

Background: IFNβ constitutes an approved drug to treat multiple sclerosis (MS), but it has limited efficacy.
Results: A modified human IFN variant, which exhibits both superagonist properties and 10-fold increased lifespan, outperforms IFNβ in an animal MS model.
Conclusion: This drug candidate has potential to supersede IFNβ for the treatment of MS.
Significance: Protein engineering allows development of more effective drugs to treat autoimmune diseases.
Keywords: Autoimmune Disease, Biomarker, Drug Development, Interferon, Multiple Sclerosis, Transgenic Mice
Abstract
IFNβ is a common therapeutic option to treat multiple sclerosis. It is unique among the family of type I IFNs in that it binds to the interferon receptors with high affinity, conferring exceptional biological properties. We have previously reported the generation of an interferon superagonist (dubbed YNSα8) that is built on the backbone of a low affinity IFNα but modified to exhibit higher receptor affinity than even for IFNβ. Here, YNSα8 was fused with a 600-residue hydrophilic, unstructured N-terminal polypeptide chain comprising proline, alanine, and serine (PAS) to prolong its plasma half-life via “PASylation.” PAS-YNSα8 exhibited a 10-fold increased half-life in both pharmacodynamic and pharmacokinetic assays in a transgenic mouse model harboring the human receptors, notably without any detectable loss in biological potency or bioavailability. This long-lived superagonist conferred significantly improved protection from MOG35–55-induced experimental autoimmune encephalomyelitis compared with IFNβ, despite being injected with a 4-fold less frequency and at an overall 16-fold lower dosage. These data were corroborated by FACS measurements showing a decrease of CD11b+/CD45hi myeloid lineage cells detectable in the CNS, as well as a decrease in IBA+ cells in spinal cord sections determined by immunohistochemistry for PAS-YNSα8-treated animals. Importantly, PAS-YNSα8 did not induce antibodies upon repeated administration, and its biological efficacy remained unchanged after 21 days of treatment. A striking correlation between increased levels of CD274 (PD-L1) transcripts from spleen-derived CD4+ cells and improved clinical response to autoimmune encephalomyelitis was observed, indicating that, at least in this mouse model of multiple sclerosis, CD274 may serve as a biomarker to predict the effectiveness of IFN therapy to treat this complex disease.
Introduction
Multiple Sclerosis (MS)5 is a chronic immune disease of the CNS characterized by inflammation and demyelination within the brain and the spinal cord. This can lead to pathologies including visual disturbance, cognitive impairment, and paralysis (1). Epidemiological evidence supports that environmental factors play a significant role in triggering or facilitating the development of MS, although a genetic component is also evident for a subset of individuals (2). Genome-wide association studies have identified more than 100 potential MS risk alleles with a strong predisposition to genes involved in immune function, revealing a bias of genes relating to human leukocyte antigen and T-helper cell development or signaling (3, 4). These genetic findings provide support to a large body of immunological data that define MS as largely a T-cell-driven immune disease of the CNS (5).
IFNβ was the first drug to be approved for the treatment of relapsing-remitting MS and, to this day, remains a major therapeutic option for MS patients. Meta-analyses of clinical trials indicate that IFNβ therapy results in a significant reduction of MS relapses, and although its efficacy is less optimal than some of the newer drugs that have recently entered the market, its safety profile is good. Although efficacy correlates with higher administered dose and increased injection frequency (6, 7), there is clear evidence that the response to IFNβ therapy differs greatly between individual patients. Therefore, much effort has been invested in determining predictive factors to separate good from bad responders but with limited success (8). IFNβ is a member of the type I interferon (IFN-I) family, which includes also 13 IFNα subtypes as well as IFNω, IFNκ, and IFNϵ in humans. All IFN-I subtypes activate signaling through binding to a heterodimeric receptor complex comprising the cell surface subunits of IFNAR1 and IFNAR2 (9–12). IFNβ is unique in that it binds to the IFNARs with much higher affinity than the other IFN-I counterparts, conferring superior biological properties to this cytokine. For example, in certain cancer cell lines, IFNβ can induce potent antiproliferative and proapoptotic responses at concentrations 2 or 3 orders of magnitude lower than required for IFNαs (13). It seems that this high affinity interaction of IFNβ with its receptor is critical for its anti-inflammatory function in MS; in fact, clinical trials using a low affinity IFNα failed to suppress clinical disease progression (14).
