, CCND3, CCNE1, CDK2 and CDK4 3′UTRs. The mutant sequences (Mu) are identical to the wild-type (WT) except the mutated nucleotides are shown in red. Luciferase vectors were generated by inserting WT or Mu 3′UTRs of target genes into pmirGLO plasmid. The reporter vectors were co-transfected with miR-188 or miR-NC. Relative luciferase activity was determined at 24 h after transfection. Results shown are means ± SD of three independent experiments (Student t test, **p < 0.01).
MiR-188 suppresses tumor formation and progression in nude mice
In order to determine the anti-cancer potential of miR-188 in vivo, nude mice were injected with CNE cells that stably expressed miR-188 or miR-NC. Strikingly, tumor induction was dramatically prevented in miR-188 injected mice (Figure 5A). Tumor mass and tumor volume were also significantly lower in mice injected with miR-188 (Figure 5B and C). We then investigated the expression of miR-188 targets in tumor tissues using western blot. The protein levels of Cyclin E1, Cyclin D3, Cyclin D1,Cyclin A2, CDK2 and CDK4 were significantly decreased in tumor tissues harvested from miR-188 injected mice (Figure 5D and E). Compared with tumor tissues from control mice, an obvious reduction of Rb S811 and S780 phosphorylation was also detected in miR-188 injected mice (Figure 5F and G). These results provide strong evidence that miR-188 suppresses tumor initiation and progression in vivo, potentially through downregulation of multiple genes involved in G1/S transition.
Figure 5.

MiR-188 suppresses tumor initiation and progression in vivo . (A) CNE cells stably expressing miR-188 or miR-NC were injected into nude mice. MiR-188 (n = 12) suppressed tumorigenesis compared with miR-NC (n = 10). (B) miR-188 inhibits tumor growth in vivo. Tumor volumes were calculated by the length and width measured by vernier calipers every 2 days. (C) The weight of tumors from mice from (B). miR-NC (mean: 327.9 mg; range: 39.6 to 659.4 mg); miR-188 (mean: 83.2 mg; range: 19.5 to 136.6 mg). (D) Immunoblot analysis of cyclin D1, cyclin D3, cyclin E1, cyclin A2, CDK2, CDK4 and GAPDH in tumors treated with miR-NC or miR-188. (E) Relative protein expression was quantified by densitometric analysis, GAPDH was used as internal control (Student t test, *p < 0.05). (F) Immunoblot analysis of phosphor-Rb Ser811, phosphor-Rb Ser780 and total Rb in tumors treated with miR-NC or miR-188. (G) Relative Rb phosphorylation was quantified by densitometric analysis, total Rb was used as internal control (Student t test, *p < 0.05).
Discussion
Being capable of affecting multiple targets is one of the most important feature of miRNAs. One miRNA can regulate several targets in a signaling pathway, alternatively, several miRNAs can also converge on a single target [13,14,26]. Co-regulation of a group of functionally related genes to provoke detectable functional changes is a common mode of miRNA-mediated gene regulation. In this study, we demonstrate that miR-188 exerts anti-cancer potential via downregulating multiple G1/S related genes, including CCND1, CCND3, CCNE1, CCNA2, CDK2 and CDK4. By suppressing the activation of G1/S related Cyclin/CDKs, miR-188 suppresses Rb phosphorylation and subsequent activation of E2F transcription factor, which leads to G1/S cell cycle arrest and growth inhibition in cancer cells (Figure 6).
Figure 6.

MiR-188 targets multiple genes involved in G 1 /S cell cycle transition. MiR-188 suppresses cyclin D/CDK4, cyclin E/CDK2 and cyclin A/CDK2 activity by directly targeting their 3′UTRs. MiR-188 mediated downregulation of G1/S CDKs suppresses Rb phosphorylation and E2F transcriptional activity, and finally leads to G1/S cell cycle arrest and growth inhibition.
