

Abstract
The use of glucocorticoids (corticotherapy) in severe sepsis is one of the main controversial issues in critical care medicine. These agents were commonly used to treat sepsis until the end of the 1980s, when several randomized trials casted serious doubt on any benefit from high-dose glucocorticoids. Later, important progress in our understanding of the role played by the hypothalamic–pituitary–adrenal axis in the response to sepsis, and of the mechanisms of action of glucocorticoids led us to reconsider their use in septic shock. The present review summarizes the basics of the physiological response of the hypothalamic–pituitary–adrenal axis to stress, including regulation of glucocorticoid synthesis, the cellular mechanisms of action of glucocorticoids, and how they influence metabolism, cardiovascular homeostasis and the immune system. The concepts of adrenal insufficiency and peripheral glucocorticoid resistance are developed, and the main experimental and clinical data that support the use of low-dose glucocorticoids in septic shock are discussed. Finally, we propose a decision tree for diagnosis of adrenal insufficiency and institution of cortisol replacement therapy.
Keywords: adrenal insufficiency, glucorticoids, hormone replacement therapy, hypothalamic–pituitary–adrenal axis, sepsis
Introduction
Ever since the discovery of the Waterhouse–Friderichsen syndrome, the occurrence of acute adrenal failure during sepsis has remained controversial. The use of glucocorticoids (corticotherapy) during sepsis has also been a subject of debate. Initially used in sepsis at high doses for its anti-inflammatory properties, corticotherapy was abandoned during the 1980s after several studies of corticotherapy in septic shock showed no benefit from treatment. However, recent evidence of adrenal dysfunction during sepsis and a better understanding of the mechanisms of action of glucocorticoids have led to a reconsideration of the role of glucocorticoids in sepsis.
Major physiologic properties and actions of glucocorticoids
The adrenal gland consists of two functional units: the medulla and the cortex. Production of the sympathetic system hormones (adrenaline [epinephrine] and noradrenaline [norepinephrine]) is localized in the medulla. The adrenal cortex consists of three zones, each of which synthesizes specific groups of hormones. Zona glomerulosa, which is superficially located, produces mineralocorticoids (aldosterone and, to a lesser extent, corticosterone); whereas zona reticularis, which is deeper set, produces weak androgens (dehydroepiandrosterone, dehydroepiandrosterone sulphate, Δ4-androstenedione and 11β-hydroxyandrostenedione). Finally, glucocorticoids (cortisol and cortisone) are produced by the zona fasciculata.
Cortisol, the main glucocorticoid, is a steroid hormone of 19 carbon atoms derived from the conversion of cholesterol by an enzymatic chain that belongs to the P450 cytochrome. Cortisol circulates in plasma either in its free and active form (which accounts only for 5–10% of total cortisol) or in its inactive form, reversibly bound to proteins. The two main binding proteins are the cortisol-binding globulin (CBG) and albumin [1].
Because of its lipophylic nature, cortisol enters the cells passively and binds to a soluble cytosolic receptor – glucocorticoid receptor type II – which in its inactive form is bound to heat shock protein-90. The glucocorticoid-receptor complex enters the nucleus and interacts directly with specific DNA sites (glucocorticoid-responsive elements), exerting both inhibitory and activating actions on transcription [2]. Study of the effect of glucocorticoids on gene expression of mononuclear cells shows that glucocorticoids upregulate and down-regulate up to 2000 genes that are involved in regulation of the immune response [3].
Cortisol is metabolized by the liver (reduced and conjugated) as well as by the kidney, where it is converted into its inactive metabolite cortisone by the 11β-hydroxysteroid dehydrogenase. Steroids that possess a ketone group at position 11 have an affinity for both glucocorticoid and mineralocorticoid receptors, which accounts for the weak mineralocorticoid activity of glucocorticoids.
Regulation of glucocorticoid production
The production of glucocorticoids is regulated by the hypothalamic–pituitary axis. Cortisol production and secretion is stimulated mainly by the adrenocorticotrophic hormone (ACTH). This peptide is comprises 39 amino acids and is produced in the anterior pituitary. It is liberated by cleavage of a large precursor, the pro-opiomelanocortin, which also liberates other peptides (β-endorphin, lipotropin, melanocyte-stimulating hormone). In the short term, ACTH stimulates cortisol production and secretion (cortisol storage in adrenal glands being low); in the longer term, ACTH also stimulates the synthesis of enzymes that are involved in cortisol production, as well as their cofactors and adrenal receptors for low-density lipoprotein cholesterol. ACTH also stimulates the production of adrenal androgens and, to a lesser extent, that of mineralocorticoids [1].
The half-life of ACTH is short and its action is fast because cortisol concentration in adrenal veins rises only a few minutes after ACTH secretion [4]. ACTH secretion is regulated by several factors. The main stimulators of its production are the corticotrophin-releasing hormone (CRH) and arginine vasopressine (AVP), which are both secreted by the hypothalamus. AVP stimulates ACTH secretion only weakly but it strongly promotes CRH action. Catecholamines, angiotensin II, serotonin and vasoactive intestinal peptide are also known stimulators of ACTH secretion. Finally, some inflammatory cytokines influence ACTH secretion, exerting either a stimulatory action (IL-1, IL-2, IL-6, tumour necrosis factor [TNF]-α) or an inhibitory one (transforming growth factor-β) [4-6].
