

Background: The molecular mechanisms underlying the non-genomic activities of testosterone in keratinocytes are unknown.
Results: Testosterone stimulates Duox1 activity through GPRC6A leading to cell death in skin keratinocyte.
Conclusion: These results support an understanding of the molecular mechanism of testosterone-dependent apoptosis through Duox1-induced H2O2 generation.
Significance: These results provide a novel signaling cascade of testosterone-mediated redox regulation in keratinocytes.
Keywords: Apoptosis, Cell Signaling, NADPH Oxidase, Reactive Oxygen Species (ROS), Testosterone
Abstract
Testosterone is an endocrine hormone with functions in reproductive organs, anabolic events, and skin homeostasis. We report here that GPRC6A serves as a sensor and mediator of the rapid action of testosterone in epidermal keratinocytes. The silencing of GPRC6A inhibited testosterone-induced intracellular calcium ([Ca2+]i) mobilization and H2O2 generation. These results indicated that a testosterone-GPRC6A complex is required for activation of Gq protein, IP3 generation, and [Ca2+]i mobilization, leading to Duox1 activation. H2O2 generation by testosterone stimulated the apoptosis of keratinocytes through the activation of caspase-3. The application of testosterone into three-dimensional skin equivalents increased the apoptosis of keratinocytes between the granular and stratified corneum layers. These results support an understanding of the molecular mechanism of testosterone-dependent apoptosis in which testosterone stimulates H2O2 generation through the activation of Duox1.
Introduction
Recently, reactive oxygen species (ROS)4 have been recognized as important messengers in cell signaling (1–3). Various agonists including growth factors, hormones, and neurotransmitters stimulate ROS generation to mediate physiological responses, such as cell proliferation, differentiation, and cell death in different cell types (1–5). Transient ROS generation by agonists may occur in the plasma membrane. It has been reported that NADPH oxidase (Nox) isozymes are primarily localized in the plasma membrane, and the activity of Nox appears to be regulated by the activation of various receptors (4). The Nox family includes seven isozymes (Nox1; gp91 phox, renamed Nox2; Nox3; Nox4; Nox5; Duox1; and Duox2) and is expressed in various tissues including colon, kidney, thyroid gland, testes, salivary glands, airways, and lymphoid organs, and it mediates ROS generation to coordinate tissue-specific functions. Duox1/2 isozymes differ from the other Nox homologs. They contain an additional peroxidase-like domain in the NH3-terminal region and intracellular Ca2+-binding EF-hand domain regions, which suggests that the activity of these isozymes is regulated by intracellular calcium mobilization.
The skin is the largest organ and protects the body. This protective barrier function largely relies on the stratum corneum, the end product of the terminal differentiation of epidermal keratinocytes (6). The stratum corneum is maintained through the continuous addition of new differentiating corneocytes surrounded by intercellular lipids; this tissue is constantly forming in the basal layer and being removed at the outermost surface. The balance between the differentiation and death of keratinocytes is crucial for the homeostasis of the epidermis (7). Many cytokines and growth factors stimulate permeability barrier formation, whereas testosterone seems to inhibit formation (8). Indeed, testosterone plays an important role in reproductive physiology and anabolic biological activities in multiple tissues and regulates the various functions of skin biology, such as sebaceous gland growth, differentiation, hair growth, skin barrier homeostasis, and wound healing (9–11).
Testosterone and dihydrotestosterone (DHT) activate the androgen receptor (AR) as nuclear receptor. The association of androgen with soluble AR results in its translocation to the nucleus and the stimulation of androgen-regulated gene expression. Recently, rapid non-genomic effects of testosterone have been reported in various cell types including smooth muscle cells, endothelial cells, Sertoli cells, prostate cells, and immune cells (12–15). However, the molecular mechanisms underlying the non-genomic activities of testosterone in keratinocytes are unknown.
Recently, emerging evidence has indicated that the orphan G protein-coupled receptor GPRC6A serves as a multiligand receptor for l-amino acids, calcium, osteocalcin, or testosterone and is expressed in various tissues, where it regulates many physiological processes (16–18). GPRC6A requires a positive allosteric modulator such as calcium for the rapid and transmembrane activity of testosterone. Indeed, a testosterone-GPRC6A complex mediates insulin secretion from β-cells, regulating glucose homeostasis, and confers a rapid response in prostate cells (19). Previous reports indicated that testosterone regulates ROS generation through the activation of Nox isozymes, suggesting a molecular connection between testosterone and cellular redox status (20). Although the Duox1 isozyme is expressed in epidermal keratinocytes, the molecular function of this isozyme is undefined. Here, we show that testosterone stimulates GPRC6A resulting in the sequential activation of a Gq-PLCβ-IP3-Ca2+ cascade in epidermal keratinocytes. Additionally, intracellular calcium mobilization mediated by testosterone induces the activation of Duox1 and the generation of H2O2, which leads to keratinocyte apoptosis. This report provides a molecular mechanism in which the non-genomic action of testosterone mediates the regulation of the cellular redox status.
EXPERIMENTAL PROCEDURES
Chemical Reagents
Testosterone (4-androsten-17β-ol-3-one, T1500), flutamide (4-nitro-3-trifluoromethylisobutyranilide, F9397), diphenyleneiodonium chloride (DPI, D2926), BAPTA-AM (A1076), and xestospongin C (X2628) were purchased from Sigma. NPS2143 (SB262470) was purchased from Selleckchem. Fura-2-acetoxymethyl ester (Fura-2/AM, F-1201), pluronic-F127 (P3000MP), and 2′,7′-dichlorofluorescin diacetate (DCF-DA, D-399) were obtained from Invitrogen. Peroxy-Orange 1 was obtained from Christopher J. Chang.
Cell Culture
Epidermal keratinocytes (MPEK-BL6) were purchased from CellnTec (Bern, Switzerland) and subcultured in keratinocyte growth medium (CellnTec). Cells were cultured in KBM medium (0.15 mm Ca2+) with keratinocyte growth medium growth supplements (Lonza, CC-3111).
Quantitative RT-PCR
The RNAs were extracted with 1 ml of TRIzol reagent (Molecular Research Center, Inc.) and 0.2 ml of chloroform and precipitated with 0.5 ml of isopropyl alcohol. The suspension was centrifuged at 12,000 × g at 4 °C for 15 min. One microgram of total RNA was reverse transcripted by the Reverse Transcription System (Promega, Madison, WI). Quantitative real-time PCR was performed using TaqMan® universal PCR master mix or SYBR® master mix. Pre-designed gene specific TaqMan probe and SYBR Green primers were purchased from Applied Biosystems. The temperature profile for the reaction was: 50 °C for 2 min, 95 °C for 20 s, and then 95 °C for 3 s and 60 °C for 30 s for 45 cycles. The relative quantity was obtained using the comparative threshold method and results were normalized against 18 S rRNA as an endogenous control.
