

Background: GABAB receptors (GABABR) and glutamine synthetase (GS), are co-expressed in astrocytes.
Results: GABABRs bind to, stabilize, and target GS to the plasma membrane.
Conclusion: GABABRs are major determinants of GS subcellular targeting and stability.
Significance: Astrocytic GABABRs may play an unexpected role in regulating neurotransmission by promoting GS stability.
Keywords: Astrocyte, Glutamine Synthase, Neuroscience, Protein-Protein Interaction, Trafficking, Astrocytes, GABAB Receptors, Glutamine Synthetase, Neurosciences, Protein-Protein Interaction, traff
Abstract
Emerging evidence suggests that functional γ-aminobutyric acid B receptors (GABABRs) are expressed by astrocytes within the mammalian brain. GABABRs are heterodimeric G-protein-coupled receptors that are composed of R1/R2 subunits. To date, they have been characterized in neurons as the principal mediators of sustained inhibitory signaling; however their roles in astrocytic physiology have been ill defined. Here we reveal that the cytoplasmic tail of the GABABR2 subunit binds directly to the astrocytic protein glutamine synthetase (GS) and that this interaction determines the subcellular localization of GS. We further demonstrate that the binding of GS to GABABR2 increases the steady state expression levels of GS in heterologous cells and in mouse primary astrocyte culture. Mechanistically this increased stability of GS in the presence of GABABR2 occurs via reduced proteasomal degradation. Collectively, our results suggest a novel role for GABABRs as regulators of GS stability. Given the critical role that GS plays in the glutamine-glutamate cycle, astrocytic GABABRs may play a critical role in supporting both inhibitory and excitatory neurotransmission.
Introduction
γ-Aminobutyric acid (GABA)3 is the major inhibitory neurotransmitter in the central nervous system (CNS). GABAB receptors (GABABRs) are metabotropic receptors that are widely expressed in the brain, which mediate the slow and prolonged inhibitory neurotransmission (1, 2). Functional GABABRs are obligatory heterodimers and are composed of GABABR1 (R1) and GABABR2 (R2) subunits (3). They are localized both in pre- and post-synaptic sites where they respectively inhibit neurotransmitter release and activate potassium channels (2, 4).
Glutamine synthetase (GS) is an essential enzyme that catalyzes the conversion of glutamate and ammonium ions to glutamine, and therefore plays a critical role in nitrogen detoxification (5). In the brain, GS expression is restricted to glial with high levels being evident in astrocytes (5, 6, 7, 8).
GS plays an essential role in brain function. Disrupting GS expression specifically in astrocytes leads to neonatal death in mice. In humans deficits in GS activity are believed to contribute to numerous neuropsychiatric disorders, including temporal lobe epilepsy, Alzheimer disease, and schizophrenia (9, 10–15, 16).
While the consequences of GS activity for regulating excitatory glutamatergic neurotransmission are self-evident, this enzyme also plays a critical role in supporting inhibitory neurotransmission since glutamine is the major metabolic precursor for neuronal GABA synthesis (17). Thus locally decreasing GS activity in the brain leads to selective deficits in GABAergic inhibition and global inhibition of GS leads to gross deficits in neuronal inhibition as reflected by the appearance of seizures that precede death (18, 19). In accordance with these animal studies the expression levels of GS and its subcellular distribution are modified in temporal lobe epilepsy (20–22). Given the critical role that GS plays in facilitating neurotransmission, it is of fundamental importance to understand how astrocytes regulate GS expression level. GABABRs are also expressed in astrocytes however their physiological significance remains elusive (23–26).
In this study, we demonstrate that astrocytic GABABRs are important determinants of GS expression levels. Specifically, we reveal that GS binds directly to the cytoplasmic tail of the R2 subunit. This interaction determines the subcellular localization of GS and enhances its stability by limiting its proteasomal degradation. Collectively these results suggest that astrocytic GABABRs are key regulators of GS stability. Given the essential role GS plays in supporting GABA synthesis, astrocytic GABABRs may play a central role in determining the efficacy of GABAergic inhibition.
MATERIALS AND METHODS
Antibodies and Expression Constructs
Anti-glutamine synthetase mouse antibody (Millipore), anti-glutamine synthetase rabbit antibody (Sigma), anti-GFAP mouse antibody (Millipore), anti-actin mouse antibody (Sigma), anti-GABABR1 subunit mouse antibody (Neuromab), anti-GABABR2 subunit mouse antibody (Neuromab), anti-GABABR1 subunit goat antibody (Millipore), anti-GABAAβ3 subunit rabbit antibody, anti-ubiquitin that recognizes poly-ubiquitin (Santa Cruz Biotechnology) anti-mouse HRP, anti-rabbit HRP (Jackson Laboratories), TRITC donkey anti-mouse, Cy5 donkey anti-rabbit, FITC donkey anti-goat (Invitrogen), N-ethylmaleimide (Sigma), Flag-R2 matrix (Sigma), ImmunoCruz B (Santa Cruz Biotechnology), Lactacystin (Tocris), Leupeptin (Amresco). The following constructs were used: The MYC-GABABR1 and FLAG-GABABR2 expression vectors as well as the GST fusion protein vectors pGEX-CR1 (CR1, containing the C-terminal domain of GABABR1) and pGEX-CR2 (CR2, containing the C-terminal domain of GABABR2) have been previously described (1). Glutamine synthetase was cloned into prK5 from mouse genome (ATTG), and a MYC tag was inserted between amino acids 372 and 373.
Cell Culture and Transfection
COS-7 cells were maintained in Dulbecco's modified Eagle's medium/F12 (1:1) nutrient mix with 10% fetal bovine serum and 1% of PenStrep (Invitrogen). COS-7 cells were transfected with glutamine synthetase (GS) with empty plasmid pRK5, prK5-mycGABABR1, pRK5β3, and pRK5-flagGABABR2 using electroporation (1). Astrocytes were prepared from the cortex and hippocampus of 3-day-old mouse pups of either sex as previously described (27). Cells were grown in Advanced modified Eagle's medium (MEM), containing 10% fetal bovine serum (FBS), 1 mm pyruvate, 2 mm glutamine in 5% CO2. Confluent cultures were shaken at 225 rpm overnight, and the medium was changed the next morning; this process was repeated a total of three times. Cells were trypsinized and cultured for 24h in 10 μm cytosine arabinoside and allowed to grow to confluence. Cells were then transferred to 6 well-plates and used 48 h after transfection.