CRH is a 41-amino-acid peptide secreted by the hypothalamus. Liberated in the hypothalamic–pituitary portal system, it stimulates the production and the secretion of pro-opiomelanocortin. Many factors influence CRH secretion. Adrenergic agonists (noradrenaline) and serotonin stimulate its production whereas substance P, opioids and γ-aminobutyric acid inhibit it. Inflammation cytokines (IL-1, IL-2, IL-6, TNF-α) also influence production of CRH [4,7].
Finally, glucocorticoids exert a negative feedback on the hypothalamic–pituitary axis, inhibiting ACTH production as well as pro-opiomelanocortin gene transcription, and CRH and AVP production.
Secretion of the hypothalamic-pituitary axis hormones (ACTH, CRH and AVP) follows a pulsatile course with a circadian rhythm. The amplitude of the secretory pulses varies throughout the day and is greatest in the morning between 6 and 8 AM, rapidly decreasing until noon and decreasing more slowly until midnight [1].
Main actions of glucocorticoids
Since the discovery of cortisol by Kendall and Reichstein in 1937, the actions of glucocorticoids have been progressively identified and defined in many areas.
Metabolic effects
Glucocorticoids play a major role in glucose metabolism. They stimulate liver gluconeogenesis and glycogenolysis, promote the action of the other hormones that are involved in gluconeogenesis (glucagon and adrenaline), and inhibit cellular uptake of glucose by inducing peripheral insulin resistance. The main consequence of these actions is a rise in blood glucose concentration [8]. Glucocorticoids also influence fat metabolism, activating lipolysis and inhibiting glucose uptake by the adipocytes. They inhibit protein synthesis and activate proteinolysis in muscles, liberating amino acids that can serve as substrates for gluconeogenesis. Finally, they are involved in bone and mineral metabolism, activating osteoclasts, inhibiting osteoblasts, decreasing intestinal calcium uptake and increasing calcium urinary secretion by decreasing its renal reabsorption [1].
Immunological and anti-inflammatory effects
Immune cells present high-affinity receptors for glucocorticoids. Many effects of glucocorticoids on the immune and inflammatory responses have been described in vitro but their clinical relevance remains controversial. An anti-inflammatory effect is clinically observed when the hormones are given at supra-physiological doses. However, glucocorticoids influence the main mediators of the inflammatory response, namely lymphocytes, natural killer lymphocytess, monocytes, macrophages, eosinophils, neutrophils, mast cells and basophils [9]. Glucocorticoid administration is followed by a fall in circulating lymphocytes, which results from the passage of lymphocytes from main circulation toward lymphoid organs (e.g. spleen, adenopathies, thoracic canal). The opposite effect is observed with granulocytes, which accumulate in blood circulation, whereas neutrophil migration toward inflammatory sites is inhibited (resulting from decreased secretion of chemokines), which contributes to a decreased local inflammatory reaction. Macrophage secretion is inhibited by the production of migration inhibitory factor [10]. Finally, glucocorticoids stimulate eosinophil apoptosis [11].
Glucocorticoids are involved in the immune response by inhibiting the production of IL-12 by macrophages and monocytes, and therefore influencing lymphocyte differentiation by acting on the Th1/Th2 balance. IL-12 is a strong stimulator of interferon-γ gynthesis and inhibitor of IL-4 secretion. Inhibition of IL-12 secretion and of the expression of its receptors on T and natural killer lymphocytes favours IL-4 production and lifts the suppressive effects of IL-12 on Th2 activity. Th1 and Th2 lymphocytes are mutually inhibitory, and therefore the promotion of Th2 activity and humoral immunity is associated with a suppression of cellular immunity [9]. However, these in vitro observations must be confirmed in vivo. A recent study of cytokines expressed by 40 patients with septic shock, 20 of whom were treated with low doses of glucocorticoids, showed an increase in IL-12 secretion and did not show an increase in Th2 differentiation while under glucocorticoid treatment [12].
Glucocorticoids modulate the cytokine response observed during inflammation (Table 1). On the cellular level, this action is mediated by the inhibition of the production and of the activity of proinflammatory cytokines (IL-1, IL-2, IL-3, IL-6, interferon-γ, TNF-α), chemokines, eicosanoids, bradykinin and migration inhibitory factor [5,10,13,14]. This inhibition results both from the direct interaction between the glucocorticoid-receptor complex and the glucocorticoid responsive elements located on DNA, and from the inhibition of transcriptions factors such as nuclear factor-κB (mediated by the inhibitory factor IκB) and activator protein-1 [2]. Simultaneously, glucocorticoids stimulate the production of anti-inflammatory factors such as IL-1 receptor agonist, the soluble TNF receptor, IL-10 and transforming growth factor-β [15,16]. This anti-inflammatory activity is completed by inhibition of the production of cyclo-oxygenase-2 and of the inducible nitric oxide (NO) synthase, which are key enzymes in inflammation. Glucocorticoids also induce the production of lipocortin-1, which in turn inhibits the synthesis of leucotrienes and of phopholipase A2 – an important enzyme that is involved in the arachidonic acid cascade [4,17].
Table 1.