Cell cycle is driven by the alternative expression or degradation of different cyclins which exert their function by binding to different CDKs [19,40]. There are two main groups of cyclins, G1/S Cyclins are essential for the control of cell cycle at the G1/S transition, G2/M Cyclins are essential for the control of cell cycle at G2/M transition and mitosis. Cyclin D is the first cyclin activated by binding to CDK4/6 in the cell cycle progression. The active Cyclin D/CDK4/6 complex phosphorylates Rb protein [41]. The phosphorylated Rb dissociates from Rb-E2F complex and activates E2F transcription factor. E2F drives the expression of multiple genes that are involved in regulating cell cycle progression, such as CCNE, CCNA, DHFR, MYB and DNA polymerase, etc. [42]. Cyclin E/CDK2 complex pushes cell cycle through G1/S transition and Cyclin A/CDK2 complex drives cells through S phase. A fundamental aspect of cancer is the dysregulation of cell cycle control. Alterations in the machinery that controls G1/S transition are frequently observed in many types of human cancers [43-45]. For instance, amplification of CCND1 gene or overexpression of Cyclin D1 protein has been found in a wide spectrum of human cancers [46,47]. CCND2 and CCND3 genes and their encoded proteins are also overexpressed in many cancer cases in humans [47-51]. Hyperactivation of CDK4 or CDK6 is also found in several human malignancies, which can be achieved through deregulated expression of D-type Cyclins, or loss of CDK inhibitor, p16INK4a [47,52-54]. Due to their critical role in cell cycle progression and tumorigenesis, the Cyclin/CDKs have been proposed as attractive therapeutic targets for anti-cancer treatment.
Accelerated cell cycle progression is the common feature of most cancers. Several lines of evidence implicate miRNAs as oncogenes or tumor suppressors through directly modulating cell cycle machinery [55]. For instance, the oncogenic miR-17/92 and miR-221/222 clusters can downregulate CDK inhibitors and Rb family members, leading to the activation of Cyclin/CDK complexes and cell cycle progression [56-59]. On the other hand, tumor suppressive miRNAs, such as let-7 and miR-15 families, downregulate a wide spectrum of positive regulators of the cell cycle machinery [55]. Our study provides strong evidence supporting miR-188 may function as a tumor suppressor and it is a potential candidate for anti-cancer therapy.
Conclusions
Our data demonstrate a novel mechanism of miR-188 control on cell proliferation and cell cycle progression. Through targeting multiple cyclin/CDK complexes, miR-188 blocks G1/S transition, suppresses Rb phosphorylation and E2F transcriptional activity. The expression of miR-188 also inversely correlates with expression of its targets in NPC tissues. Moreover, miR-188 is capable of inhibiting tumor initiation and progression in xenograft mouse model, indicating that miR-188 may have anti-cancer potential in human nasopharyngeal cancer.
Materials and methods
Cell culture and transfection
CNE cell line was purchased from Kunming Cell Bank (China) and cultured in Dulbecco’s modified Eagle’s medium (Gibco/Invitrogen, 12800–017) supplemented with 10% fetal bovine serum (PAA, A15-101) at 37°C in a humidified 5% CO2 incubator. Cells were transfected with siRNA or miRNAs duplexes using Lipofectaime 2000 (Invitrogen Corp., Carlsbad, CA, USA) according to manufacturer’s instructions. MiR-188 mimics and miRNA inhibitors were synthesized and purified by GenePharma Co. (Shanghai, China).
miRNA and mRNA expression
Total RNA from cultured cell were extracted with RNAiso plus (Takara, 9108) following the manufacture’s protocol. For miRNA expression assay, total RNA was reversely transcribed using Taqman MicroRNA Reverse Transcription Kit (Life Technologies, 4366597), then miRNA real-time PCR was performed using Taqman MicroRNA Assay Kit (Life Technologies). For mRNA expression assay, total RNA was reversely transcribed using M-MLV Reverse Transcription System (Takara) and SYBE Green PCR master mix (Toyobo, QPK-201) was purchased for mRNA real-time PCR. All quantitative real-time PCR were performed on an ABI 7300 Real Time System, RNU6 or GAPDH was used as internal control for miRNA and mRNA assay respectively. Relative gene expression was calculated by mean of relative quantification (2-ΔΔCt) as previously described [60]. The specific real-time PCR primers for each gene were listed in Table S2 (Additional file 5: Table S2).