Immunoprecipitation and Metabolic Labeling
Cells were lysed in lysis buffer consisting of Tris, pH 8 20 mm, NaCl 150 mm, Triton 1%, EDTA 5 mm, NaF 10 mm, Na3VO4 2 mm, and Na pyrophosphate 10 mm for 1 h at 4 °C on a circular rotor. Lysates were precleared with mouse IgG attached to prot-G beads for 4 h at 4 °C. Lysates were then incubated over-night with 50 μl of Flag-R2 matrix. Precipitated immunocomplexes were washed, alternatively, in low salt (NaCl 150 mm) and high salt (500 mm NaCl) and very stringent buffer (NaCl 2 m and Triton 2%) and PBS 1× at 4 °C. Immunoprecipitated material antigen was resolved on a 8% SDS-PAGE gel and immunoblotted with Flag and/or GS antibodies and visualized using detection ECL (Super-Signal®; Pierce). Signals were then quantified using a LAS-3000 imager (Fujifilm). To measure ubiquitination cells were lysed in 1% SDS, NaF 50 mm, and EDTA 1 mm and then sonicated. Lysates were then diluted with lysis buffer supplemented with 10 mm of N-ethylmaleimide and immunoprecipitated with GS antibody. Precipitated material was then immunoblotted with anti-ubiquitin and GS antibodies. For metabolic labeling, COS-7 cells were incubated in methionine-free Dulbecco's modified Eagle's medium containing 100 μCi of [35S] for 30 min. Cells were then washed and incubated in complete media for up to 8 h at 37 °C. GS stability was then measured using immunoprecipitation as outlined above and [35S]methionine incorporation was quantified using a phospho-imager and Quantity One software (Bio-Rad). For the analysis of ubiquitination SDS-PAGE was performed under non-reducing conditions. These conditions were employed to prevent masking of GS immunoreactivity (45 kDa) by IgG heavy chain (50 kDa) on subsequent immunoblots.
In Vitro Binding Assays
GST (gluthathione S-transferase) fusion proteins were purified in BL21 (DE3) Escherichia coli (28) and bound to glutathione-agarose beads. 20 μg of those beads were incubated overnight at 4 °C with hippocampal lysates (lysis buffer: NaCl 2 m, Triton 2%, Tris pH8 20 mm, EDTA 5 mm, NaF 10 mm, Na3VO4 2 mm, Na pyrophosphate 10 mm). The beads were washed three times with lysis buffer, and samples were resolved by 10% SDS-PAGE and immunoblotted with GS antibody.
Biotinylation
To isolate cell surface proteins, COS-7 cells or astrocytes were labeled with 1 mg/ml NHS-Biotin (Pierce) at 4 °C for 30 min. Cultures were then lysed as outlined above, and detergent-soluble extracts were exposed to avidin beads (Pierce). Cell surface and total fractions were then subject to immunoblotting for GS, actin, or R2 antibody.
Immunofluorescence
48 h after transfection, cells were washed in PBS 1× at 4 °C and then fixed in PFA 4% followed exposure to 50 mm NH4Cl. After permeabilizing with 0.3% Triton, cells were incubated overnight with anti-glutamine synthetase mouse, anti-β3 rabbit, and anti-R2 goat antibodies. Cells were washed and then incubated with and TRITC anti-rabbit mouse (1:1000), Cy5 anti-rabbit rabbit (1:1000), and FITC anti-goat (1:1000) antibodies for 1 h at room temperature and mounted with Dako. Immunofluorescence was visualized with an invert confocal microscope (Nikon). Image acquisition was performed with NIS-Element software. Images were analyzed with ImageJ software. To measure colocalization we created regions of interest (ROI) corresponding to one cell expressing both R2 and GS. The program highlighted the colocalized points of two 8-bits images. The colocalized points appeared white by default. Two points were considered as colocalized if their respective intensities were strictly higher than the threshold of their channels. Percentage of colocalized pixels per area were then compared for R2 and GS immunoreactivity between treatments.
Membrane Fractionation
Mice hippocampi were homogenized with a glass Teflon homogenizer in ten times volume of ice-cold homogenization buffer (sucrose 0.32 m, HEPES 10 mm pH 7.4, EDTA 2 mm, EGTA 2 mm, NaF 50 mm, Na pyrophosphate 10 mm, and a mixture of protease inhibitors). Nuclei were removed by centrifugation at 1000 × g for 15 min. A crude membrane fraction was then isolate via centrifugation at 17,000. Following 2 washes in the above buffer membranes were solubilized in SDS-sample buffer. Membrane and cytosol fractions were then subject to immunoblotting for GS, tubulin, or R2 antibody.
Data Analysis
Data were analyzed using GraphPad PRISM, and statistical significance was determined at p < 0.05 using one-way ANOVA followed by Dunnett's multiple comparison post hoc test or Student's t test for two groups.
RESULTS
Glutamine Synthetase Binds to the GABABR2 Subunit
Prior mass spectroscopy analysis of GABABRs purified from rodent brain revealed the presence of GS, which in the adult brain is predominantly expressed in astrocytes (29). Given that glia express functional GABABRs, we sought to determine if these GPCRs were associated with GS, an enzyme that is predominantly localized to the cytoplasm. We compared the subcellular distribution of GABABRs and GS in crude membrane and cytosolic fractions prepared from rodent brain. The GABABR2 subunit and N-cadherin were both localized to the membrane fraction while tubulin was localized to cytosolic fractions. Comparatively, nearly equivalent levels of GS were found both in the cytosolic and membrane fractions (Fig. 1A).
FIGURE 1.