Main anti-inflammatory effects of glucocorticoids
| Anti-inflammatory effect | Details |
| Proinflammatory cytokine production | Inhibition of IL-2, IL-3, IL-4(?), IL-5, IFN-γ, GM-CSF synthesis by T lymphocytes |
| Inhibition of IL-1, TNF-α, IL-6, IL-8, IL-12, MIF synthesis by macrophages/monocytes | |
| Inhibition of IL-8 synthesis by neutrophils | |
| Anti-inflammatory cytokine production | Increase in IL-10, TGF-β, IL-1 receptor antagonist synthesis |
| Inflammatory cell migration | Inhibition of chemokine production (MCP-1, IL-8, MIP-1α) |
| Stimulation of MIF and lipocortine-1 production by macrophages | |
| Inflammation mediator expression | Inhibition of soluble PLA2, inducible COX-2 and inducible NOS synthesis |
| Cell membrane markers expression | Inhibition of CD14 expression on macrophages/monocytes |
| Inhibition of adhesion molecule expression (ICAM-1, ECAM-1, LFA-1, CD2) on endothelial cells | |
| Apoptosis | Activation of eosinophils and mature T lymphocyte apoptosis |
COX, cylo-oxygenase; ECAM, endothelial cell adhesion molecule; GM-CSF, granulocyte–macrophage colony-stimulating factor; ICAM, intercellular adhesion molecule; IFN, interferon; IL, interleukin; LFA, leucocyte function associated antigen; MCP, monocyte chemoattractant protein; MIF, migration inhibitory factor; MIP, macrophage inflammatory peptide; NOS, nitric oxide synthase; PL, phospholipase; TGF, transforming growth factor; TNF, tumour necrosis factor.
Cardiovascular effects
Glucocorticoids are involved in vascular reactivity. Indeed, although hypertension is a common complication of corticotherapy, hypotension is a key symptom of adrenal failure. Blocking the effects of endogenous cortisol in animals results in arterial hypotension, which seems secondary to an effect on peripheral resistances, whereas cardiac output is unaffected. This effect of cortisol appears independent of mineralocorticoid activity. Although the mechanisms of the vascular effects are not fully understood, glucocorticoids modulate vascular reactivity to angiotensin II and to catecholamines (adrenaline and noradrenaline). The increase in transcription and expression of glucocorticoid receptors might be one of the mechanisms involved [18]. Glucocorticoids also modulate vascular permeability and decrease production of NO as well as of other vasodilator factors [1,7].
Glucocorticoids and stress
The main role of the stress response is to maintain homeostasis. The hypothalamic–pituitary axis, along with the adrenergic and sympathetic nervous systems, are the main mediators of the stress response.
Almost all forms of stress (whether physical or psychological) are followed by an immediate increase in ACTH secretion, which is followed a few minutes later by an important rise in cortisol blood levels [19]. Stress also results in a decrease in CBG, leading to an increase in cortisol blood levels [20]. Moreover, free cortisol concentration can be enhanced at the inflammatory site by an increase in neutrophil elastase activity, which will contribute to cleavage of cortisol and CBG. Finally, cytokines may also increase the affinity of receptors for glucocorticoids [15]. During stress adrenal hormone production is characterized by a shift in the production of mineralocorticoids with a drop in aldosterone production, while renin levels rise. However, the clinical significance and consequences of this fall in mineralocorticoid production is unknown, and mineralocorticoid treatment in sepsis remains controversial. These events are associated with a loss of the circadian rhythm of cortisol secretion secondary to an increase in CRH and ACTH production, stimulated by inflammatory cytokines, vagal stimulation and reduction in cortisol negative feedback [9,19].
As described above, a rise in glucocorticoid concentration results in multiple effects with the aim of maintaining homeostasis during stress. Metabolic effects, especially hyperglycaemia, contribute to increasing energetic substrates during a period that requires increased metabolism and transfer of available glucose to insulin-independent cells (e.g. central nervous system, inflammatory cells). Cardiovascular effects occur to maintain normal vascular reactivity during the stress period. Finally, glucocorticoids counteract almost every step of the inflammatory cascade, modulating the immune response. These different mechanisms are integrated in the adaptive response to stress. Inflammatory cytokines (e.g. TNF-α, IL-6, IL-1) acutely activate the hypothalamic–pituitary axis, which reduces the inflammatory response; cortisol, in turn, exerts a negative feedback on the hypothalamic–pituitary axis, which allows the body to regulate the period in which the immunosuppressive and catabolic effects of glucocorticoids are active [6]. However, over time, prolonged exposure to cytokines might lead to an altered response of the hypothalamic–pituitary axis. Thus, low levels of ACTH have been described in patients presenting with severe sepsis or systemic inflammatory response syndrome [14,21]. Likewise, chronic increase in IL-6 can lead to a decrease in ACTH production, while TNF-α may cause a reduction in adrenal function, CRH stimulation and ACTH production [7,22].
The concentration of circulating cortisol that is 'normal' for the response to stress remains controversial. Various levels have been proposed to define normal cortisol concentration, ranging from 15 to 20 μg/dl [7,23-26]. These values are based on the response observed after stimulation with exogenous ACTH (250 μg) or with insulin-induced hypoglycaemia in stable individuals. The relevance of these values in patients subjected to acute stress remains to be demonstrated. A rise in serum cortisol levels is observed in patients presenting with severe sepsis as well as in patients undergoing surgery [14,27-30]. Peak cortisol levels correlate with the severity of infection. Thus, Rothwell and Lawler [31], measuring serum cortisol levels on intensive care unit admission in 260 patients, observed significantly higher levels in patients who did not survive. Serum cortisol level was an independent predictive factor for outcome, reflecting the intensity of the activation of the hypothalamic–pituitary axis as well as the severity of the 'stressful' factor.