MiR-188 stable cell line
Plasmid carrying GFP and miR-188 hairpin was purchased from GenePharma Co. (Shanghai, China). CNE cells were transiently transfected with miR-188 plasmid, followed by Blasticidin S (YEASEN, 60218ES10) selection at final concentration of 20 ug/ml. Stable clones were obtained based on GFP expression.
Colony formation and cell proliferation assays
CNE cells were plated at a concentration of 3 × 103 cells per well in 6-well plate and transfected with miR-188 or miR-NC every three day after cells adhesion. MiR-NC or miR-188 stable cells were plated at 4 × 103 cells per well in 6-well plate. After culturing for 10 days, cells were fixed with cold methanol and stained with 1% crystal violet. The number of colonies was counted using Gel-Pro analyzer software. For cell growth assay, cells were plated at a concentration of 2 × 104 cells per well in 24-well plate and transfected with miR-NC or miR-188, the number of viable cells was determined by the trypan-blue exclusion assay.
Bioinformatics analysis
In order to figure out the potential targets of miR-188, we used Targetscan [61] (http://www.targetscan.org/) and Findtar [62] (http://bio.sz.tsinghua.edu.cn/) algorithm to search human genome based on NCBI mRNA database (http://www.ncbi.nlm.nih.gov/). All predicted targets were uploaded to DAVID Bioinformatics Resources Functional Annotation Tool [38,39] (http://david.abcc.ncifcrf.gov/home.jsp) for pathway annotation and functional annotation clustering.
Western blotting
Cells were lysed in ice-cold whole cell lysis buffer(50 mM Tris–HCl, pH 8.0, 4 M urea and 1% Triton X-100) supplemented with complete protease inhibitor Cocktail (Roche Diagnostics, 04693132001). Whole cell lysates with equal protein were resolved by SDS-PAGE and transferred to nitrocellulose membrane. After blocking with 5% milk in Tris-buffered saline plus 0.02% Tween-20 (TBST), membranes were incubated with the following antibodies: cyclin D1 (Epitomics, 1677–1), cyclin D3 (Epitomics, 1846–1), E2F1 (Cell Signaling, 3742), cyclin E (Santa Cruz, sc-481), cyclin A (Santa Cruz, sc-596), CDK2 (Cell Signaling, 2546), CDK4 (Cell Signaling, 2906); phosphor-Rb (Ser 780) (Cell Signaling, 3590), phosphor-Rb (Ser-811) (ABclonal Biotechnology, AP0089), total Rb (Cell Signaling, 9309) and GAPDH (Proteintech, 10494-1-AP). Membranes were then incubated with horseradish peroxidase-coupled specific secondary anti-mouse (KPL, 074–1806) or anti-rabbit antibodies (KPL, 474–1506). Protein bands were visualized using ECL blotting detection reagents (KPL, 54-61-00).
Clinical specimens
All nasopharyngeal carcinoma tissues were collected from tumor resection in the Peking University Shenzhen Hospital (Shenzhen, China). Informed consent was obtained from each patient and this study was approved by the local ethics committee.
Luciferase activity assay
The 3′ UTRs of CCND1, CCND3, CDK4, CCNA2, CDK2 and CCNE1 containing predicted targets of miR-188 and their corresponding mutants were amplified from reverse transcribed cDNA and cloned to pmirGLO Dual-Luciferase reporter vector (Promega, E1330). The primer sequences for the wild-type (WT) and mutated (Mu) constructs were listed in Table S3 (Additional file 5: Table S3). For luciferase activity assay, WT or Mu constructs were cotransfected with miR-NC or miR-188 into HeLa cell using Lipofectamine 2000. Firefly and Renilla luciferase activity were measured by Dual-Luciferase Reporter Assay System (Promega, E1960). Relative luciferase activity was calculated by normalization the ratio of firefly and Renilla luciferase to that of negative control transfected cells. For E2F activity assay, 200 ng of E2F reporter vector pE2F-TA-Luc (Clontech, 63914) was cotransfected with miR-NC or miR-188 into CNE cells [63]. Luciferase activity was determined at 24 h after transfection.