GS binds to the cytoplasmic tail of GABABR2. A, soluble and membrane fractions prepared from hippocampus were immunoblotted with N-cadherin (N-cadh), R2, tubulin, and GS antibodies. B, fusion proteins were exposed to hipppocampal extracts, subject to SDS-PAGE, transferred to a membrane stained with Ponceau S (upper panel) or immunoblotted with GS antibody (lower panel). GS levels were then compared with those seen with GST. Data represent mean ± S.E. (p < 0.01; ANOVA, n = 3). C, a schematic of the GST-R2 deletion constructs used for experimentation. D, fusion proteins were exposed to hippocampal extracts and processed as detailed above. The levels of GS binding were normalized to values for GST-R2. Data represent mean ± S.E. (p < 0.05; ANOVA, n = 4). *, significantly different from control, (p < 0.05; **, p < 0.01).
To assess if GS and GABABRs were capable of association we examined if the cytoplasmic intracellular domains of the GABABR1 and R2 subunits were capable of binding to GS. To do this, we expressed these regions as glutathione S-transferase fusion proteins in E. coli (GST, GST/R1 and GST/R2, respectively (1). Purified fusion proteins incubated with hippocampal lysates showed significantly higher levels of GS binding to GST-R2 but not GST-R1 (Fig. 1B; 3.4 ± 0.3 AU, p = 0.0093). To further delineate the binding site within the R2 intracellular domain for GS, we used constructs in which amino acids 762–828 (Δ1), 779–899 (Δ2), and 898–941 (Δ3) were deleted (Fig. 1C). Compared with GST-R2, Δ1 and Δ2 bound similar levels of GS (Δ1 = 1.013 ± 0.32 and Δ2 = 1.3 ± 0.55). However, binding to Δ3 was reduced to 0.06 ± 0.04 of control (Fig. 1D, p = 0.018). Collectively, these results suggest that residues 898–941 within GABABR2 are critical for GS binding.
To assess whether GS and R2 were capable of interacting in a cellular environment we used COS-7 cells, which do not express significant levels of either protein when compared with brain (Fig. 2A). Therefore, we expressed GS alone or together with a FLAG-tagged version of the GABABR2 subunit (30). Detergent soluble extracts from expressing cells were subject to immunoprecipitation with Flag beads and precipitated material was immunoblotted with GS and R2 antibodies. Under these conditions, co-immunoprecipitation of GS with R2 was evident (Fig. 2B). Our in vitro studies suggested that the interaction of GS with GABABR2 was dependent upon residues within the cytoplasmic C terminus. Importantly, deletion of the entire C-terminal intracellular domain did not compromise either the membrane trafficking of the R2 subunit or its ability to form functional GABABRs on co-expression with GABABR1 (31, 32). Therefore, we examined the effects of deleting 891–941 (R2ΔCT) on the ability of GS to immunoprecipitate with R2. The deletion included the hypothetical binding site of GS (Fig. 2C) and the reported phosphorylation site of PKA to exclude its eventual effect (1). We observed that using equimolar amounts of the respective plasmids that R2ΔCT was more stable than R2. To control for differences in expression level between R2 and R2ΔCT constructs, we compared the immunoreactivity ratios of GS:R2 and GS:R2ΔCT. The ratio of GS:R2ΔCT immunoreactivity was significantly reduced to 29 ± 12% compared with GS:R2 (Fig. 2C, p = 0.047). Together, these results suggest that the association between GS and GABABRs is dependent upon residues 891–941 in the R2 subunit.
FIGURE 2.
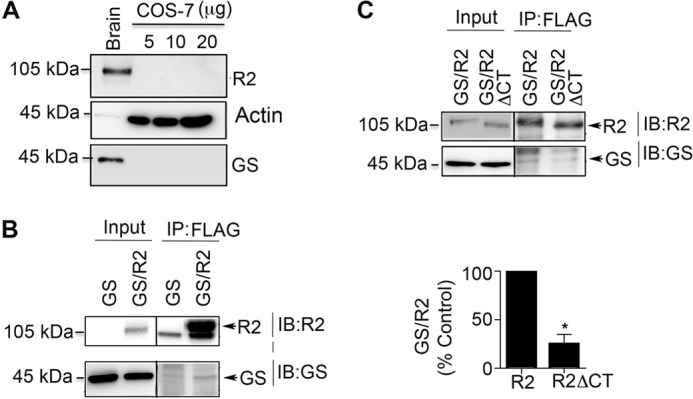
Association of GS and GABABRs is mediated via residues 898–941 in GABABR2. A, lysates of hippocampus (5 μg) and COS-7 cells were immunoblotted with R2, actin and GS antibodies. B, lysates from COS-7 cells expressing R2/GS or GS were immunoprecipitated with Flag beads and immunoblotted with R2 and GS antibodies. C, lysates from cells expressing GS/R2 or GS/R2ΔCT were immunoprecipiated with Flag antibody and immunoblotted with GS and R2 antibodies. The ratio of GS:R2 immunoreactivity was determined and normalized to values seen in wild type R2. Data represent mean ± S.E. (unpaired t test; p < 0.05; n = 3). *, significantly different from control (p < 0.05).
GABABRs Increase the Total Expression Levels of GS
To analyze the significance of GABABRs to GS expression we tested their role in regulating enzyme levels. Compared with cells expressing GS alone, co-expression with R2 increased GS levels to 180 ± 20% of control (Fig. 3A, p = 0.0032). This effect was also seen in cells expressing functional GABABRs composed of R1 and R2 (187 ± 19% of control; p = 0.0033).
FIGURE 3.