Gene Knockdown by siRNA Transfection
For knockdown of Duox1, GPRC6A, and Gq, cells were transfected with control siRNA (ON-TARGETplus Non-targeting siRNA #2, D-001810-02-20), Duox1 (siGENOME Duox1 siRNA, D-047172-01-0005), GPRC6A (siGENOME Gprc6a siRNA- SMARTpool, M-054451-00-0005, mixtures of four highly functional, individual siRNAs), siRNA (Dharmacon), or Gq (FlexiTube GeneSolution for Gnaq SI02708713) siRNA (Qiagen) according to the manufacturer's protocol.
Immunoblotting
Cells were chilled in lysis buffer (50 mm Tris-HCl (pH 7.4), 1% Triton X-100, 0.5% Nonidet P-40, 150 mm NaCl, 1 mm EDTA, 0.1 m 4-(2-aminoethyl)benzenesulfonyl fluoride, 1 mm Na3VO4, 1 mm sodium fluoride, 1 μg/ml of aprotinin, 1 μg/ml of leupeptin, and 10% glycerol) for 30 min, then sonicated (at 10% amplitude for 1 s in a Branson sonifier equipped with a microtip), followed by a 30-min 14,000 × g centrifugation. After boiling in 5× SDS-PAGE sample buffer, the samples were subjected to 10% SDS-PAGE. The resolved proteins were electrotransferred to a nitrocellulose membrane. The membrane was immunoblotted with anti-Duox1 and GPRC6A (Santa Cruz Biotechnology), followed by horseradish peroxidase-conjugated goat anti-rabbit IgG antibody. Bands were visualized by chemiluminescence (Fujifilm LAS-3000).
Measurement of Intracellular ROS
After the confluent cells were stimulated, cells were washed with Hanks' balanced salt solution and incubated for 10 min in the dark at 37 °C with the same solution containing 10 μm DCF-DA (Molecular Probes). The cells were then examined with a laser-scanning confocal microscope (model LSM 510, Carl Zeiss) equipped with an argon laser tuned to an excitation wavelength of 488 nm, an LP505 emission filter (515 to 540 nm), and a Zeiss Axiovert 100× objective lens. Images were digitized and stored at a resolution of 512 by 512 pixels. Five groups of cells were randomly selected from each sample, and the mean relative fluorescence intensity for each group of cells was measured with a Zeiss vision system (LSM510, version 2.3) and then averaged for all groups. All experiments were repeated at least three times.
Measurement of Intracellular H2O2 by Peroxy-Orange 1
After the cell culture media was swapped out, cells were incubated for 15 min in the dark at 37 °C with 5 μm PO-1 in Dulbecco's PBS, and then stimulants were added to the dye/Dulbecco's PBS cell mixture. The cells were then examined with a laser scanning confocal microscope (model LSM 510, Carl Zeiss) equipped with an argon laser tuned to an excitation wavelength of 540 nm. Five groups of cells were randomly selected from each sample, and the mean relative fluorescence intensity for each group of cells was measured with a Zeiss vision system (LSM510, version 2.3) and then averaged for all groups. All experiments were repeated at least three times.
Measurement of Intracellular Calcium
Keratinocytes (106 cells) were seeded onto 22-mm glass coverslips and cultured for 24 h, and then incubated for 16 h in serum-free medium. Cells were loaded with 5 μm Fura-2 AM and 0.05% pluronic acid for 1 h at room temperature in HEPES buffer (140 mm NaCl, 5 mm KCl, 1 mm CaCl2, 1 mm MgCl2, 10 mm glucose, 10 mm HEPES, adjusted to pH 7.4 with HCl). Cells were then washed twice with HEPES buffer. The coverslips were next attached to a chamber slide mounted onto the microscope. HEPES buffer (140 mm NaCl, 5 mm KCl, 1 mm MgCl2, 10 mm glucose, 1 mm EDTA, 1 mm EGTA, 10 mm HEPES, adjusted to pH 7.4 with HCl) was perfused at slow speed through the chamber with 100 nm testosterone applied locally to cells by pressure ejection via a micropipette. Calcium measurements were obtained by direct imaging via a real time fluorescent confocal imaging system. Images were acquired at 340/380 nm for excitation and 510 nm for emission. Fura 2 fluorescence was calibrated according to the method described by Grynkiewicz et al. (21). For this purpose, the cells were exposed to 5 μm ionomycin in modified HEPES solution containing either 3 mm Ca2+ or 5 mm EGTA to obtain the maximum (Rmax) and minimum (Rmin) ratio of fluorescence (R), respectively. [Ca2+]i was calculated according to the equation [Ca2+]i = Kd × β × (R − Rmin)|(Rmax − R), with use of the dissociation constant (Kd) of Fura 2, where β is the ratio of the 380-nm excitation signals of ionomycin-treated cells at 5 mm EGTA and 3 mm Ca2+. Experiments were repeated at least three times with similar results.
Measurement of IP1
IP1 ELISA (72IP1PEA, Cisbio) was used according to the manufacturer's instructions to measure the IP1 produced in the cell after G protein-coupled receptor activation. Intracellular IP1, a measure of the degradation products of InsP3 and a surrogate for InsP3 levels, was measured after LiCl (50 mm) treatment to prevent the degradation of IP1 into myo-inositol. Briefly, 5 × 105 cells were plated in a 24-well culture plate and incubated overnight, and then incubated for 16 h in serum-free medium. Cells were stimulated with testosterone for the indicated times and lysed for 30 min. The lysates were then sonicated and centrifuged. After transfer of the supernatant into the ELISA plate supplied with the kit, samples were incubated for 3 h with IP1-HRP conjugate and anti-IP1 mAb. After a washing step, the revelation is carried out by addition of the HRP substrate 3,3′,5,5′-tetramethylbenzidine. The reaction was stopped and the optical density (OD) was read at 450 nm with an optional correction between 610 and 650 nm (correction for optical imperfections in the plate). Experiments were repeated at least three times with similar results.
Mitochondrial Membrane Potential
The measurement of mitochondrial transmembrane potential was performed using the JC-1-based assay (Invitrogen). For each sample, cells were suspended in 1 ml of warm medium at ∼1 × 106 cells/ml. Then 1 μl of 5 mm JC-1 (5 μm final concentration, Molecular Probes) was added and the cells were incubated at 37 °C, in 5% CO2, for 15 to 30 min. The cells were analyzed on a flow cytometer (FACS Calibur, BD Bioscience) with 488 nm excitation using emission filters appropriate for Alexa Fluor® 488 dye. A plot of green fluorescence (FL1) from cells with loss of mitochondrial membrane potential was recorded.
Caspase-3 Activity Assay
The caspase-3 fluorometric assay kit (Calbiochem) was used according to the manufacturer's instructions to determine the amount of apoptosis in keratinocytes. The caspase-3 Fluorometric Assay Kit is based on the hydrolysis of acetyl-Asp-Glu-Val-Asp-7-amido-4-methylcoumarin (Ac-DEVD-AMC) by caspase-3, resulting in the release of the fluorescent 7-amino-4-methylcoumarin. The excitation and emission wavelengths of 7-amino-4-methylcoumarin were 360 and 460 nm, respectively. Briefly, the total amount of cells including floating cells were collected after a 24-h incubation in serum-free medium with and without testosterone treatment, and then the lysates were sonicated and incubated on ice for 10 min, followed by centrifugation at 13,000 × g for 10 min at 4 °C. Total protein was normalized to sample to protein concentrations. The samples were added to assay buffer in a 96-well plate, including caspase-3 substrate. The samples were incubated at 37 °C for 60 min, and then assayed for caspase-3 activity. Experiments were repeated at least three times with similar results.