Cell synchronization and flow cytometry
For synchronization, cells transfected with miR-NC or miR-188 were treated with 2 mM hydroxyurea (Sigma-Aldrich, H8627) for 16 h to block cell at G1 phase. The synchronized cells were released by washing the cells 3 times with pre-warmed PBS. At 6 h after release, the cells were fixed in 70% ethanol in PBS overnight. Cells were then stained with 10 μg/ml Propidium Iodide (Sigma-Aldrich, P4170) for DNA content analysis by use of a BD Influx™ Flow Cytometer (BD Biosciences).
BrdU Elisa assay
BrdU elisa assay was performed by use of a Cell proliferation Brdu Elisa Assay kit (Roche, 11647229001). CNE cells transfected with indicated small RNAs were synchronized at G1 phase by hydroxyurea treatment. Cells were released and cultured with 10 μM BrdU for 2 h. After removing labeling medium, cells were fixed and incubated with anti-BrdU-POD working solution for 90 min. Cells were washed 3 times with washing solution and added substrate solution and incubated for 3–10 min at room temperature. The incorporation of BrdU during DNA synthesis in proliferating cells was determined by measuring the light emission of the samples in a microplate luminometer.
EdU imaging assay
CNE cells transfected with indicated small RNAs were synchronized at G1 phase by hydroxyurea treatment. Cells were released and incubated with 25 μM EdU and subsequently stained using a Click-iT EdU Alexa Fluor 488 Imaging kit (Life Technologies, C10337). Images were taken with an Olympus FV1000 confocal microscope (Olympus, Tokyo, Japan). The percentage of cells labeled with EdU was quantified using Image J software.
Tumor formation assay
Athymic nude mice aged 4 weeks were purchased from the Experimental Animal Center of Guang Zhou University of Chinese Medicine (Guang Zhou, China) and kept in pathogen-free animal facilities for 2 weeks at Tsinghua University Shenzhen Graduate School. All experimental procedures involving animals were approved by the Institutional Laboratory Animal Care and Use Committee of Experimental Animal Center of Guang Zhou University of Chinese Medicine. Approximately 5 × 105 viable CNE cells stably expressing miR-NC or miR-188 were injected subcutaneously into the dorsal flank of nude mice. Three weeks after tumor implantation, the mice were sacrificed and the tumor incidence and tumor weight of each animal was analyzed. The expression of miR-188 target genes of each tumor was detected by western blot.
Statistical analysis
Data presented as bar graphs were the means ± S.D. of at least three independent experiments. The student’s t test was used to evaluate the significant difference between 2 groups of data. A P value less than 0.05 was considered as statistically significant.
Acknowledgement
This work was supported by the National Natural Science Foundation of China and Canadian Institutes of Health Research (No. 81261120556), National Natural Science Foundation of China (No. 31100976), Basic Research Grant of Shenzhen (No. JCYJ20120616213930060) and China Postdoctoral Science Foundation funded project (No. 2013 M530604).
Abbreviations
- CCND1
Cyclin D1
- CCND3
Cyclin D3
- CDK
Cyclin dependent kinase
- CCNA2
Cyclin A2
- CCNE1
Cyclin E1
- Rb
Retinoblastoma protein
- DHFR
Dihydrofolate reductase
- ELISA
Enzyme-linked immunosorbent assay
- qRT-PCR
Quantitative real-time PCR
- HU
Hydroxyurea
- EdU
5-Ethynyl-2′-deoxyuridine
- BrdU
5-bromo-2′-deoxyuridine
- UTR
Untranslated region
Additional files
The expression of miR-188 in CNE cells. (A) The relative expression level of miR-188 in CNE cells transiently transfected with miR-188 or miR-NC. (B) The relative expression level of miR-188 in CNE cells stably expressing miR-188 or miR-NC (C1, clone 1; C2, clone 2).
DAVID annotation categories the most enrichment KEGG pathway. MAPK signaling pathway, ErbB signaling pathway, Axon guidance and Cell cycle were the first four pathways according to their p values.