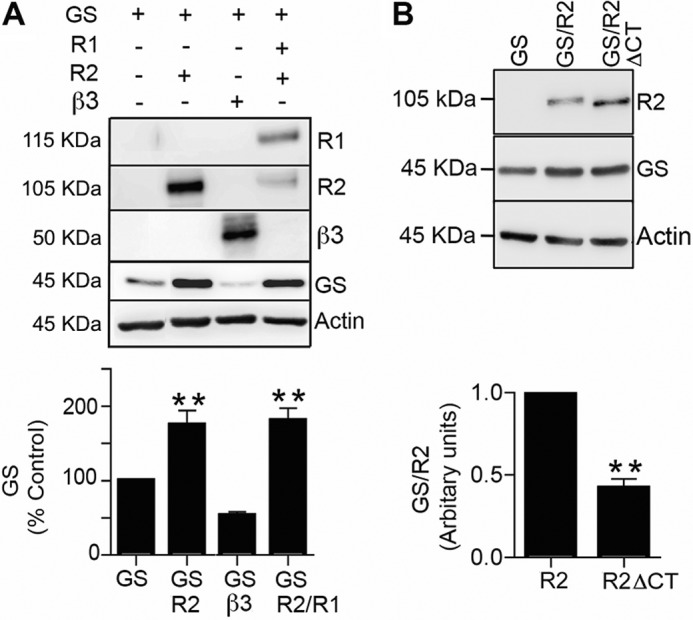
GABABRs increase GS expression levels. A, COS-7 cells expressing GS, GS/R2, GS/β3 and GS/R1/R2 were immunoblotted with R1, R2, β3, GS, and actin antibodies. The levels of GS expression were normalized to cells expressing this protein alone. Data represent mean ± S.E. (p < 0.01; ANOVA, n = 4). B, cells expressing GS, GS/R2 and GS/R2ΔCT were immunoblotted with R2 and GS antibodies. The ratio of GS:R2 immunoreactivity was determined and normalized to cells expressing wild type R2. Data represent mean ± S.E. (p < 0.01; unpaired t test, n = 3). *, significantly different from control (p < 0.001).
To test if the ability of the R2 subunit to stabilize GS expression is shared with other proteins we examined the effects of the GABAAR β3 subunit on GS stability. In common with GABABR2, GABAAR β3 is able to exit the endoplasmic reticulum and stably accumulate on the plasma membrane without the requirement for oligomerization with other GABAA receptor subunits or accessory proteins (33, 34). In contrast to R2, co-expression with β3 did not significantly modify the steady state accumulation of GS (Fig. 3A, p = 0.093).
The ability of GABABRs to interact with GS is dependent upon amino acids 891–941. Therefore we created a construct in which these resides were deleted (R2ΔCT). We observed that for the same amount of transfected DNA, R2ΔCT was more stable than R2. In order to control for variations in the expression levels of R2 and R2ΔCT we measured the ratio of GS:R2 immunoreactivity. Deletion of amino acids 891–941 significantly decreased this value to 45 ± 7% of control (Fig. 3B, p = 0.005). Collectively, these results suggest that GABABRs increase expression levels of GS, which is dependent on residues 891–941 in GABABR2.
GABABRs Enhance the Targeting of GS to the Plasma Membrane
To verify our immunoblotting data, we used immunocytochemistry to compare the expression levels and subcellular localization of GS when expressed alone or with GABABRs. To do so we compare the number of GS pixels/unit area under each condition. Compared with cells expressing GS alone co-expression with R2 increased GS levels from 41.7 to 50 ± 1.2 pixel/unit area (Fig. 4A; p = 0.0001). In contrast, co-expression with the β3 subunit reduced this value to 23.5 ± 1.7 (Fig. 4).
FIGURE 4.
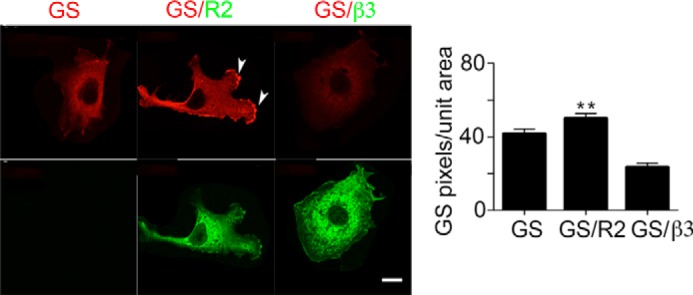
Analyzing the effects of GABABRs on the subcellular distribution of GS. COS-7 expressing GS, GS/R2 and GS/β3 were stained with the respective antibodies followed by confocal microscopy, scale bar = 20 microns, the arrows indicate GS staining associated with the membrane. The number of GS positive pixels was compared between treatments as shown in the righthand panel. Data represent mean ± S.E. (p < 0.01; ANOVA, n = 4). **, significantly different from control, (p < 0.01).
We noted in that in some of our images the presence of R2 appeared to lead to translocation of GS to the plasma membrane (see the arrows in Fig. 4). To further investigate this observation, cytosolic and membrane fractions were subject to immunoblotting. In cells expressing R2 there was a significant increase in the level of GS in the membrane fraction when compared with cells expressing GS alone (Fig. 5A, GS/R2; 179 ± 23% of control; p = 0.043). By comparing the ratio of GS:R2 and GS:R2ΔCT immunoreactivity it was evident that the accumulation of GS in the membrane fractions was dependent upon residues 891–941 in R2 (Fig. 5A, GS:R2ΔCT; 63.2 ± 8% of control; p = 0.021).
FIGURE 5.