It has long been recognized that the short half-life of interferons in the blood may hamper their efficacy as injected drugs. IFN PEGylation has been developed to counter this problem and has been widely used in several clinical applications. In particular, two long-lived IFNα2 variants conjugated with 12 and 40 kDa PEGs (PegIntronTM and PegasysTM) have been approved to treat hepatitis C infections. Both are prescribed as once weekly injections in patients, demonstrating improved efficacy and patient compliance compared with their non-PEGylated counterparts (the latter are typically injected three times per week) (15, 16). More recently, a 20-kDa N-terminal PEG conjugate of human IFNβ (PLEGRIDYTM) with prolonged pharmacodynamics has been developed for MS therapy (17–19). This new PEGylated IFNβ variant injected either once every 2 weeks or once every 4 weeks was shown to significantly reduce disease relapse rates over a 48-week trial period in MS patients compared with placebo controls (20). However, this trial was not designed to test for relative drug efficacy in comparison with conventional IFNβ therapies for MS treatment, and accordingly, relative effectiveness of PEG-IFNβ in providing improved clinical response still remains to be established.
In our laboratory, we have used structural insight to engineer a tight receptor-binding variant named YNSα8 from the backbone of low affinity IFNα2 (21–23). This variant is an extremely potent IFNβ-like molecule according to its very similar gene activation profile, supporting the notion that the tightness of binding to the IFNAR receptor complex is the overwhelming biophysical factor that distinguishes different interferons with regard to their biological activities. However, in studying the effect of these superagonists in a mouse model, we are faced with a dilemma, because the human and mouse IFNAR sequences have diverged considerably, and cross-species activation of human IFN-I subtypes in mouse cells is weak and nonphysiological. To allow the study of human IFN-I in mice, we have recently generated transgenic mice harboring humanized IFNAR receptors (HyBNAR) that sensitively transduce human IFN-I signaling (24). Here, we examine a human IFN-I superagonist with a greatly improved pharmacological lifespan through PASylation (25), a biological alternative to PEGylation, using the HyBNAR transgenic mouse model. We compared the pharmacodynamic properties of PAS-YNSα8 with the non-PASylated counterpart and with that of human IFNβ, demonstrating superior efficacy in a mouse EAE model that mimics MS in humans.
EXPERIMENTAL PROCEDURES
Construction of Expression Plasmids for the Production of PASylated YNSα8
The previously described plasmid pASK-IBA4-IFN (25), which encodes human IFNα2b as fusion protein with an N-terminal Strep-tag II, was subjected to QuikChange site-directed mutagenesis (Agilent Technologies, Waldbronn, Germany) using appropriate oligodeoxynucleotide primers to introduce the following six amino acid substitutions: H57Y, E58N, Q61S, E159K, S160R, and R162K. After that, the N-terminal Strep-tag II was replaced by a His6 tag, and three PAS#1(200) cassettes were inserted in a consecutive manner downstream of the His6 tag using the single SapI restriction site directly in front of the IFNα2b coding region as described (25), thus yielding the expression plasmid pASK75-His6-PAS#1(600)-YNSα8. An equivalent vector was prepared for the human wild type IFNα2b (pASK75-His6-PAS#1(600)-IFN).