TdT-UDP Nick End Labeling (TUNEL) Assay
Apoptotic cells were detected in paraffin sections by the TUNEL technique using an In Situ Cell Death Detection Kit (Roche Applied Science, Basel, Switzerland) to determine the amount of apoptosis in keratinocytes and epidermis. Keratinocytes were incubated with testosterone or 70% ethanol in serum-free medium for 24 h. Samples were fixed with 4% paraformaldehyde in PBS for 1 h at room temperature. After fixation, samples were washed with PBS and permeabilized with 0.1% sodium citrate, 0.1% Triton X-100 for 2 min on ice. Samples were washed with PBS and then incubated with enzyme and labeling solution (Roche Diagnostics) for 1 h at 37 °C in a humidified chamber. The enzyme and labeling solutions catalyze the polymerization of labeled nucleotides to free 3′-OH DNA ends. Samples were then washed 3 times with PBS and stained with DAPI for 10 min, and then mounted with mounting solution (Sigma) after washing 3 times with PBS. Detection of fragmented DNA was then observed with fluorescence microscopy. TUNEL staining was examined by confocal microscopy. The percentage of positively stained cells was determined by counting the numbers of TUNEL-labeled and DAPI-stained cells using ImageJ software.
Human Reconstituted Skin (EpiDermTM FT-200)
The skin equivalents (SEs), Epiderm FT-200 (MatTek Corp.), were removed from agarose-containing 24-well plates and moved to 6-well plates filled with EpiDerm Full Thickness medium. Epiderm FT-200 SEs were cultured at 37 °C, in 5% CO2 for 1 day and then transferred to a new medium-containing dish for subsequent testosterone (Sigma) treatment every 24 h for 48 h.
Immunostaining
SEs were cryopreserved in frozen section compound (FSC 22®, Leica Microsystems), sectioned at 6 μm, and stained with hematoxylin and eosin for standard evaluation. To compare the number of apoptotic cells, replicate cryosections were incubated with the In Situ Cell Death Detection Kit, Fluorescein (Roche Diagnostics) for 40 min at room temperature. Nuclei were counterstained with Fluorescent Mounting Medium with 4,6-diamidino-2-phenylindole (E19-18, Golden Bridge International, Inc., Mukilteo, WA) for 5 min. The reactivity was evaluated under a microscope (Bx-41, Olympus, Japan) equipped with reflected fluorescence system, and photomicrographs were taken using a microscope digital camera (DP72, Olympus, Japan).
Statistic Analysis
All data are presented as mean ± S.E. Significant differences between treatment groups were identified with a t test. p Values less than 0.05 were considered statistically significant.
RESULTS
Connection between the Non-genomic Activity of Testosterone and ROS Generation in Epidermal Keratinocytes
The rapid, non-genomic activity of testosterone has been documented in various tissues. Specifically, testosterone induces the mobilization of intracellular calcium ([Ca2+]i) within a few minutes in Sertoli cells, prostate cells, and smooth muscle cells, which indicates that the rapid response to testosterone is mediated by an AR-independent cascade (12, 13).
To evaluate the non-genomic activity of testosterone in epidermal keratinocytes, we first measured [Ca2+]i mobilization as a hallmark of the non-genomic action of testosterone. The stimulation of epidermal keratinocytes with testosterone resulted in rapid and transient [Ca2+]i mobilization within 1 min (Fig. 1A). To test whether testosterone stimulates ROS generation in keratinocytes, we measured the intracellular levels of ROS by measuring the fluorescence of 2′,7′-DCF-DA using a confocal microscope (22). The stimulation of epidermal keratinocytes with testosterone revealed significantly increased ROS generation within 5 min, which then decreased after 30 min (Fig. 1, B and C). It well known that physiological testosterone concentrations are 15–30 nm (23). We tested the effect of various concentrations of testosterone on ROS generation in epidermal keratinocytes. Testosterone stimulated the ROS production of epidermal keratinocytes in a concentration-dependent manner (Fig. 1D).
FIGURE 1.

Testosterone (T)-induced mobilization of intracellular calcium and H2O2 generation in keratinocytes. A, testosterone-induced calcium influx. Cytosolic Ca2+ concentration ([Ca2+]i) of keratinocytes loaded with Fura-2/AM after exposure to testosterone, 10 s after detection. The representative trace shown is the intracellular Ca2+ influx in single cells treated with 100 nm testosterone. Data represent three repeated experiments and mean ± S.E. of three independent experiments. B, time course for ROS production was monitored by confocal microscopic analysis of DCF fluorescence in keratinocytes stimulated with 100 nm testosterone. C, data are presented as the mean ± S.E. (n = 3). D, ROS generation was increased after 5 min of testosterone treatment in a concentration-dependent manner. Data are presented as the mean ± S.E. (n = 3). E, H2O2 generation was monitored by confocal microscopic analysis of Peroxy-Orange 1 fluorescence in keratinocytes stimulated with 100 nm testosterone for 5 min. Data are presented as the mean ± S.E. (n = 3). F, cells were pretreated with the androgen receptor inhibitor, flutamide (100 nm), for 2 h and ROS generation was monitored after 5 min of testosterone treatment. Data are the mean ± S.E. (n = 4).
To identify the ROS species in the cells, we used PO-1 dye, which is specifically sensitive to H2O2 (24). Increasing PO-1 fluorescence, indicating H2O2 generation, was detected in epidermal keratinocytes in response to testosterone (Fig. 1E). Therefore, the predominant species of ROS induced by testosterone in epidermal keratinocytes is H2O2.
To evaluate whether testosterone-mediated H2O2 generation is dependent on non-genomic activity, we used flutamide as an AR antagonist (25). The pre-treatment of epidermal keratinocytes with flutamide failed to inhibit testosterone-induced H2O2 (Fig. 1F). These results indicate that the H2O2 generation is independent from AR activation.
Testosterone-mediated [Ca2+]i Mobilization Induces the Activation of Duox1 in Keratinocytes
We explored whether testosterone-mediated H2O2 generation is regulated by Nox activity. The pretreatment of the keratinocytes with DPI as a Nox inhibitor resulted in a significant reduction of H2O2 generation in response to testosterone (Fig. 2A). The result indicated that testosterone-mediated H2O2 generation might be from enhancing Nox activity instead of an AR-dependent pathway. We next tested the expression levels of Nox isozymes in epidermal keratinocytes. Quantitative real-time PCR indicated that Duox1 was the predominant Nox isozyme in epidermal keratinocytes; other Nox isozymes were barely detectable (Fig. 2B). This result indicated that the Duox1 isozyme may be responsible for testosterone-induced ROS generation in epidermal keratinocytes.