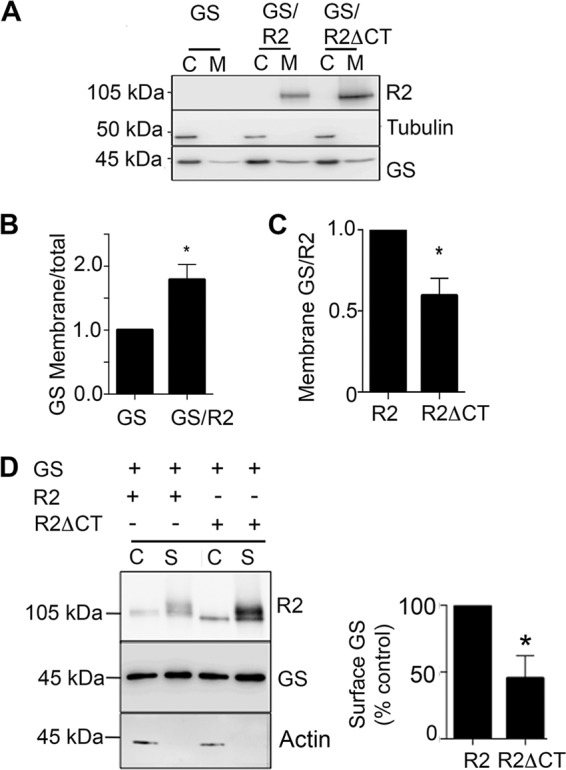
GABABRs target GS to the plasma membrane. A, cytosolic (C) and membrane fractions (M) from COS-7 cells expressing GS, GS/R2, and GS/R2ΔCT were immunoblotted with R2, tubulin, and GS antibodies. B, GS levels in the membrane fractions of cells expressing GS or GS/R2 were determined by the ratio membrane/total immunoreactivity (total = membrane+cytosolic) and then normalized to cells expressing GS. Data represent mean ± S.E. (unpaired t test, p < 0.05, n = 4). C, GS levels in the membrane fractions of cells expressing GS/R2 or GS/R2ΔCT were determined by GS:R2. The levels were then normalized to cells expressing R2. Data represent mean ± S.E. (unpaired t test; p < 0.05, n = 4). B, cells expressing GS/R2, or GS/R2ΔCT, were labeled with 1 mg/ml NHS-Biotin. Cells were then lysed and after purification on avidin the resulting cytosolic (C) and surface (S) fractions were immunoblotted with R2, GS, and actin antibodies. GS levels in surface fractions were then normalized to cells expressing R2 subunits. Data represent mean ± S.E. (unpaired t test; p < 0.05, n = 4). *, significantly different from control, (p < 0.05).
As an independent means of verifying this result cells we performed biotinylation experiments to compare the amount of GS at the plasma membrane in presence of either R2 or R2ΔCT. GS immunoreactivity was detected in the surface fraction with R2 and it was dependent on residues 891–941 (Fig. 5B, R2ΔCT = 48 ± 12% of control; p = 0.019). Together, these results suggest that the association between GS and GABABR2 subunit determines GS subcellular localization.
GABABRs Enhance GS Stability by Modulating Ubiquitination and Proteasomal Degradation
To assess the mechanisms by which GABABRs increase the steady state accumulation of GS, we compared its stability in the presence and absence of R2. To do so, COS-7 cells were labeled for 30 min with [35S]methionine washed and incubated at 37 °C for 8 h. The amount of remaining [35S]GS was then compared between treatments (Fig. 6A). When expressed alone 55 ± 10% of GS remained after 8 h. In contrast when co-expressed with R2, 96 ± 3% of GS remained (Fig. 6A, p = 0.033)
FIGURE 6.

GABABRs increase GS stability by reducing its ubiquitination and proteasomal degradation. A, COS-7 cells expressing GS or GS/R2 were labeled for 30 min with 100 μCi/ml [35S]methionine and incubated for 8 h. The level of 35S-labeled GS remains at 0, 4, and 8 h was measured using immunoprecipitation followed by SDS-PAGE (top lefthand panel). After quantification using a phospho-imager the amount of 35S-labeled GS was normalized to 0 time as shown in the lefthand panel Data represent mean ± S.E., (paired t test; p < 0.05, n = 3). B, cells expressing GS, GS/R2, or control non-transfected cells (NT) were subject to immunoprecipitated with GS antibody. Precipitated material was subject to SDS-PAGE and immunoblotted with GS and UB antibodies. The ratios of UB:GS immunoreactivity were determined and normalized to values seen in cells expressing GS alone. Data represent mean ± S.E., (unpaired t test; p < 0.01, n = 4). C, cells expressing GS and GS/R2 were treated with vehicle (−) or lactacystin (+) for 20 μm for 8 h. Lysates were subject to immunoblotting with R2, GS, and actin antibodies and GS levels were normalized to those seen in vehicle-treated controls (dotted line). Data represent mean ± S.E. (p < 0.01, paired t test, n = 4). *, significantly different from control, (p < 0.05); **, p < 0.01.
In Schwann cells GS is subject to ubiquitination and proteasomal degradation (35). Thus, we examined if GABABRs stabilize GS by regulating its ubiquitination. To do so cells expressing GS alone or with R2 were immunoprecipitated with GS antibody and immunoblotted with antibodies against poly-ubiquitin in addition to GS. To quantify ubiquitination gels were cut below 100 kDa to occlude IgG cross reactivity, and HRP reaction product was measured between 90–40 kDa, and corrected for nonspecific binding subtracting values seen from non-transfected cells. Using this approach it was evident that GS ubiquitination was significantly decreased to 59 ± 11% of control when expressed with R2 (Fig. 6B; p = 0.0098).
To further analyze GS degradation, expressing cells were treated with the proteasome inhibitor lactacystin for 8 h. Treatment of cells with lactacystin increased GS levels to 139 ± 9% of control (Fig. 6C, p = 0.004). The effects of lactacystin on GS stability were occluded by GABABR2 (Fig. 6C). Collectively, these results suggest that GABABRs increase GS stability by reducing its ubiquitination and subsequent proteasomal degradation.
GABABRs Are Determinants of GS Expression Levels in Cultured Astrocytes and the Brain
To examine the relevance of our studies in COS-7 cells, we assessed the role that GABABRs play in determining the stability of GS in cultured astrocytes. Previous studies have shown that cultured astrocytes express both GABABR receptor subunits and GS (26, 36). To test the significance of GABABRs for GS stability, we used lipofection to introduce R2 and R2ΔCT subunits into cultured astrocytes (a procedure that leads to transfection efficiencies between 10 and 30%). Similar to COS-7 cells, the level of endogenous GS expression in astrocytes transfected with R2ΔCT was reduced to 60 ± 7% of control (Fig. 7A, p = 0.007). In order to determine the effect of R2 subunit on GS trafficking, the transfected astrocytes were subject to biotinylation. GS was observed in the surface fraction in the presence of the R2 subunit. This effect was dependent on residues 891–941 (Fig. 7B, R2ΔCT = 19 ± 4%, p = 0.002).