Stress response is associated with a rise in serum cortisol levels, but the absolute value that represents an appropriate response is not known. It probably varies according to the underlying cause (depending on the importance of the surgical procedure, whether the patient presents with trauma or severe sepsis) [19,32]. Moreover, a dynamic evaluation of adrenal function is necessary in order to appreciate the integrity of the hypothalamic–pituitary axis. This evaluation relies on ACTH stimulation tests, which explore ACTH receptor efficiency and the ability of adrenal cells to produce glucocorticoid. However, inducing hypoglycaemia with an insulin test may be dangerous in unstable patients. The dynamic response of serum cortisol levels after stimulation defines whether the hypothalamic–pituitary axis is responding appropriately. The 'normal' value after stress exposure is not known, but a rise of 9 μg/dl or more is commonly accepted as an appropriate response [33,34].
Glucocorticoids and sepsis
The initial phase of sepsis is associated with intense inflammatory activity, secondary to the identification of infectious components by the immune system. The major anti-inflammatory role played by glucocorticoids naturally led to consideration of their use in sepsis. The first evaluations of corticotherapy in severe sepsis were done with high doses of glucocorticoids and did not show any benefit on duration of shock or on outcome [35,36]. A meta-analysis of nine prospective randomized controlled studies [37] concluded that glucocorticoids have no favourable effects on morbidity and mortality in severe sepsis, and even suggested an increased risk for superinfection-related death.
The emerging concept that transient adrenal failure represents an aggravating factor during sepsis and septic shock led to reconsideration of the use of glucocorticoids in sepsis. Numerous factors interfere with the hypothalamic–pituitary axis response during sepsis. The presence of an underlying pituitary or adrenal pathology can lead to an acute adrenal failure, which can be triggered by sepsis. The use of specific drugs can interfere with adrenal function either by inhibiting the enzymes that are involved in cortisol synthesis (e.g. etomidate, ketokenazole) or by increasing cortisol metabolism (phenytoin, phenobarbital) [19]. Finally, in postmortem studies conducted in individuals who died from septic shock, bilateral adrenal haemorrhages or necrosis was observed in up to 30% of cases [14,19].
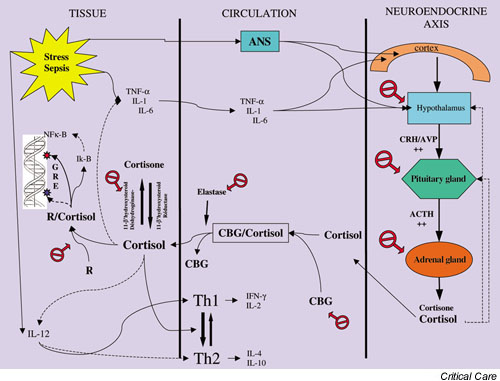
Several studies have shown that the frequency of adrenal failure, defined as an inappropriate glucocorticoid response during sepsis, depends largely on the threshold used [14,21,28,34,38-41]. Dysfunction of the adrenal–hypothalamic–pituitary axis during severe illness can result from a combination of different mechanisms, mainly adrenal failure and peripheral resistance to glucocorticoids (Fig. 1).
Figure 1.
Activation of the hypothalamic–pituitary–adrenal axis during acute stress. Activating effects are shown with plain arrows, and inhibitory effects with dotted arrows. The round symbols indicate the potential mechanisms that are involved in the axis dysfunction that occurs during sepsis either by failure of production or by tissue resistance to glucocorticoids. ACTH, adrenocorticotrophic hormone; AVP, arginine vasopressine; CBG, cortisol-binding globulin; CRH, corticotropin-releasing hormone; GRE, glucocorticoid responsive element; IFN, interferon; IL, interleukin; R, glucocorticoid receptor; ANS, autonomic nervous system; Th, T helper; TNF, tumour necrosis factor.
Adrenal failure
Briegel and coworkers [42] studied adrenal function during septic shock and after recovery in 20 patients. They showed that 13 of the patients presented with adrenal failure, which regressed after recovery from the sepsis. In 59 septic patients, Marik and Zaloga [39] also found primary adrenal failure in 25% and hypothalamic–pituitary axis failure in 17% of cases. In a study conducted in 189 patients presenting with severe sepsis [34], about 10% of patients had adrenal failure (defined in that study as serum cortisol levels < 20 μg/dl) whereas 50% of the patients had reversible adrenal failure defined as high basal serum cortisol levels but a blunted response to ACTH stimulation (increment in cortisol levels after 250 μg of ACTH of <9 μg/dl). The presence of adrenal failure was associated with a significant increase in mortality.