FIGURE 2.

Testosterone (T)-mediated intracellular calcium mobilization induces the activation of Duox1 in keratinocytes. A, effects of a Nox inhibitor on testosterone-induced ROS generation. Cells were pretreated with the Nox inhibitor, DPI (10 μm), for 30 min and ROS generation was monitored after 5 min of testosterone treatment. Data are presented as mean ± S.E. (n = 5). B, expression of Nox isoforms in keratinocytes measured by real-time PCR analysis with primers specific for each Nox isoform and 18 S rRNA as the reference gene. C, testosterone stimulates H2O2 generation by Duox1 in keratinocytes. Cells were transfected with either Duox1 siRNA or control siRNA with RNAiMAX. After 100 nm testosterone treatment, the generation of H2O2 was monitored by confocal microscopic analysis of DCF fluorescence. Data are presented as mean ± S.E. (n = 3). D, the protein level of Duox1 was analyzed by immunoblot. E, effects of intracellular Ca2+ chelation on testosterone-induced ROS generation in keratinocytes. Cells were pretreated with the intracellular Ca2+ chelator, BAPTA/AM (10 μm), for 30 min, and ROS generation was monitored after 5 min of testosterone treatment by laser-scanning confocal microscopy. Data are presented as mean ± S.E. (n = 5).
To explore the function of Duox1 in testosterone-induced H2O2 generation, Duox1-targeting siRNA was transfected into keratinocytes and then H2O2 generation was measured with DCF-DA fluorescence. The stimulation of Duox1-silenced keratinocytes with testosterone failed to generate H2O2, whereas control siRNA-transfected cells responded normally (Fig. 2, C and D). It has been reported that Duox isozymes contain canonical calcium-binding EF-hands, indicating that intracellular calcium plays a critical role in the activation of Duox (3). We hypothesized that transient [Ca2+]i mobilization by the non-genomic activity of testosterone affects Duox1-dependent H2O2 generation. BAPTA-AM is known to scavenge intracellular Ca2+ (26). The pretreatment of keratinocytes with BAPTA-AM as an intracellular Ca2+ scavenger resulted in significantly reduced testosterone-induced H2O2 generation, compared with control cells (Fig. 2E). This result indicated that [Ca2+]i mobilization by the non-genomic activity of testosterone is coupled with Duox-dependent intracellular H2O2 generation.
A previous report indicated that the orphan G protein-coupled receptor GPRC6A acts as a testosterone receptor in prostate cells (18). Here, we found that GPRC6A was expressed in keratinocytes as well as outer root sheath cells and dermal papilla cells in hair follicles (data not shown). To validate the function of GPRC6A as a testosterone receptor in the mobilization of [Ca2+]i, the expression of GPRC6A in epidermal keratinocytes was reduced by transfection of GPRC6A siRNA. Epidermal keratinocytes transfected with control siRNA showed increased [Ca2+]i mobilization in response to testosterone, whereas knockdown of GPRC6A prevented [Ca2+]i release (Fig. 3, A, B, and D). This result indicates that GPRC6A mediates the rapid testosterone-mediated mobilization of [Ca2+]i in epidermal keratinocytes.
FIGURE 3.

Testosterone (T) stimulates intracellular calcium and H2O2 generation through GPRC6A in keratinocytes. A–D, keratinocytes were transfected with control siRNA or GPRC6A siRNA with RNAiMAX. A, cytosolic Ca2+ concentration ([Ca2+]i) of keratinocytes loaded with Fura-2/AM after exposure to testosterone at 10 s after detection in control cells, but there was no increase in GPRC6A-silenced cells (B). C, testosterone stimulates H2O2 generation through GPRC6A in keratinocytes. After 100 nm testosterone treatment, the generation of H2O2 was monitored by confocal microscopic analysis of DCF fluorescence. Data are presented as mean ± S.E. (n = 4). D, the protein level of GPRC6A was analyzed by immunoblot. E, the effects of a GPRC6A inhibitor on testosterone-induced ROS generation. Cells were pretreated with the GPRC6A inhibitor, NPS2143 (1 μm), for 30 min and ROS generation was monitored after 5 min of testosterone treatment. Data are presented as mean ± S.E. (n = 4).
We evaluated the effect of GPRC6A-mediated [Ca2+]i mobilization on the activation of Duox containing a calcium binding domain in the NH3-terminal region. The silencing of GPRC6A by transfection of GPRC6A siRNA resulted in decreased H2O2 generation in response to testosterone, suggesting that GPRC6A-dependent [Ca2+]i mobilization stimulates the activity of Duox1 (Fig. 3C). Moreover, the pretreatment of epidermal keratinocytes with NPS2143, a non-competitive antagonist of GPRC6A (27, 28), resulted in significantly reduced testosterone-induced H2O2 generation (Fig. 3E). These results indicate that GPRC6A plays an important role in testosterone-dependent H2O2 generation in epidermal keratinocytes.
Effect of Downstream Signaling Networks of GPRC6A on H2O2 Generation in Keratinocytes
GPRC6A is known to be coupled with the Gq protein leading to IP3 generation and intracellular calcium mobilization (29). We tested the effect of Gq on H2O2 generation by the stimulation of testosterone. Transfection of epidermal keratinocytes with Gq-targeted siRNA resulted in decreased H2O2 generation compared with control siRNA in response to testosterone (Fig. 4, A and B). Next we measured testosterone-induced IP3 generation. Gq-dependent PLCβ stimulates the hydrolysis of phosphatidylinositol 4,5-bisphosphate into diacylglycerol, which activates PKC and IP3, which stimulates [Ca2+]i mobilization through binding to the IP3 receptor (IP3R) in the endoplasmic reticulum (30). The metabolism of IP3 by phosphatase is too fast to detect in cells. Here, we measured inositol monophosphate (IP1) as the end product of IP3 in epidermal keratinocytes (31). The stimulation of epidermal keratinocytes with testosterone resulted in rapid IP1 production (Fig. 4C).
FIGURE 4.

Effect of downstream signaling networks of GPRC6A on H2O2 generation in keratinocytes. A, testosterone (T) stimulates H2O2 generation by Gq in keratinocytes. Cells were transfected with control siRNA or Gq siRNA with RNAiMAX. After 100 nm testosterone treatment, the generation of H2O2 was then monitored by confocal microscopic analysis of DCF fluorescence. Data are presented as mean ± S.E. (n = 4). B, Gq mRNA expression was assessed in total RNA by RT-PCR as described under ”Experimental Procedures.“ C, testosterone increases the IP3 level in keratinocytes. PLC activity was tested by IP1 ELISA (72IP1PEA, Cisbio), in which keratinocytes were stimulated with testosterone for the indicated times. Intracellular IP1, a measure of the degradation products of InsP3 and a surrogate for InsP3 levels, was measured after LiCl (50 mm) treatment to prevent the degradation of IP1 into myo-inositol. D, testosterone stimulates H2O2 generation through the IP3 pathway in keratinocytes. The effects of blocking IP3R on testosterone-induced ROS generation in keratinocytes is shown. Cells were pretreated with the IP3R inhibitor, xestospongin C (2 μm), for 2 h and ROS generation was monitored after 5 min of testosterone treatment by laser-scanning confocal microscopy. Data are presented as mean ± S.E. (n = 4).