Densitometric analysis of miR-188 target genes expression. (A) Relative protein levels of cyclin E1, CDK2, CDK4, cyclin A2, cyclin D1 and cyclin D3 in CNE cells transiently transfected with miR-NC or miR-188. (B) Relative protein levels of miR-188 targets in CNE cells stably expressing miR-NC or miR-188 (C1, clone 1; C2, clone 2). (C) Relative protein levels of miR-188 target genes in CNE cells transfected with Ant-NC or Ant-199. GAPDH was used as internal control. Student t test, *p < 0.05, **p < 0.01, ***p < 0.001.
Stable expression of miR-188 inhibits G1/S transition and Rb phosphorylation. (A) Flow Cytometry analysis of CNE cells stably expressing miR-NC or miR-188 released from hydroxyurea for 6 h. (B) Relative levels of Rb phosphorylation were quantified by densitometric analysis. Total Rb was used as internal control. Student t test, ** p < 0.01, ***p < 0.001. (C) Immunoblot analysis of phosphor-Rb S811, phosphor-Rb S780, total Rb and GAPDH in CNE cells stably expressing miR-NC or miR-188. (D) Relative levels of Rb phosphorylation in (C) were quantified by densitometric analysis, total Rb was used as internal control. Student t test, *p < 0.05, ** p < 0.01.
Primers used for qRT-PCR (Table S2) and luciferase activity assay (Table S3).
Footnotes
Jiangbin Wu, Qing Lv and Jie He contributed equally to this work.
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
JW, QL and JH carried out laboratory work including plasmid construction, biochemical studies, flow cytometry and statistical analysis. HZ, XM, WX and NH participated in design of some experiments. XM and KC helped in collection of clinical specimens and experiments in these specimens. JW, NX and YZ planned the study and drafted the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jiangbin Wu, Email: wyh0794@gmail.com.
Qing Lv, Email: lvtsing@gmail.com.
Jie He, Email: jie.he2004@gmail.com.
Haoxiang Zhang, Email: 380847315@qq.com.
Xueshuang Mei, Email: xueshuang_mei@163.com.
Kai Cui, Email: cui.kai1107@gmail.com.
Nunu Huang, Email: huangnunu2781781@126.com.
Weidong Xie, Email: xiewd@sz.tsinghua.edu.cn.
Naihan Xu, Email: xu.naihan@sz.tsinghua.edu.cn.
Yaou Zhang, Email: zhangyo@sz.tsinghua.edu.cn.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shyh-Chang N, Daley GQ. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell. 2013;12:395–406. doi: 10.1016/j.stem.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou J, Wang W, Gao Z, Peng X, Chen X, Chen W, Xu W, Xu H, Lin MC, Jiang S. MicroRNA-155 promotes glioma cell proliferation via the regulation of MXI1. PLoS One. 2013;8:e83055. doi: 10.1371/journal.pone.0083055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang J, Zhao H, Xin Y, Fan L. MicroRNA-198 inhibits proliferation and induces apoptosis of lung cancer cells via targeting FGFR1. J Cell Biochem. 2014;115:987–995. doi: 10.1002/jcb.24742. [DOI] [PubMed] [Google Scholar]
- 5.Huang HN, Chen SY, Hwang SM, Yu CC, Su MW, Mai W, Wang HW, Cheng WC, Schuyler SC, Ma N, Lu FL, Lu J. miR-200c and GATA binding protein 4 regulate human embryonic stem cell renewal and differentiation. Stem Cell Res. 2013;12:338–353. doi: 10.1016/j.scr.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 6.Yi C, Xie WD, Li F, Lv Q, He J, Wu J, Gu D, Xu N, Zhang Y. MiR-143 enhances adipogenic differentiation of 3 T3-L1 cells through targeting the coding region of mouse pleiotrophin. FEBS Lett. 2011;585:3303–3309. doi: 10.1016/j.febslet.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Sun Q, Zhang Z, Ge S, Han ZG, Chen WT. Loss of microRNA-143/145 disturbs cellular growth and apoptosis of human epithelial cancers by impairing the MDM2-p53 feedback loop. Oncogene. 2013;32:61–69. doi: 10.1038/onc.2012.28. [DOI] [PubMed] [Google Scholar]