Peripheral resistance to glucorticoids
Inappropriate response to inflammation can be enhanced by tissue resistance to glucocorticoids. In the 59 septic patients they studied, Marik and Zaloga [39] found an incidence of 19% of ACTH resistance. Several factors may be involved and these probably interact: decreased access of cortisol to the inflammatory site secondary to the reduction in circulating CBG; modulation of local cortisol level by a reduction in the cleavage of CBG–cortisol complex (antielastase activity); reduction in the number and affinity of glucocorticoid receptors, shown on lymphocytes treated with different cytokines; and a rise in the conversion of cortisol in inactive cortisone by increased activity of the 11 β-hydroxyseroid dehydrogenase stimulated by IL-2, IL-4 and IL-13. These different mechanisms can account for decreased activity of glucocorticoids while serum cortisol level is apparently appropriate.
Effects of low-dose corticotherapy during sepsis
Anti-inflammatory effects
Patients in septic shock treated with low doses of hydrocortisone (300 mg/day over 5 days) exhibit a fall in temperature and heart rate associated with a decrease in inflammatory response (phospholipase A2 and C-reactive protein), proinflammatory cytokines and soluble adhesion complex, as well as an increase in anti-inflammatory cytokines [17]. Keh and coworkers [12] recently studied the effects of low-dose treatment with hydrocortisone (240 mg/day after a bolus dose of 100 mg) in 40 patients in septic shock and demonstrated a reduction in the production of inflammatory cytokines (IL-6 and IL-8), which was associated with a reduction in endothelial and neutrophil activation. The production of IL-10 and TNF-α soluble receptors (anti-inflammatory factors) was also decreased. After withdrawal of treatment, a rebound effect was observed in all of those mediators.
Cardiovascular effects
In healthy individuals, the local administration of lipopolysaccharide (endotoxin) is followed by a reduction in the contractile response to noradrenaline, which is fully prevented by pretreatment with hydrocortisone. Treatment with hydrocortisone simultaneously with or just before administration of lipopolysaccharide also prevents the occurrence of arterial hypotension and the rise of both heart rate and plasma adrenaline levels [43,44].
In numerous cases of septic shock, administration of low-dose corticotherapy was followed by an improvement in haemodynamic status and in response to vasopressor drugs [17,26,45,46]. Administration of a single dose of 50 mg hydrocortisone in septic shock was followed by a significant increase in blood pressure in patients treated with catecholamines [47,48]. A multicentre study of 300 patients with septic shock comparing the effects of low-dose treatment with hydrocortisone (50 mg every 6 hours) plus fludrocortisone with those of placebo over 7 days [26] showed a reduction in the duration of shock in treated patients, who exhibited blunted responses to the ACTH stimulation test. These effects were associated with a significant improvement in survival in treated patients. Keh and coworkers [12], in a double-blind study of the effects of low doses of hydrocortisone (240 mg/day after a 100 mg bolus) in 40 patients presenting with septic shock, also found an improvement in haemodynamic status in treated patients, with a reduction in the duration of shock and with recurrence of catecholamine dependency after withdrawal of glucocorticoid treatment. The increase in mean arterial blood pressure was associated with an increase in systemic resistance and a reduction in cardiac index and heart rate, suggesting that the effects of glucocorticoids mainly concern peripheral vascular tone. Several mechanisms for the vascular effects of glucocorticoids might be involved. The inhibition of NO production – a vasodilator – may result from direct inhibition of the inducible NO synthase by glucocorticoids [12]. Glucocorticoids might also increase the expression of catecholamine receptors, desensitized by the negative feedback of high circulating levels of catecholamines [49]. Finally, inhibition of the local production of inflammation factors or of the direct stimulation by guanylate cyclase might also be involved.
Effects on outcome
While studies concerning corticotherapy given at pharmalogical doses showed no benefit in terms of survival in patients presenting with severe sepsis or septic shock, there are now data favouring the use of low doses of hydrocortisone for at least 5 days in patients with septic shock.
In 18 critically ill patients with adrenal failure, daily doses of 200 mg hydrocortisone significantly increased survival rate compared with conventional treatment (90% versus 13%) [23]. In a randomized, placebo-controlled, double-blind trial of 41 septic shock patients [46], 100 mg hydrocortisone every 8 hours for 5 days significantly improved 28-day survival compared with placebo (63% versus 32%). These favourable effects of low-dose glucocorticoids in septic shock were confirmed in a phase III trial [26]. However, in that study only those patients with septic shock and a poor response to the ACTH test benefited from glucocorticoids.
Therapeutic strategies
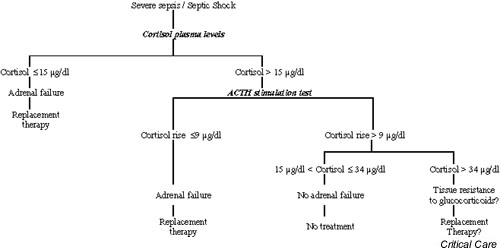
Adrenal failure can contribute to haemodynamic instability and can perpetuate inflammation, and should therefore be sought out in patients presenting with severe sepsis. Haemodynamic instability, high dependency on catecholamines despite control of infection, and occurrence of hypoglycaemia or of hypereosinophilia should lead to suspicion of adrenal failure. Basal serum cortisol levels of 15 μg/dl or less indicate adrenal insufficiency [24,26]. When cortisol levels are greater than 15 μg/dl, an ACTH stimulation test is required to rule out adrenal insufficiency. An increment of 9 μg/dl or less strongly suggests adrenal failure. Finally, an increase in cortisol levels in excess of 9 μg/dl when the basal level is greater than 34 μg/dl suggests tissue resistance to glucocorticoid (Fig. 2).