Xestospongin C has been reported as a membrane-permeable inhibitor of the IP3-mediated [Ca2+]i mobilization by blocking the IP3 receptor (32). However, 20 μm xestospongin C has no effect on ryanodine receptor-mediated Ca2+ release (33). We evaluated the function of xestospongin C in testosterone-dependent H2O2 generation. The pretreatment of epidermal keratinocytes with xestospongin C prevented the generation of H2O2 in response to testosterone (Fig. 4D). These results indicated that the GPRC6A-Gq-IP3-[Ca2+]i signaling cascade induced by testosterone is essential for the Duox1 activation leading to H2O2 generation in epidermal keratinocytes.
Testosterone-mediated H2O2 Stimulates the Apoptosis of Epidermal Keratinocytes
Several lines of evidence indicated that testosterone induces the apoptosis of various cell types including smooth muscle cells, endothelial cells, neuronal cells, renal cells, and dermal papilla cells (34–38). Here, we showed that testosterone induces their signaling network resulting in H2O2 generation in epidermal keratinocytes. Therefore, we hypothesized that testosterone-dependent H2O2 generation is involved in the apoptosis of keratinocytes. It has been well established that disruption of mitochondrial integrity was affected in the early apoptotic process (39). Therefore, we measured depolarization of the outer mitochondrial membrane as a marker of apoptosis in response to testosterone in keratinocytes. Stimulation of keratinocytes with testosterone resulted in decreased mitochondrial membrane potential (Fig. 5A). To determine whether testosterone induced keratinocyte apoptosis, we performed the TUNEL assay. The stimulation of epidermal keratinocytes with testosterone resulted in significantly increased TUNEL labeling compared with unstimulated cells (Fig. 5B). Many reports have indicated that testosterone induces apoptosis through AR (10, 35, 36). To validate the effect of the AR on testosterone-induced apoptosis, epidermal keratinocytes were pretreated with flutamide and then subjected into TUNEL labeling. Flutamide did not inhibit testosterone-induced apoptosis in keratinocytes indicating that the AR is not involved in apoptosis (Fig. 5C). The pretreatment of keratinocytes with DPI (Nox inhibitor) or BAPTA-AM ([Ca2+]i chelator) resulted in a significant inhibition of testosterone-induced apoptosis (Fig. 5C).
FIGURE 5.

Testosterone-mediated H2O2 stimulates apoptosis of epidermal keratinocytes. A, depolarization of mitochondrial membrane potential. Keratinocytes were incubated with or without testosterone (100 nm) for 24 or 36 h, and then assessed as described under ”Experimental Procedures.“ B, TUNEL assay. Keratinocytes were incubated with or without testosterone (100 nm) for 24 h, and then fixed, washed in PBS, and stained with TdT-UDP nick end labeling for 1 h at 37 °C. Apoptotic cells were visualized by fluorescence microscopy. C, cells were pretreated with flutamide (100 nm) for 2 h, DPI (10 μm) for 30 min, and BAPTA/AM (10 μm) for 30 min and incubated for 24 h in the absence or presence of testosterone. Samples were stained with TUNEL and analyzed. The percentage of fluorescent cells in 10 random high-power fields was determined. Data were analyzed by Student's t test. D, to evaluate the role of Duox1 and GPRC6A on DNA damage, siRNA was used against Duox1 and GPRC6A. Duox1 and GPRC6A siRNA-treated and untreated keratinocytes were incubated for 24 h in the absence or presence of testosterone. Samples were stained with TUNEL and analyzed. B-D, the percentage of fluorescent cells in 10 random high-power fields was determined. Data were analyzed by Student's t test. *, comparison with testosterone-treated control group, p < 0.0000005; **, p < 0.000005. E, caspase-3 activity assay. Testosterone (100 nm) was added to serum-free keratinocyte basal medium (KBM, Lonza) for 24 h. Lysates were assayed for caspase-3 activity using a caspase-3 fluorometric assay kit (Calbiochem). F, to evaluate the role of Duox1 and GPRC6A on caspase-3 activity, siRNA was used against Duox1 and GPRC6A. Duox1 and GPRC6A siRNA-treated and untreated keratinocytes were incubated for 24 h in the absence or presence of testosterone. Lysates were assayed for caspase-3 activity. E and F, data are presented as mean ± S.E. (n = 3). Silences of Duox1 and GPRC6A are as described in the legends to Figs. 2D and 3D.
To evaluate the function of Duox1 or GPRC6A in testosterone-dependent apoptosis, knockdown of Duox1 or GPRC6A was performed. Interestingly, silencing Duox1 or GPRC6A failed to induce testosterone-dependent apoptosis in epidermal keratinocytes (Fig. 5D). Furthermore, pretreatment of keratinocytes with NPS2143, an inhibitor of GPRC6A, completely blocked testosterone-induced apoptosis (data not shown). Because activation of caspase-3 is a hallmark of apoptosis, we measured caspase-3 activity in response to testosterone (34, 36, 40). The incubation of epidermal keratinocytes with testosterone revealed increasing caspase-3 activity in response to testosterone (Fig. 5E). To examine whether activation of caspase-3 is induced by GPRC6A and Duox1 activity, we tested the effect of knocking down GPRC6A or Duox1 on the activation of caspase-3. The transfection of epidermal keratinocytes with GPRC6A siRNA or Duox1 siRNA prevented caspase-3 activation (Fig. 5F).
To confirm the testosterone-induced apoptosis of keratinocytes, we examined the effects of testosterone on three-dimensional in vitro SEs models. Treatment of SEs with testosterone increased TUNEL-positive keratinocytes between the granular and stratified corneum layers (Fig. 6A, right lower panel). The number of apoptotic cells with flattened nuclei was increased in the same region based on H&E staining of testosterone-treated SEs (Fig. 6A, left lower panel). An immature cornified layer was increased in testosterone-treated SEs compared with control SEs (Fig. 6A, left lower panel). Statistical analysis revealed that testosterone-treated SEs had ∼3-fold more TUNEL-positive cells than control SEs (Fig. 6B).
FIGURE 6.