- 8.Grueter CE, van Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassel-Duby R, Olson EN. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149:671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, Einhorn WS, Takeuchi A, Engreitz JM, Hagan JP, Kharas MG, Urbach A, Thornton JE, Triboulet R, Gregory RI, Altshuler D, Daley GQ, DIAGRAM Consortium; MAGIC Investigators The Lin28/let-7 axis regulates glucose metabolism. Cell. 2011;147:81–94. doi: 10.1016/j.cell.2011.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen HC, Chen GH, Chen YH, Liao WL, Liu CY, Chang KP, Chang YS, Chen SJ. MicroRNA deregulation and pathway alterations in nasopharyngeal carcinoma. Br J Cancer. 2009;100:1002–1011. doi: 10.1038/sj.bjc.6604948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foekens JA, Sieuwerts AM, Smid M, Look MP, de Weerd V, Boersma AW, Klijn JG, Wiemer EA, Martens JW. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A. 2008;105:13021–13026. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat Rev Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 13.He J, Wu J, Xu N, Xie W, Li M, Li J, Jiang Y, Yang BB, Zhang Y. MiR-210 disturbs mitotic progression through regulating a group of mitosis-related genes. Nucleic Acids Res. 2013;41:498–508. doi: 10.1093/nar/gks995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pencheva N, Tran H, Buss C, Huh D, Drobnjak M, Busam K, Tavazoie SF. Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent melanoma metastasis and angiogenesis. Cell. 2012;151:1068–1082. doi: 10.1016/j.cell.2012.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 16.Frankel LB, Lund AH. MicroRNA regulation of autophagy. Carcinogenesis. 2012;33:2018–2025. doi: 10.1093/carcin/bgs266. [DOI] [PubMed] [Google Scholar]
- 17.Dumortier O, Hinault C, Van Obberghen E. MicroRNAs and metabolism crosstalk in energy homeostasis. Cell Metab. 2013;18:312–324. doi: 10.1016/j.cmet.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 18.Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harbor Perspect Biol. 2013;5:a008904. doi: 10.1101/cshperspect.a008904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- 20.Pagano M, Pepperkok R, Verde F, Ansorge W, Draetta G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992;11:961–971. doi: 10.1002/j.1460-2075.1992.tb05135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murphree AL, Benedict WF. Retinoblastoma: clues to human oncogenesis. Science. 1984;223:1028–1033. doi: 10.1126/science.6320372. [DOI] [PubMed] [Google Scholar]
- 22.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 23.Neganova I, Lako M. G1 to S phase cell cycle transition in somatic and embryonic stem cells. J Anat. 2008;213:30–44. doi: 10.1111/j.1469-7580.2008.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- 25.Bonci D, Coppola V, Musumeci M, Addario A, Giuffrida R, Memeo L, D’Urso L, Pagliuca A, Biffoni M, Labbaye C, Bartucci M, Muto G, Peschle C, De MR. The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med. 2008;14:1271–1277. doi: 10.1038/nm.1880. [DOI] [PubMed] [Google Scholar]
- 26.Liu Q, Fu H, Sun F, Zhang H, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. miR-16 family induces cell cycle arrest by regulating multiple cell cycle genes. Nucleic Acids Res. 2008;36:5391–5404. doi: 10.1093/nar/gkn522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun F, Fu H, Liu Q, Tie Y, Zhu J, Xing R, Sun Z, Zheng X. Downregulation of CCND1 and CDK6 by miR-34a induces cell cycle arrest. FEBS Lett. 2008;582:1564–1568. doi: 10.1016/j.febslet.2008.03.057. [DOI] [PubMed] [Google Scholar]
- 28.Kozaki K, Imoto I, Mogi S, Omura K, Inazawa J. Exploration of tumor-suppressive microRNAs silenced by DNA hypermethylation in oral cancer. Cancer Res. 2008;68:2094–2105. doi: 10.1158/0008-5472.CAN-07-5194. [DOI] [PubMed] [Google Scholar]