Figure 2.
Strategy for detection and treatment of adrenal failure during sepsis. ACTH, adrenocorticotrophic hormone.
The presence of adrenal failure requires prompt initiation of replacement therapy with low doses of corticosteroids. Replacement treatment with hydrocortisone (200–300 mg/day) can be combined with 9α-fludrocortisone (50 μg/day) and administered for 7 days in order to improve haemodynamic status and response to vasopressor treatment, allowing reduction in the duration of shock and improvements in short-term and long-term survival. The ongoing Corticotherapy for Septic Shock (CORTICUS) phase III trial, which is evaluating hydrocortisone alone in septic shock, may help in deciding whether to combine hydrocortisone with fludrocortisone.
Given the high frequency of adrenal dysfunction in septic shock, replacement therapy should be started immediately after the ACTH test is done, and be continued only in patients with adrenal failure.
Further studies are needed to better identify adrenal failure in septic shock. These studies should compare the low dose (1 μg) ACTH test with the traditional 250 μg ACTH test, using the metopirone test or induced hypoglycaemia as 'gold standards' for the diagnosis of adrenal failure.
Conclusion
Adrenal dysfunction is commonly observed in severely ill patients, especially in sepsis, and can increase morbidity and mortality. The diagnosis of adrenal failure in the critically ill remains a challenge and its criteria need to be improved. While the use of high-dose corticoid treatment in sepsis is not justified, replacement therapy by glucocorticoids can improve outcomes in patients in septic shock. Detecting and treating adrenal failure during severe sepsis or septic shock seem bound to become a major part in the management of the critically ill.
Competing interests
None declared.
Abbreviations
ACTH = adrenocorticotrophic hormone; AVP = arginine vasopressin; CBG = cortisol-binding globulin; CRH = corticotrophin-releasing hormone; IL = interleukin; NO = nitric oxide; Th = T helper; TNF = tumour necrosis factor.
References
- Orth DN, Kovacs WJ, DeBold CR. The Adrenal Cortex. In: Wilson JD, Foster DW, editor. In Williams Textbook of Endocrinology. Philadelphia: W.B. Saunders Company; 1992. pp. 489–531. [Google Scholar]
- Almawi WY. Molecular mechanisms of glucocorticoid effects. Mod Asp Immunobiol. 2001;2:78–82. [Google Scholar]
- Galon J, Franchimont D, Hiroi N, Frey G, Boettner A, Ehrhart-Bornstein M, O'Shea JJ, Chrousos GP, Bornstein SR. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- Cavaillon JM. Action of glucocorticoids in the inflammatory cascade [in French] Réanim Urgences. 2000;9:605–612. doi: 10.1016/S1164-6756(00)90035-5. [DOI] [Google Scholar]
- Tsigos C, Chrousos GP. Hypothalamic-pituitary-adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53:865–871. doi: 10.1016/S0022-3999(02)00429-4. [DOI] [PubMed] [Google Scholar]
- Marik PE, Zaloga GP. Adrenal insufficiency in the critically ill: a new look at an old problem. Chest. 2002;122:1784–1796. doi: 10.1378/chest.122.5.1784. [DOI] [PubMed] [Google Scholar]
- Pilkis SJ, Granner DK. Molecular physiology of the regulation of hepatic gluconeogenesis and glycolysis. Annu Rev Physiol. 1992;54:885–909. doi: 10.1146/annurev.ph.54.030192.004321. [DOI] [PubMed] [Google Scholar]
- Chrousos GP. The stress response and immune function: clinical implications. The 1999 Novera H. Spector Lecture. In: Conti A, Maestroni JM, McCann SM, Sternberg EM, Lipton JM, Smith CC, editor. In Neuroimmunomodulation Perspectives at the New Millennium. New York: Ann NY Acad Sci; 2000. pp. 38–67. [DOI] [PubMed] [Google Scholar]
- Beishuizen A, Thijs LG, Haanen C, Vermes I. Macrophage migration inhibitory factor and hypothalamo-pituitary-adrenal function during critical illness. J Clin Endocrinol Metab. 2001;86:2811–2816. doi: 10.1210/jc.86.6.2811. [DOI] [PubMed] [Google Scholar]
- Beishuizen A, Vermes I, Hylkema BS, Haanen C. Relative eosinophilia and functional adrenal insufficiency in critically ill patients. Lancet. 1999;353:1675–1676. doi: 10.1016/S0140-6736(99)01346-X. [DOI] [PubMed] [Google Scholar]
- Keh D, Boehnke T, Weber-Cartens S, Schulz C, Ahlers O, Bercker S, Volk HD, Doecke WD, Falke KJ, Gerlach H. Immunologic and hemodynamic effects of 'low-dose' hydrocortisone in septic shock: a double-blind, randomized, placebo-controlled, crossover study. Am J Respir Crit Care Med. 2003;167:512–520. doi: 10.1164/rccm.200205-446OC. [DOI] [PubMed] [Google Scholar]