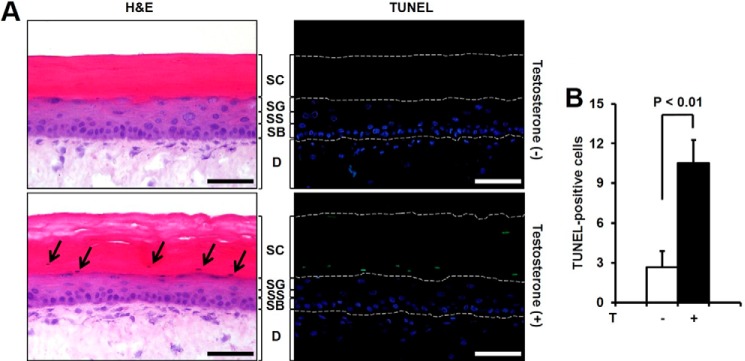
Effect of testosterone (T) on apoptosis of skin equivalents and keratinocytes in C57BL6. Skin equivalents were treated with testosterone (10 μm) every 24 h for 48 h. A, SEs were stained with H&E (left panel) or TUNEL (right panel). The upper panel shows an untreated S.E. and the lower panel shows a testosterone-treated SE. Many TUNEL-positive cells are observed only in the testosterone-treated SE. Arrows indicate the cells showing the characteristic morphology of apoptosis in H&E staining. Scale bar = 50 μm (SC, stratum corneum; SG, stratum granulosum; SS, stratum spinosum; SB, stratum basale; D, dermis). B, TUNEL-positive cells are counted in SEs treated with or without testosterone. The data shown are the mean ± S.E. of the results from 4 independent sample preparations; p < 0.01.
DISCUSSION
It is well established that testosterone is converted to DHT by 5α-reductase and that DHT then binds to the androgen receptor. This complex translocates into the nucleus, which leads to activating the expression of various genes. The genomic activity of testosterone regulates the development of reproductive organs and stimulates muscle and bone formation. Although the expression of the androgen receptor in keratinocytes is low (41), testosterone is involved in epidermal permeability barrier formation and wound healing, which indicates that testosterone regulates skin homeostasis through an AR-independent pathway (Fig. 5C).
Because intracellular calcium mobilization within minutes is a hallmark of the non-genomic activity of testosterone in many cell types (12–15), we first revealed that the orphan G protein-coupled receptor GPRC6A mediates the activity of testosterone in epidermal keratinocytes (Fig. 3). Our interest in this report focused on the molecular connection between the rapid, non-classical activity of testosterone and regulation of the cellular redox state. Because the Duox1 isozyme, the major isoform of Nox in epidermal keratinocytes, contains an EF-hand calcium-binding site, the connection of Duox1 isozyme activation with [Ca2+]i mobilization by testosterone was explored. Silencing either GPRC6A or Gq in epidermal keratinocytes completely inhibited testosterone-mediated H2O2 generation (Figs. 3C and 4A). Moreover, pretreatment of epidermal keratinocytes with BAPTA, an intracellular Ca2+ scavenger, resulted in suppressed H2O2 generation in response to testosterone (Fig. 2E). Thus, our results indicated that the testosterone-GPRC6A-mediated signaling cascade, including rapid [Ca2+]i release, resulted in increased H2O2 generation through Duox1 activation (Fig. 7).
FIGURE 7.

Proposed model for non-genomic activity of testosterone in keratinocytes. GPRC6A mediates the non-genomic effects of testosterone on intracellular calcium mobilization, suggesting the involvement of PLCβ-mediated phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis via the Gq protein in the testosterone-mediated pathway. Eventually, testosterone generates H2O2 through Duox1 in skin keratinocytes.
Several lines of evidence indicate that various agonists stimulate intracellular ROS as second messengers in cells. It has been shown that Nox/Duox isozymes in the plasma membrane induce intracellular ROS generation. Nox and Duox isozymes share highly similarity. Their NH3-terminal regions contain a heme group and their COOH-terminal regions bear NADPH- and FAD-binding sites. Transferring electrons from NADPH to O2 through FAD and heme groups leads to ROS generation. Based on the structure of the Nox/Duox isozymes, the direction of ROS generation is inside to out. Recently, growth factors and cytokines were shown to stimulate intracellular ROS generation through the formation of redox-active endosomes (42, 43). IL-1 binding to IL-1 receptor induces endosome formation including a Nox2 complex for the generation of intracellular ROS generation, which results in the activation of a cell signaling network. In this report, we did not provide information that testosterone stimulates endosome formation including the Duox1 isozyme. However, it is known that the COOH-terminal region of G protein-coupled receptors bears machinery to induce endocytosis (44). Therefore, it is likely that the testosterone-GPRC6A-Duox1 complex stimulates redox-active endosome formation, which results in intracellular H2O2 production in epidermal keratinocytes.
It has been reported that receptor-mediated cell signaling triggers ROS generation for cell growth and differentiation through activation of Nox isozymes (45). Nox-mediated ROS generation functions as second messenger in cell signaling and influences the activity of various downstream cytosolic proteins. For example, protein-tyrosine phosphatases are well known downstream targets of Nox-mediated ROS generation. Specifically, the sulfhydryl group of cysteine residues in the active center is oxidized to sulfenic acid by ROS converting the enzyme to an inactive form that in turn promotes tyrosine phosphorylation of cytosolic proteins (46, 47). Another example is the activation of apoptosis signal-regulating kinase-1 (ASK-1) by ROS. It is well known that ASK-1 interacts with thioredoxin as a repressor. Nox-mediated ROS by various stresses oxidize thioredoxin allowing dissociation of ASK-1, which mediates cell death (47, 48). In these events, oxidation of protein-tyrosine phosphatase and dissociation of ASK-1 indeed takes place within a short time span (minutes) but activation of their downstream signaling cascades, disruption of mitochondrial integrity, and gene expression changes require longer (hours).
It has been generally accepted that the generation of ROS by exogenous and endogenous stimuli plays an important role in the homeostasis of skin (49, 50). Specifically, the long-term effect of oxidative stress induced by exogenous stimuli such as UV is implicated in skin aging. UV-induced ROS generation is known to create DNA adduct and accelerate the aging process (49). Moreover, the topical application of linolein hydroperoxide stimulates the apoptosis of keratinocytes by inducing apoptosis-related genes (51). Even if agonist-dependent H2O2 generation acts as a second messenger in cell signaling, uncontrolled ROS generation induces apoptosis through the activation of caspase, expression of the Bcl2 family, or dysfunction of mitochondria (52). These previous reports indicated that ROS-induced apoptosis is related to skin cell aging. According to the notion that oxidative stress induces skin cell aging, the signaling cascade induced by testosterone stimulates Duox1-mediated H2O2 generation, which results in the apoptosis of keratinocytes. In testosterone-treated SE models, TUNEL-positive cells were significantly increased between the granular and stratified corneum layers, which indicates that testosterone regulates homeostasis of keratinocytes through the generation of H2O2 (Fig. 6). It has been reported that testosterone can interfere with the epidermal permeability barrier. Moreover, a replacement for the testosterone-castrated group showed a functional alteration of the epidermal barrier, including the formation of an abnormal cornified envelope leading to hyperkeratosis. Increased DHT has been reported to play a role in follicular hyperkeratinization in acne vulgaris (53). Increased caspase-3 is also known to be involved in the pathogenesis of various skin diseases, such as lichen planus (54). Taken together, these previous reports and our results provide a new concept for understanding the homeostasis of keratinocytes.
Acknowledgment
We thank Dr. Christopher Chang (University of California at San Francisco) for PO-1.