FIGURE 7.

GABABRs regulate GS expression levels in cultured astrocytes and the brain. A, astrocytes expressing R2 or R2ΔCT were immunoblotted with R2, GS, and actin antibodies. The ratios of GS:R2 and GS:R2ΔCT immunoreactivity were determined and normalized to values for GS:R2. Data represent mean ± S.E., p < 0.01; unpaired t test, n = 5. B, astrocytes transfected with R2, or R2ΔCT were labeled with NHS-Biotin. After purification on avidin the resulting cytosolic (C) and surface (S) fractions were immunoblotted with R2, GS, and actin antibodies. GS levels in surface fractions were normalized to levels seen for cells expressing R2. Data represent mean ± S.E., (p < 0.01; unpaired t test, n = 4). C, astrocytes transfected with R2 or R2ΔCT and stained with Flag (green) and GS (red) antibodies followed by confocal microscopy; scale bar, 20 microns. Arrows indicate co-localized puncta of GS/R2 immunoreactivity, and in the lower panel co-localization as determined using ImageJ is shown in white. The level of endogenous GS in R2-positive puncta was then determined and expressed as mean gray value (p < 0.001; unpaired t test, n = 4). In the lower panel, co-localization between GS and R2, or GS and R2ΔCT were determined. Data represent mean ± S.E., p < 0.01; unpaired t test, n = 3. D, detergent solubilized hippocampal extracts were from WT and R2KO mice were immunoblotted with R2, GS, and actin antibodies. The levels of GS were determined and normalized to values in WT mice (p < 0.05; unpaired t test, n = 6). *, significantly different from control, p < 0.05; **, p < 0.01; ***, p < 0.001.
To confirm these results, we performed immunocytochemistry to compare expression levels and co-localization between R2 and GS (Fig. 7C). In R2-transfected astrocytes, there was a significant increase in the level of endogenous GS expression compared with cells expressing R2ΔCT (Fig. 7C, R2 = 1.35 ± 0.1, R2ΔCT = 0.89 ± 0.08, p = 0.001, Mean Gray Value, AU). Moreover, we observed higher levels of co-localization between R2 and GS (R2 = 17.47 ± 2.35) compared with GS/R2ΔCT (6.97 ± 1, p = 0.0002). Taken together, these results suggest that GABABR2 is a critical determinant of GS stability and subcellular localization in cultured astrocytes.
It is well accepted that the properties of astrocytes in culture are often distinct to their counterparts within the brain. Therefore, we assessed whether modifying GABABR expression levels in the brain would influenced GS expression levels, using GABABR2 knockout mice (R2-KO) (38). In these mice R2 expression is ablated and R1 subunit levels are greatly reduced (37). R2 homozygotes die shortly after birth and thus we measured GS expression levels in p2–4 mice R2-KO and WT littermate controls. Consistent with previously published studies, R2 subunit expression was not detected in R2-KO mice (38). Furthermore, GS expression levels were reduced in R2-KO mice to 39 ± 9% of WT controls (Fig. 7D, p = 0.015). These results are consistent with our studies in COS-7 cells and further support the role of the GABABR2 subunit in determining the steady state expression levels of GS in both cultured astrocytes and the brain.
DISCUSSION
In the brain GS expression is restricted to astrocytes and other types of glia. GS plays an essential role in regulating neuronal excitability by providing neurons with glutamine the major metabolic precursor of GABA. In parallel with this GS plays an important role in limiting excitoxicity by reducing glutamate accumulation. Consistent with an essential role for GS in limiting neuronal excitability inhibition of GS activity leads to seizures and a selective reduction in the efficacy of inhibitory neurotransmission.
GS has long been considered as a cytoplasmic enzyme, but recent work has shown a vesicular-like labeling in astrocytes (39). Here we have examined the cellular mechanisms that underlie the stability and subcellular localization of GS. Our studies focused on the possible role of GABABRs, which are co-expressed only in astrocytes with GS in physiological conditions. Our results demonstrate that GS from brain extracts selectively binds to residues 891–941 within the intracellular domain of GABABR2. Importantly, these residues lie outside the coil-coiled domain in R2, which is essential for dimerization with GABABR1. Thus binding of GS to GABABR2 will not preclude the formation of functional receptor heterodimers. To assess whether these respective proteins were associated in a cellular environment we used COS-7 cells. This initial expression system allowed us to limit possible confounds of protein overexpression, as they do not express significant levels of either protein. As revealed by our immunoprecipitation experiment, binding of GS to GABABR2 was evident, and it was an interaction that was stable in 2 m NaCl. Consistent with our in vitro assays binding of GS to GABABRs as measured using immunoprecipitation was also dependent upon residues 891–941 within the R2 subunit.
To assess the physiological significance of this protein-protein interaction, we examined the effects of GABABRs on the stability of GS. Strikingly, co-expression with GABABRs increased the total expression levels of GS, a phenomenon that was dependent upon 891–941 in GABABR2. Moreover, this effect was not replicated by co-expression with the β3 subunit of GABAAR, which is able to access the plasma membrane like GABABR2. In keeping with increasing steady state expression levels, metabolic labeling indicated that GABABRs increased the stability of GS. Past studies in Schwann cells suggested that GS is subject to proteasomal degradation. In agreement with this, GABABRs decreased GS ubiquitination, and occluded the effects of proteasome inhibitors on GS stability. Collectively, these results suggest that GABABRs act to stabilize GS by reducing its ubiquitination and subsequent proteasomal degradation. In addition to our biochemical experiments, we used imaging to assess the effects of GABABRs on GS stability. This approach confirmed our biochemical measurements and also revealed that GABABRs may play a role in targeting GS to the plasma membrane. To further investigate this possibility, we used subcellular fractionation coupled with biotinylation. These approaches revealed that targeting of GS to the cell surface was dependent upon residues 891–941 in GABABRs.