- Beishuizen A, Thijs LG. Endotoxin and the hypothalamo-pituitary-adrenal (HPA) axis. J Endotoxin Res. 2003;9:3–24. doi: 10.1179/096805103125001298. [DOI] [PubMed] [Google Scholar]
- Soni A, Pepper GM, Wyrwinski PM, Ramirez NE, Simon R, Pina T, Gruenspan H, Vaca CE. Adrenal insufficiency occurring during septic shock: incidence, outcome, and relationship to peripheral cytokine levels. Am J Med. 1995;98:266–271. doi: 10.1016/S0002-9343(99)80373-8. [DOI] [PubMed] [Google Scholar]
- Franchimont D, Martens H, Hagelstein MT, Louis E, Dewe W, Chrousos GP, Belaiche J, Geenen V. Tumor necrosis factor alpha decreases, and interleukin-10 increases, the sensitivity of human monocytes to dexamethasone: potential regulation of the glucocorticoid receptor. J Clin Endocrinol Metab. 1999;84:2834–2839. doi: 10.1210/jc.84.8.2834. [DOI] [PubMed] [Google Scholar]
- Muller B, Peri G, Doni A, Perruchoud AP, Landmann R, Pasqualini F, Mantovani A. High circulating levels of the IL-1 type II decoy receptor in critically ill patients with sepsis: association of high decoy receptor levels with glucocorticoid administration. J Leukoc Biol. 2002;72:643–649. [PubMed] [Google Scholar]
- Briegel J, Kellermann W, Forst H, Haller M, Bittl M, Hoffmann GE, Buchler M, Uhl W, Peter K. Low-dose hydrocortisone infusion attenuates the systemic inflammatory response syndrome. The Phospholipase A2 Study Group. Clin Invest. 1994;72:782–787. doi: 10.1007/BF00180547. [DOI] [PubMed] [Google Scholar]
- Annane D, Bellissant E. Impact of corticosteroids on the vascular response to catecholamines in septic shock. Réanimation. 2002;11:111–116. [Google Scholar]
- Lamberts SW, Bruining HA, de Jong FH. Corticosteroid therapy in severe illness. N Engl J Med. 1997;337:1285–1292. doi: 10.1056/NEJM199710303371807. [DOI] [PubMed] [Google Scholar]
- Beishuizen A, Thijs LG, Vermes I. Patterns of corticosteroid-binding globulin and the free cortisol index during septic shock and multitrauma. Intensive Care Med. 2001;27:1584–1591. doi: 10.1007/s001340101073. [DOI] [PubMed] [Google Scholar]
- Schroeder S, Wichers M, Klingmuller D, Hofer M, Lehmann LE, von Spiegel T, Hering R, Putensen C, Hoeft A, Stuber F. The hypothalamic-pituitary-adrenal axis of patients with severe sepsis: altered response to corticotropin-releasing hormone. Crit Care Med. 2001;29:310–316. doi: 10.1097/00003246-200102000-00017. [DOI] [PubMed] [Google Scholar]
- Mastorakos G, Chrousos GP, Weber JS. Recombinant interleukin-6 activates the hypothalamic-pituitary-adrenal axis in humans. J Clin Endocrinol Metab. 1993;77:1690–1694. doi: 10.1210/jc.77.6.1690. [DOI] [PubMed] [Google Scholar]
- McKee JI, Finlay WE. Cortisol replacement in severely stressed patients [letter] Lancet. 1983;1:484. doi: 10.1016/S0140-6736(83)91489-7. [DOI] [PubMed] [Google Scholar]
- Cooper MS, Stewart PM. Corticosteroid insufficiency in acutely ill patients. N Engl J Med. 2003;348:727–734. doi: 10.1056/NEJMra020529. [DOI] [PubMed] [Google Scholar]
- Streeten DH. What test for hypothalamic-pituitary-adrenocortical insufficiency? Lancet. 1999;354:179–180. doi: 10.1016/S0140-6736(98)00318-3. [DOI] [PubMed] [Google Scholar]
- Annane D, Sebille V, Charpentier C, Bollaert PE, Francois B, Korach JM, Capellier G, Cohen Y, Azoulay E, Troche G, Chaumet-Riffaut P, Bellissant E. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. JAMA. 2002;288:862–871. doi: 10.1001/jama.288.7.862. [DOI] [PubMed] [Google Scholar]
- Jurney TH, Cockrell JL, Jr, Lindberg JS, Lamiell JM, Wade CE. Spectrum of serum cortisol response to ACTH in ICU patients. Correlation with degree of illness and mortality. Chest. 1987;92:292–295. doi: 10.1378/chest.92.2.292. [DOI] [PubMed] [Google Scholar]
- Loisa P, Rinne T, Kaukinen S. Adrenocortical function and multiple organ failure in severe sepsis. Acta Anaesthesiol Scand. 2002;46:145–151. doi: 10.1034/j.1399-6576.2002.460204.x. [DOI] [PubMed] [Google Scholar]
- Span LF, Hermus AR, Bartelink AK, Hoitsma AJ, Gimbrere JS, Smals AG, Kloppenborg PW. Adrenocortical function: an indicator of severity of disease and survival in chronic critically ill patients. Intensive Care Med. 1992;18:93–96. doi: 10.1007/BF01705039. [DOI] [PubMed] [Google Scholar]