- 29.Pierson J, Hostager B, Fan R, Vibhakar R. Regulation of cyclin dependent kinase 6 by microRNA 124 in medulloblastoma. J Neuro-Oncol. 2008;90:1–7. doi: 10.1007/s11060-008-9624-3. [DOI] [PubMed] [Google Scholar]
- 30.Lujambio A, Ropero S, Ballestar E, Fraga MF, Cerrato C, Setien F, Casado S, Suarez-Gauthier A, Sanchez-Cespedes M, Git A, Spiteri I, Das PP, Caldas C, Miska E, Esteller M. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67:1424–1429. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 31.Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M, Vandenberg SR, Ginzinger DG, James CD, Costello JF, Bergers G, Weiss WA, Alvarez-Buylla A, Hodgson JG. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alteri A, De Vito F, Messina G, Pompili M, Calconi A, Visca P, Mottolese M, Presutti C, Grossi M. Cyclin D1 is a major target of miR-206 in cell differentiation and transformation. Cell Cycle. 2013;12:3781–3790. doi: 10.4161/cc.26674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen D, Farwell MA, Zhang B. MicroRNA as a new player in the cell cycle. J Cell Physiol. 2010;225:296–301. doi: 10.1002/jcp.22234. [DOI] [PubMed] [Google Scholar]
- 34.Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003;9:175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee K, Kim JH, Kwon OB, An K, Ryu J, Cho K, Suh YH, Kim HS. An activity-regulated microRNA, miR-188, controls dendritic plasticity and synaptic transmission by downregulating neuropilin-2. J Neurosci Off J Soc Neurosci. 2012;32:5678–5687. doi: 10.1523/JNEUROSCI.6471-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Y, Zhou B, Wu D, Yin Z, Luo D. Baicalin modulates microRNA expression in UVB irradiated mouse skin. J Biomed Res. 2012;26:125–134. doi: 10.1016/S1674-8301(12)60022-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS One. 2006;1:e116. doi: 10.1371/journal.pone.0000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 39.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murray AW. Recycling the cell cycle: cyclins revisited. Cell. 2004;116:221–234. doi: 10.1016/S0092-8674(03)01080-8. [DOI] [PubMed] [Google Scholar]
- 41.Munro S, Carr SM, La Thangue NB. Diversity within the pRb pathway: is there a code of conduct? Oncogene. 2012;31:4343–4352. doi: 10.1038/onc.2011.603. [DOI] [PubMed] [Google Scholar]
- 42.Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/S0092-8674(03)00401-X. [DOI] [PubMed] [Google Scholar]
- 43.Bukholm IR, Bukholm G, Nesland JM. Over-expression of cyclin A is highly associated with early relapse and reduced survival in patients with primary breast carcinomas. Int J Cancer. 2001;93:283–287. doi: 10.1002/ijc.1311. [DOI] [PubMed] [Google Scholar]
- 44.Taran K, Owecka A, Kobos J. Prognostic importance of cyclin E1 expression in neuroblastic tumors in children. Pol J Pathol. 2013;64:149–152. doi: 10.5114/pjp.2013.36016. [DOI] [PubMed] [Google Scholar]
- 45.Baker SJ, Reddy EP. CDK4: a key player in the cell cycle, development, and cancer. Genes Cancer. 2012;3:658–669. doi: 10.1177/1947601913478972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi YJ, Li X, Hydbring P, Sanda T, Stefano J, Christie AL, Signoretti S, Look AT, Kung AL, von Boehmer H, Sicinski P. The requirement for cyclin D function in tumor maintenance. Cancer Cell. 2012;22:438–451. doi: 10.1016/j.ccr.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deshpande A, Sicinski P, Hinds PW. Cyclins and cdks in development and cancer: a perspective. Oncogene. 2005;24:2909–2915. doi: 10.1038/sj.onc.1208618. [DOI] [PubMed] [Google Scholar]