As a means of assessing the relevance of our studies in COS-7 cells, we assessed the role that GABABRs play in regulating the stability of endogenous GS expression in cultured astrocytes. This approach revealed that the presence of R2 subunit lacking residues 891 to 941 selectively reduced total GS expression levels and its expression at the plasma membrane. Given the limitations of working with cultured astrocytes, we further assessed the effects of GABABRs on GS stability in the brain using R2-KO mice. Consistent with our studies in COS-7 cells and astrocytes, GS expression levels were reduced in mice deficient in GABABR2, strongly supporting our in vitro measurements.
In summary, our studies suggest a significant role for GABABRs, both in determining the stability of GS and in regulating its targeting to the plasma membrane. Given the role GS plays in regulating GABAergic inhibition, this interaction may be significant to in ensure efficient glutamine synthesis to support GABA synthesis by local interneurons.
This work was supported by National Institutes of Health-NINDS Grants NS051195, NS056359, NS081735 (to S. J. M.), NIH-NIMH Grant MH097446, (to P. D. and S. J. M.), and a grant from the Simons Foundation (No. 206026 to S. J. M.).
- GABA
- γ-aminobutyric acid
- GABABR
- γ-aminobutyric acid B receptor
- GS
- glutamine synthetase
- KO
- knockout.
REFERENCES
- 1. Couve A., Thomas P., Calver A. R., Hirst W. D., Pangalos M. N., Walsh F. S., Smart T. G., Moss S. J. (2002) Cyclic AMP-dependent protein kinase phosphorylation facilitates GABA(B) receptor-effector coupling. Nat. Neurosci. 5, 415–424 [DOI] [PubMed] [Google Scholar]
- 2. Bettler B., Tiao J. Y. (2006) Molecular diversity, trafficking and subcellular localization of GABAB receptors. Pharmacol. Ther. 110, 533–543 [DOI] [PubMed] [Google Scholar]
- 3. White J. H., Wise A., Main M. J., Green A., Fraser N. J., Disney G. H., Barnes A. A., Emson P., Foord S. M., Marshall F. H. (1998) Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature 396, 679–682 [DOI] [PubMed] [Google Scholar]
- 4. Chalifoux J. R., Carter A. G. (2011) GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 21, 339–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Suárez I., Bodega G., Fernández B. (2002) Glutamine synthetase in brain: effect of ammonia. Neurochem. Int. 41, 123–142 [DOI] [PubMed] [Google Scholar]
- 6. Norenberg M. D. (1979) Distribution of glutamine synthetase in the rat central nervous system. The Journal of Histochemistry and Cytochemistry: Official Journal of the Histochemistry Society 27, 756–762 [DOI] [PubMed] [Google Scholar]
- 7. Martinez-Hernandez A., Bell K. P., Norenberg M. D. (1977) Glutamine synthetase: glial localization in brain. Science 195, 1356–1358 [DOI] [PubMed] [Google Scholar]
- 8. Derouiche A., Frotscher M. (1991) Astroglial processes around identified glutamatergic synapses contain glutamine synthetase: evidence for transmitter degradation. Brain Res. 552, 346–350 [DOI] [PubMed] [Google Scholar]
- 9. He Y., Hakvoort T. B., Vermeulen J. L., Labruyère W. T., De Waart D. R., Van Der Hel W. S., Ruijter J. M., Uylings H. B., Lamers W. H. (2010) Glutamine synthetase deficiency in murine astrocytes results in neonatal death. Glia 58, 741–754 [DOI] [PubMed] [Google Scholar]
- 10. Häberle J., Shahbeck N., Ibrahim K., Hoffmann G. F., Ben-Omran T. (2011) Natural course of glutamine synthetase deficiency in a 3 year old patient. Mol. Genet. Metab. 103, 89–91 [DOI] [PubMed] [Google Scholar]
- 11. Häberle J., Gorg B., Toutain A., Rutsch F., Benoist J. F., Gelot A., Suc A. L., Koch H. G., Schliess F., Häussinger D. (2006) Inborn error of amino acid synthesis: human glutamine synthetase deficiency. J. Inh. Metab. Dis. 29, 352–358 [DOI] [PubMed] [Google Scholar]
- 12. Häberle J., Görg B., Rutsch F., Schmidt E., Toutain A., Benoist J. F., Gelot A., Suc A. L., Höhne W., Schliess F., Häussinger D., Koch H. G. (2005) Congenital glutamine deficiency with glutamine synthetase mutations. New Engl. J. Med. 353, 1926–1933 [DOI] [PubMed] [Google Scholar]
- 13. Smith C. D., Carney J. M., Starke-Reed P. E., Oliver C. N., Stadtman E. R., Floyd R. A., Markesbery W. R. (1991) Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 88, 10540–10543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burbaeva G., Boksha I. S., Tereshkina E. B., Savushkina O. K., Starodubtseva L. I., Turishcheva M. S. (2005) Glutamate metabolizing enzymes in prefrontal cortex of Alzheimer's disease patients. Neurochem. Res. 30, 1443–1451 [DOI] [PubMed] [Google Scholar]
- 15. Steffek A. E., McCullumsmith R. E., Haroutunian V., Meador-Woodruff J. H. (2008) Cortical expression of glial fibrillary acidic protein and glutamine synthetase is decreased in schizophrenia. Schizophrenia Research 103, 71–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Eid T., Behar K., Dhaher R., Bumanglag A. V., Lee T. S. (2012) Roles of glutamine synthetase inhibition in epilepsy. Neurochem. Res. 37, 2339–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schousboe A., Bak L. K., Waagepetersen H. S. (2013) Astrocytic Control of Biosynthesis and Turnover of the Neurotransmitters Glutamate and GABA. Front. Endocrinol. 4, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liang S. L., Carlson G. C., Coulter D. A. (2006) Dynamic regulation of synaptic GABA release by the glutamate-glutamine cycle in hippocampal area CA1. J. Neurosci. 26, 8537–8548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ortinski P. I., Dong J., Mungenast A., Yue C., Takano H., Watson D. J., Haydon P. G., Coulter D. A. (2010) Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat. Neurosci. 13, 584–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Papageorgiou I. E., Gabriel S., Fetani A. F., Kann O., Heinemann U. (2011) Redistribution of astrocytic glutamine synthetase in the hippocampus of chronic epileptic rats. Glia 59, 1706–1718 [DOI] [PubMed] [Google Scholar]