- Schein RM, Sprung CL, Marcial E, Napolitano L, Chernow B. Plasma cortisol levels in patients with septic shock. Crit Care Med. 1990;18:259–263. doi: 10.1097/00003246-199003000-00002. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Lawler PG. Prediction of outcome in intensive care patients using endocrine parameters. Crit Care Med. 1995;23:78–83. doi: 10.1097/00003246-199501000-00015. [DOI] [PubMed] [Google Scholar]
- Chernow B, Alexander HR, Smallridge RC, Thompson WR, Cook D, Beardsley D, Fink MP, Lake CR, Fletcher JR. Hormonal responses to graded surgical stress. Arch Intern Med. 1987;147:1273–1278. doi: 10.1001/archinte.147.7.1273. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Udwadia ZF, Lawler PG. Cortisol response to corticotropin and survival in septic shock. Lancet. 1991;337:582–583. doi: 10.1016/0140-6736(91)91641-7. [DOI] [PubMed] [Google Scholar]
- Annane D, Sebille V, Troche G, Raphael JC, Gajdos P, Bellissant E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283:1038–1045. doi: 10.1001/jama.283.8.1038. [DOI] [PubMed] [Google Scholar]
- Anonymous Effect of high-dose glucocorticoid therapy on mortality in patients with clinical signs of systemic sepsis. The Veterans Administration Systemic Sepsis Cooperative Study Group. N Engl J Med. 1987;317:659–665. doi: 10.1056/NEJM198709103171102. [DOI] [PubMed] [Google Scholar]
- Bone RC, Fisher CJ, Jr, Clemmer TP, Slotman GJ, Metz CA, Balk RA. A controlled clinical trial of high-dose methylprednisolone in the treatment of severe sepsis and septic shock. N Engl J Med. 1987;317:653–658. doi: 10.1056/NEJM198709103171101. [DOI] [PubMed] [Google Scholar]
- Cronin L, Cook DJ, Carlet J, Heyland DK, King D, Lansang MA, Fisher CJ., Jr Corticosteroid treatment for sepsis: a critical appraisal and meta-analysis of the literature. Crit Care Med. 1995;23:1430–1439. doi: 10.1097/00003246-199508000-00019. [DOI] [PubMed] [Google Scholar]
- Annane D, Raphael JC, Gajdos P. Are endogenous glucocorticoid levels adequate in septic shock? Intensive Care Med. 1996;22:711–712. doi: 10.1007/BF01709752. [DOI] [PubMed] [Google Scholar]
- Marik PE, Zaloga GP. Adrenal insufficiency during septic shock. Crit Care Med. 2003;31:141–145. doi: 10.1097/00003246-200301000-00022. [DOI] [PubMed] [Google Scholar]
- Moran JL, Chapman MJ, O'Fathartaigh MS, Peisach AR, Pannall PR, Leppard P. Hypocortisolaemia and adrenocortical responsiveness at onset of septic shock. Intensive Care Med. 1994;20:489–495. doi: 10.1007/BF01711901. [DOI] [PubMed] [Google Scholar]
- Rothwell PM, Udwadia ZF, Jackson EA, Lawler PJ. Plasma cortisol levels in patients with septic shock. Crit Care Med. 1991;19:589–590. [PubMed] [Google Scholar]
- Briegel J, Schelling G, Haller M, Mraz W, Forst H, Peter K. A comparison of the adrenocortical response during septic shock and after complete recovery. Intensive Care Med. 1996;22:894–899. doi: 10.1007/s001340050184. [DOI] [PubMed] [Google Scholar]
- Barber AE, Coyle SM, Marano MA, Fischer E, Calvano SE, Fong Y, Moldawer LL, Lowry SF. Glucocorticoid therapy alters hormonal and cytokine responses to endotoxin in man. J Immunol. 1993;150:1999–2006. [PubMed] [Google Scholar]
- Bhagat K, Collier J, Vallance P. Local venous responses to endotoxin in humans. Circulation. 1996;94:490–497. doi: 10.1161/01.cir.94.3.490. [DOI] [PubMed] [Google Scholar]
- Briegel J, Forst H, Haller M, Schelling G, Kilger E, Kuprat G, Hemmer B, Hummel T, Lenhart A, Heyduck M, Stoll C, Peter K. Stress doses of hydrocortisone reverse hyperdynamic septic shock: a prospective, randomized, double-blind, single-center study. Crit Care Med. 1999;27:723–732. doi: 10.1097/00003246-199904000-00025. [DOI] [PubMed] [Google Scholar]
- Bollaert PE, Charpentier C, Levy B, Debouverie M, Audibert G, Larcan A. Reversal of late septic shock with supraphysiologic doses of hydrocortisone. Crit Care Med. 1998;26:645–650. doi: 10.1097/00003246-199804000-00010. [DOI] [PubMed] [Google Scholar]
- Bellissant E, Annane D. Effect of hydrocortisone on phenylephrine: mean arterial pressure dose-response relationship in septic shock. Clin Pharmacol Ther. 2000;68:293–303. doi: 10.1067/mcp.2000.109354. [DOI] [PubMed] [Google Scholar]
- Annane D, Bellissant E, Sebille V, Lesieur O, Mathieu B, Raphael JC, Gajdos P. Impaired pressor sensitivity to noradrenaline in septic shock patients with and without impaired adrenal function reserve. Br J Clin Pharmacol. 1998;46:589–597. doi: 10.1046/j.1365-2125.1998.00833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RS, Karl IE. The pathophysiology and treatment of sepsis. N Engl J Med. 2003;348:138–150. doi: 10.1056/NEJMra021333. [DOI] [PubMed] [Google Scholar]