This work was supported by National Research Foundation of Korea (NRF) Grant 2012R1A5A1048236, Bio & Medical Technology Development Program Grant 2012M3A9B4028785, and Redoxomics Grant 2012M3A9C5048708 funded by the Ministry of Science, ICT & Future Planning.
- ROS
- reactive oxygen species
- NOX
- NADPH oxidase
- DHT
- dihydrotestosterone
- AR
- androgen receptor
- [Ca2+]i
- intracellular calcium
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis-acetoxymethyl ester
- DCF-DA
- 2′,7′-dichlorofluorescin diacetate
- SEs
- skin equivalents
- IP1
- inositol monophosphate
- ASK-1
- apoptosis signal-regulating kinase-1
- PO-1
- Peroxy-Orange 1
- DPI
- diphenyleneiodonium chloride
- IP3
- inositol 1,4,5-trisphosphate.
REFERENCES
- 1. Block K., Gorin Y. (2012) Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer 12, 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lassègue B., San Martín A., Griendling K. K. (2012) Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 110, 1364–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bae Y. S., Oh H., Rhee S. G., Yoo Y. D. (2011) Regulation of reactive oxygen species generation in cell signaling. Mol. Cells 32, 491–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petry A., Weitnauer M., Görlach A. (2010) Receptor activation of NADPH oxidases. Antioxid. Redox. Signal. 13, 467–487 [DOI] [PubMed] [Google Scholar]
- 5. Aguirre J., Lambeth J. D. (2010) Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic. Biol. Med. 49, 1342–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elias P. M. (1983) Epidermal lipids, barrier function, and desquamation. J. Invest. Dermatol. 80, 44s–49s [PubMed] [Google Scholar]
- 7. Candi E., Schmidt R., Melino G. (2005) The cornified envelope: a model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 6, 328–340 [DOI] [PubMed] [Google Scholar]
- 8. Feingold K. R., Denda M. (2012) Regulation of permeability barrier homeostasis. Clin. Dermatol. 30, 263–268 [DOI] [PubMed] [Google Scholar]
- 9. Zouboulis C. C., Degitz K. (2004) Androgen action on human skin: from basic research to clinical significance. Exp. Dermatol. 13, 5–10 [DOI] [PubMed] [Google Scholar]
- 10. De Gendt K., Verhoeven G. (2012) Tissue- and cell-specific functions of the androgen receptor revealed through conditional knockout models in mice. Mol. Cell. Endocrinol. 352, 13–25 [DOI] [PubMed] [Google Scholar]
- 11. Zouboulis C. C., Chen W. C., Thornton M. J., Qin K., Rosenfield R. (2007) Sexual hormones in human skin. Horm. Metab. Res. 39, 85–95 [DOI] [PubMed] [Google Scholar]
- 12. Loss E. S., Jacobus A. P., Wassermann G. F. (2011) Rapid signaling responses in Sertoli cell membranes induced by follicle stimulating hormone and testosterone: calcium inflow and electrophysiological changes. Life Sci. 89, 577–583 [DOI] [PubMed] [Google Scholar]
- 13. Foradori C. D., Weiser M. J., Handa R. J. (2008) Non-genomic actions of androgens. Front. Neuroendocrinol. 29, 169–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rahman F., Christian H. C. (2007) Non-classical actions of testosterone: an update. Trends Endocrinol. Metab. 18, 371–378 [DOI] [PubMed] [Google Scholar]
- 15. Simoncini T., Genazzani A. R. (2003) Non-genomic actions of sex steroid hormones. Eur. J. Endocrinol. 148, 281–292 [DOI] [PubMed] [Google Scholar]
- 16. Pi M., Quarles L. D. (2012) Multiligand specificity and wide tissue expression of GPRC6A reveals new endocrine networks. Endocrinology 153, 2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pi M., Chen L., Huang M. Z., Zhu W., Ringhofer B., Luo J., Christenson L., Li B., Zhang J., Jackson P. D., Faber P., Brunden K. R., Harrington J. J., Quarles L. D. (2008) GPRC6A null mice exhibit osteopenia, feminization and metabolic syndrome. PLoS One 3, e3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pi M., Parrill A. L., Quarles L. D. (2010) GPRC6A mediates the non-genomic effects of steroids. J. Biol. Chem. 285, 39953–39964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pi M., Wu Y., Lenchik N. I., Gerling I., Quarles L. D. (2012) GPRC6A mediates the effects of l-arginine on insulin secretion in mouse pancreatic islets. Endocrinology 153, 4608–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chignalia A. Z., Schuldt E. Z., Camargo L. L., Montezano A. C., Callera G. E., Laurindo F. R., Lopes L. R., Avellar M. C., Carvalho M. H., Fortes Z. B., Touyz R. M., Tostes R. C. (2012) Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension 59, 1263–1271 [DOI] [PubMed] [Google Scholar]
- 21. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 22. Joo J. H., Ryu J. H., Kim C. H., Kim H. J., Suh M. S., Kim J. O., Chung S. Y., Lee S. N., Kim H. M., Bae Y. S., Yoon J. H. (2012) Dual oxidase 2 is essential for the Toll-like receptor 5-mediated inflammatory response in airway mucosa. Antioxid. Redox. Signal. 16, 57–70 [DOI] [PubMed] [Google Scholar]
- 23. Taieb J., Mathian B., Millot F., Patricot M. C., Mathieu E., Queyrel N., Lacroix I., Somma-Delpero C., Boudou P. (2003) Testosterone measured by 10 immunoassays and by isotope-dilution gas chromatography-mass spectrometry in sera from 116 men, women, and children. Clin. Chem. 49, 1381–1395 [DOI] [PubMed] [Google Scholar]
- 24. Dickinson B. C., Huynh C., Chang C. J. (2010) A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. J. Am. Chem. Soc. 132, 5906–5915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hellman L., Bradlow H. L., Freed S., Levin J., Rosenfeld R. S., Whitmore W. F., Zumoff B. (1977) The effect of flutamide on testosterone metabolism and the plasma levels of androgens and gonadotropins. J. Clin. Endocrinol. Metab. 45, 1224–1229 [DOI] [PubMed] [Google Scholar]
- 26. Tymianski M., Spigelman I., Zhang L., Carlen P. L., Tator C. H., Charlton M. P., Wallace M. C. (1994) Mechanism of action and persistence of neuroprotection by cell-permeant Ca2+ chelators. J. Cereb. Blood Flow Metab. 14, 911–923 [DOI] [PubMed] [Google Scholar]
- 27. Faure H., Gorojankina T., Rice N., Dauban P., Dodd R. H., Bräuner-Osborne H., Rognan D., Ruat M. (2009) Molecular determinants of non-competitive antagonist binding to the mouse GPRC6A receptor. Cell Calcium 46, 323–332 [DOI] [PubMed] [Google Scholar]