- 21. Hammer J., Alvestad S., Osen K. K., Skare Ø., Sonnewald U., Ottersen O. P. (2008) Expression of glutamine synthetase and glutamate dehydrogenase in the latent phase and chronic phase in the kainate model of temporal lobe epilepsy. Glia 56, 856–868 [DOI] [PubMed] [Google Scholar]
- 22. van der Hel W. S., Notenboom R. G., Bos I. W., van Rijen P. C., van Veelen C. W., de Graan P. N. (2005) Reduced glutamine synthetase in hippocampal areas with neuron loss in temporal lobe epilepsy. Neurology 64, 326–333 [DOI] [PubMed] [Google Scholar]
- 23. Charles K. J., Deuchars J., Davies C. H., Pangalos M. N. (2003) GABA B receptor subunit expression in glia. Mol. Cell Neurosci. 24, 214–223 [DOI] [PubMed] [Google Scholar]
- 24. Kang J., Jiang L., Goldman S. A., Nedergaard M. (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat. Neurosci. 1, 683–692 [DOI] [PubMed] [Google Scholar]
- 25. Serrano A., Haddjeri N., Lacaille J. C., Robitaille R. (2006) GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J. Neurosci. 26, 5370–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oka M., Wada M., Wu Q., Yamamoto A., Fujita T. (2006) Functional expression of metabotropic GABAB receptors in primary cultures of astrocytes from rat cerebral cortex. Biochem. Biophys. Res. Commun. 341, 874–881 [DOI] [PubMed] [Google Scholar]
- 27. Schwartz J. P., Wilson D. J. (1992) Preparation and characterization of type 1 astrocytes cultured from adult rat cortex, cerebellum, and striatum. Glia 5, 75–80 [DOI] [PubMed] [Google Scholar]
- 28. Lunn M. L., Nassirpour R., Arrabit C., Tan J., McLeod I., Arias C. M., Sawchenko P. E., Yates J. R., 3rd, Slesinger P. A. (2007) A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat. Neurosci. 10, 1249–1259 [DOI] [PubMed] [Google Scholar]
- 29. Schwenk J., Metz M., Zolles G., Turecek R., Fritzius T., Bildl W., Tarusawa E., Kulik A., Unger A., Ivankova K., Seddik R., Tiao J. Y., Rajalu M., Trojanova J., Rohde V., Gassmann M., Schulte U., Fakler B., Bettler B. (2010) Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature 465, 231–235 [DOI] [PubMed] [Google Scholar]
- 30. Filippov A. K., Couve A., Pangalos M. N., Walsh F. S., Brown D. A., Moss S. J. (2000) Heteromeric assembly of GABA(B)R1 and GABA(B)R2 receptor subunits inhibits Ca(2+) current in sympathetic neurons. J. Neurosci. 20, 2867–2874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calver A. R., Robbins M. J., Cosio C., Rice S. Q., Babbs A. J., Hirst W. D., Boyfield I., Wood M. D., Russell R. B., Price G. W., Couve A., Moss S. J., Pangalos M. N. (2001) The C-terminal domains of the GABA(b) receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J. Neurosci. 21, 1203–1210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Robbins M. J., Calver A. R., Filippov A. K., Hirst W. D., Russell R. B., Wood M. D., Nasir S., Couve A., Brown D. A., Moss S. J., Pangalos M. N. (2001) GABA(B2) is essential for g-protein coupling of the GABA(B) receptor heterodimer. J. Neurosci. 21, 8043–8052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wooltorton J. R., Moss S. J., Smart T. G. (1997) Pharmacological and physiological characterization of murine homomeric β3 GABA(A) receptors. Eur. J. Neurosci. 9, 2225–2235 [DOI] [PubMed] [Google Scholar]
- 34. Taylor P. M., Thomas P., Gorrie G. H., Connolly C. N., Smart T. G., Moss S. J. (1999) Identification of amino acid residues within GABA(A) receptor beta subunits that mediate both homomeric and heteromeric receptor expression. J. Neurosci. 19, 6360–6371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saitoh F., Araki T. (2010) Proteasomal degradation of glutamine synthetase regulates schwann cell differentiation. J. Neurosci. 30, 1204–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hösli E., Hösli L. (1990) Evidence for GABAB-receptors on cultured astrocytes of rat CNS: autoradiographic binding studies. Exp. Brain Res. 80, 621–625 [DOI] [PubMed] [Google Scholar]
- 37. Thuault S. J., Brown J. T., Sheardown S. A., Jourdain S., Fairfax B., Spencer J. P., Restituito S., Nation J. H., Topps S., Medhurst A. D., Randall A. D., Couve A., Moss S. J., Collingridge G. L., Pangalos M. N., Davies C. H., Calver A. R. (2004) The GABA(B2) subunit is critical for the trafficking and function of native GABA(B) receptors. Biochem. Pharmacol. 68, 1655–1666 [DOI] [PubMed] [Google Scholar]
- 38. Gassmann M., Shaban H., Vigot R., Sansig G., Haller C., Barbieri S., Humeau Y., Schuler V., Müller M., Kinzel B., Klebs K., Schmutz M., Froestl W., Heid J., Kelly P. H., Gentry C., Jaton A. L., Van der Putten H., Mombereau C., Lecourtier L., Mosbacher J., Cryan J. F., Fritschy J. M., Lüthi A., Kaupmann K., Bettler B. (2004) Redistribution of GABAB(1) protein and atypical GABAB responses in GABAB(2)-deficient mice. J. Neurosci. 24, 6086–6097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Anlauf E., Derouiche A. (2013) Glutamine Synthetase as an Astrocytic Marker: Its Cell Type and Vesicle Localization. Front. Endocrinol. 4, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]