- 48.Xu XL, Chen SZ, Chen W, Zheng WH, Xia XH, Yang HJ, Li B, Mao WM. The impact of cyclin D1 overexpression on the prognosis of ER-positive breast cancers: a meta-analysis. Breast Cancer Res Treat. 2013;139:329–339. doi: 10.1007/s10549-013-2563-5. [DOI] [PubMed] [Google Scholar]
- 49.Peurala E, Koivunen P, Haapasaari KM, Bloigu R, Jukkola-Vuorinen A. The prognostic significance and value of cyclin D1, CDK4 and p16 in human breast cancer. Breast Cancer Res. 2013;15:R5. doi: 10.1186/bcr3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li ZM, Spagnuolo L, Mensah AA, Rinaldi A, Bhagat G, Zucca E, Doglioni C, Ferreri AJ, Ponzoni M, Bertoni F. Gains of CCND3 gene in ocular adnexal MALT lymphomas: an integrated analysis. Br J Haematol. 2013;160:719–722. doi: 10.1111/bjh.12161. [DOI] [PubMed] [Google Scholar]
- 51.Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, Genesca L, Trimarchi T, Kelliher MA, Clark M, Soulier J, Chen-Kiang S, Aifantis I. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell. 2012;22:452–465. doi: 10.1016/j.ccr.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.An HX, Beckmann MW, Reifenberger G, Bender HG, Niederacher D. Gene amplification and overexpression of CDK4 in sporadic breast carcinomas is associated with high tumor cell proliferation. Am J Pathol. 1999;154:113–118. doi: 10.1016/S0002-9440(10)65257-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Costello JF, Plass C, Arap W, Chapman VM, Held WA, Berger MS, Su Huang HJ, Cavenee WK. Cyclin-dependent kinase 6 (CDK6) amplification in human gliomas identified using two-dimensional separation of genomic DNA. Cancer Res. 1997;57:1250–1254. [PubMed] [Google Scholar]
- 54.Borg A, Sandberg T, Nilsson K, Johannsson O, Klinker M, Masback A, Westerdahl J, Olsson H, Ingvar C. High frequency of multiple melanomas and breast and pancreas carcinomas in CDKN2A mutation-positive melanoma families. J Natl Cancer Inst. 2000;92:1260–1266. doi: 10.1093/jnci/92.15.1260. [DOI] [PubMed] [Google Scholar]
- 55.Bueno MJ, Malumbres M. MicroRNAs and the cell cycle. Biochim Biophys Acta. 1812;2011:592–601. doi: 10.1016/j.bbadis.2011.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Inomata M, Tagawa H, Guo YM, Kameoka Y, Takahashi N, Sawada K. MicroRNA-17-92 down-regulates expression of distinct targets in different B-cell lymphoma subtypes. Blood. 2009;113:396–402. doi: 10.1182/blood-2008-07-163907. [DOI] [PubMed] [Google Scholar]
- 57.Wu S, Huang S, Ding J, Zhao Y, Liang L, Liu T, Zhan R, He X. Multiple microRNAs modulate p21Cip1/Waf1 expression by directly targeting its 3′ untranslated region. Oncogene. 2010;29:2302–2308. doi: 10.1038/onc.2010.34. [DOI] [PubMed] [Google Scholar]
- 58.Fornari F, Gramantieri L, Ferracin M, Veronese A, Sabbioni S, Calin GA, Grazi GL, Giovannini C, Croce CM, Bolondi L, Negrini M. MiR-221 controls CDKN1C/p57 and CDKN1B/p27 expression in human hepatocellular carcinoma. Oncogene. 2008;27:5651–5661. doi: 10.1038/onc.2008.178. [DOI] [PubMed] [Google Scholar]
- 59.Medina R, Zaidi SK, Liu CG, Stein JL, van Wijnen AJ, Croce CM, Stein GS. MicroRNAs 221 and 222 bypass quiescence and compromise cell survival. Cancer Res. 2008;68:2773–2780. doi: 10.1158/0008-5472.CAN-07-6754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 61.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 62.Ye W, Lv Q, Wong CK, Hu S, Fu C, Hua Z, Cai G, Li G, Yang BB, Zhang Y. The effect of central loops in miRNA:MRE duplexes on the efficiency of miRNA-mediated gene regulation. PLoS One. 2008;3:e1719. doi: 10.1371/journal.pone.0001719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Helin K, Wu CL, Fattaey AR, Lees JA, Dynlacht BD, Ngwu C, Harlow E. Heterodimerization of the transcription factors E2f-1 and Dp-1 leads to cooperative transactivation. Gene Dev. 1993;7:1850–1861. doi: 10.1101/gad.7.10.1850. [DOI] [PubMed] [Google Scholar]