- 28. Rossol M., Pierer M., Raulien N., Quandt D., Meusch U., Rothe K., Schubert K., Schöneberg T., Schaefer M., Krügel U., Smajilovic S., Bräuner-Osborne H., Baerwald C., Wagner U. (2012) Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 3, 1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pi M., Faber P., Ekema G., Jackson P. D., Ting A., Wang N., Fontilla-Poole M., Mays R. W., Brunden K. R., Harrington J. J., Quarles L. D. (2005) Identification of a novel extracellular cation-sensing G-protein-coupled receptor. J. Biol. Chem. 280, 40201–40209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berridge M. J. (2009) Inositol trisphosphate and calcium signalling mechanisms. Biochim. Biophys. Acta 1793, 933–940 [DOI] [PubMed] [Google Scholar]
- 31. Blättermann S., Peters L., Ottersbach P. A., Bock A., Konya V., Weaver C. D., Gonzalez A., Schröder R., Tyagi R., Luschnig P., Gäb J., Hennen S., Ulven T., Pardo L., Mohr K., Gütschow M., Heinemann A., Kostenis E. (2012) A biased ligand for OXE-R uncouples Gα and Gβγ signaling within a heterotrimer. Nat. Chem. Biol. 8, 631–638 [DOI] [PubMed] [Google Scholar]
- 32. Gul R., Shawl A. I., Kim S. H., Kim U. H. (2012) Cooperative interaction between reactive oxygen species and Ca2+ signals contributes to angiotensin II-induced hypertrophy in adult rat cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 302, H901–H909 [DOI] [PubMed] [Google Scholar]
- 33. Gafni J., Munsch J. A., Lam T. H., Catlin M. C., Costa L. G., Molinski T. F., Pessah I. N. (1997) Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron 19, 723–733 [DOI] [PubMed] [Google Scholar]
- 34. Bowles D. K., Maddali K. K., Dhulipala V. C., Korzick D. H. (2007) PKCdelta mediates anti-proliferative, pro-apoptic effects of testosterone on coronary smooth muscle. Am. J. Physiol. Cell Physiol. 293, C805–C813 [DOI] [PubMed] [Google Scholar]
- 35. Ling S., Dai A., Williams M. R., Myles K., Dilley R. J., Komesaroff P. A., Sudhir K. (2002) Testosterone (T) enhances apoptosis-related damage in human vascular endothelial cells. Endocrinology 143, 1119–1125 [DOI] [PubMed] [Google Scholar]
- 36. Estrada M., Varshney A., Ehrlich B. E. (2006) Elevated testosterone induces apoptosis in neuronal cells. J. Biol. Chem. 281, 25492–25501 [DOI] [PubMed] [Google Scholar]
- 37. Verzola D., Gandolfo M. T., Salvatore F., Villaggio B., Gianiorio F., Traverso P., Deferrari G., Garibotto G. (2004) Testosterone promotes apoptotic damage in human renal tubular cells. Kidney Int. 65, 1252–1261 [DOI] [PubMed] [Google Scholar]
- 38. Winiarska A., Mandt N., Kamp H., Hossini A., Seltmann H., Zouboulis C. C., Blume-Peytavi U. (2006) Effect of 5α-dihydrotestosterone and testosterone on apoptosis in human dermal papilla cells. Skin Pharmacol. Physiol. 19, 311–321 [DOI] [PubMed] [Google Scholar]
- 39. Ulivieri C. (2010) Cell death: Insights into the ultrastructure of mitochondria. Tissue Cell 42, 339–347 [DOI] [PubMed] [Google Scholar]
- 40. Papadopoulou N., Charalampopoulos I., Anagnostopoulou V., Konstantinidis G., Föller M., Gravanis A., Alevizopoulos K., Lang F., Stournaras C. (2008) Membrane androgen receptor activation triggers down-regulation of PI-3K/Akt/NF-κB activity and induces apoptotic responses via Bad, FasL and caspase-3 in DU145 prostate cancer cells. Mol. Cancer 7, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Choudhry R., Hodgins M. B., Van der Kwast T. H., Brinkmann A. O., Boersma W. J. (1992) Localization of androgen receptors in human skin by immunohistochemistry: implications for the hormonal regulation of hair growth, sebaceous glands and sweat glands. J. Endocrinol. 133, 467–475 [DOI] [PubMed] [Google Scholar]
- 42. Li Q., Harraz M. M., Zhou W., Zhang L. N., Ding W., Zhang Y., Eggleston T., Yeaman C., Banfi B., Engelhardt J. F. (2006) Nox2 and Rac1 regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol. Cell. Biol. 26, 140–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller F. J., Jr., Chu X., Stanic B., Tian X., Sharma R. V., Davisson R. L., Lamb F. S. (2010) A differential role for endocytosis in receptor-mediated activation of Nox1. Antioxid. Redox. Signal. 12, 583–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Calebiro D., Nikolaev V. O., Persani L., Lohse M. J. (2010) Signaling by internalized G-protein-coupled receptors. Trends Pharmacol. Sci. 31, 221–228 [DOI] [PubMed] [Google Scholar]
- 45. Jiang F., Zhang Y., Dusting G. J. (2011) NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev. 63, 218–242 [DOI] [PubMed] [Google Scholar]
- 46. Lee S. R., Kwon K. S., Kim S. R., Rhee S. G. (1998) Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 273, 15366–15372 [DOI] [PubMed] [Google Scholar]
- 47. Janssen-Heininger Y. M., Mossman B. T., Heintz N. H., Forman H. J., Kalyanaraman B., Finkel T., Stamler J. S., Rhee S. G., van der Vliet A. (2008) Redox-based regulation of signal transduction: principles, pitfalls, and promises. Free Radic. Biol. Med. 45, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., Kawabata M., Miyazono K., Ichijo H. (1998) Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17, 2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Trüeb R. M. (2009) Oxidative stress in ageing of hair. Int. J. Trichology 1, 6–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bito T., Nishigori C. (2012) Impact of reactive oxygen species on keratinocyte signaling pathways. J. Dermatol. Sci. 68, 3–8 [DOI] [PubMed] [Google Scholar]
- 51. Naito A., Midorikawa T., Yoshino T., Ohdera M. (2008) Lipid peroxides induce early onset of catagen phase in murine hair cycles. Int. J. Mol. Med. 22, 725–729 [PubMed] [Google Scholar]
- 52. de Rivero Vaccari J. P., Sawaya M. E., Brand F., 3rd, Nusbaum B. P., Bauman A. J., Bramlett H. M., Dietrich W. D., Keane R. W. (2012) Caspase-1 level is higher in the scalp in androgenetic alopecia. Dermatol. Surg. 38, 1033–1039 [DOI] [PubMed] [Google Scholar]
- 53. Thiboutot D., Knaggs H., Gilliland K., Lin G. (1998) Activity of 5-α-reductase and 17-β-hydroxysteroid dehydrogenase in the infrainfundibulum of subjects with and without acne vulgaris. Dermatology 196, 38–42 [DOI] [PubMed] [Google Scholar]
- 54. Abdel-Latif A. M., Abuel-Ela H. A., El-Shourbagy S. H. (2009) Increased caspase-3 and altered expression of apoptosis-associated proteins, Bcl-2 and Bax in lichen planus. Clin. Exp. Dermatol. 34, 390–395 [DOI] [PubMed] [Google Scholar]