Recombinant Protein Production and Purification
Recombinant protein production was performed according to a published procedure (25). The IFN variants were produced by periplasmic secretion in Escherichia coli KS272 (26) in the presence of the helper plasmid pTUM4 (27) as needed. Bacteria were cultivated either in shake flasks containing 2 liters of LB medium supplemented with 100 mg/liter ampicillin, 30 mg/liter chloramphenicol (for pTUM4), 1 g/liter proline, and 5 g/liter glucose or, alternatively, in a 4- or 8-liter bench top fermenter with a synthetic glucose mineral salt medium supplemented with the same antibiotics, as well as proline, following a published procedure (28). In the shake flask, recombinant gene expression was induced with 200 μg/liter anhydrotetracycline at A550 = 0.5 for up to 3 h at 22 °C. In case of fermenter production, induction was achieved by addition of 500 μg/liter anhydrotetracycline as soon as the culture reached A550 = 20 for a period of up to 2.5 h. Immediately thereafter, the cells were harvested by centrifugation, and a periplasmic extract was prepared in a buffer containing 500 mm sucrose, 200 mm sodium borate, pH 8.0, 1 mm (15 mm for 8 liters fermentation) EDTA and 1 mm 2,2′-dithiodipyridine (Sigma-Aldrich). PASylated YNSα8 and IFN were initially purified from the periplasmic E. coli extract via the His6 tag using a Ni2+-charged HisTrap HP column (GE Healthcare). Then cation exchange chromatography was performed on a Resource S column (GE Healthcare) using 20 mm Tris-HCl, pH 7.0, as running buffer and a NaCl concentration gradient for elution. All proteins were finally polished by size exclusion chromatography on a Superdex 200 pg HiLoad 16/60 column (GE Healthcare) in PBS (4 mm KH2PO4, 16 mm Na2HPO4, 115 mm NaCl). Protein purity was checked by SDS-PAGE, and protein concentrations were determined via UV absorption at 280 nm using calculated extinction coefficients of 19,180 m−1 cm−1 for PAS-YNSα8 and 17,900 m−1 cm−1 for PAS-IFN. Note that the PAS sequence shows no absorption at this wavelength (25). Final endotoxin content was typically below 20 endotoxin units/mg as measured with an Endosafe-PTS system using cartridges with 0.1–10 units/ml sensitivity (Charles River Laboratories, Wilmington, MA). Protein identity was confirmed by ESI/qTOF-MS on an maXis mass spectrometer (Bruker Daltonics, Bremen, Germany) in positive ion mode after dialysis against 10 mm ammonium acetate, pH 6.6. Directly prior to measurement, the solution was supplemented with 20% (v/v) acetonitrile and 0.5% (v/v) formic acid.
Surface Plasmon Resonance (SPR) Measurements
SPR real time affinity measurements were performed on a BIAcore 2000 system (BIAcore, Uppsala, Sweden) as described (25) with a human IFNAR2-Fc chimera (R&D Systems, Minneapolis, MN) immobilized via an amine-coupled anti-Fc antibody (Jackson ImmunoResearch, West Grove, UK) on a CMDP sensor chip (Xantec, Düsseldorf, Germany). The purified PAS-YNSα8 was injected in an appropriate concentration series using PBS containing 0.05% (v/v) Tween 20 as running buffer at a flow rate of 25 μl/min. The kinetic parameters were determined by fitting the raw data to a Langmuir binding model for bimolecular complex formation using BIAevaluation software version 4.1 (BIAcore).
Cell Culture
Measurements of antiproliferation and antiviral activity of IFNs on human WISH cells were described previously (22). In both assays, the EC50 values were calculated using KaleidaGraph version 4.1, according to the formula y = A0 + A/(1 + c/EC50)^s, where y represents the absorbance corresponding to the relative number of cells, A0 is the offset, A is the amplitude, c is the IFN concentration, and s is the slope (23).
Quantitative PCR (qPCR)
Gene induction levels using qPCR were performed according to the protocol detailed in Ref. 24. Measurements were made using either an Agilent 7300 real time PCR system (96-well setup) or for some of the studies (see Figs. 7 and 9) high throughput qPCR using BioMark 96 × 96 dynamic arrays (Fluidigm Corporation) according to the manufacturer's protocol. Relative expression levels were calculated by the −ΔΔCT (cycle threshold) relative quantification method (log2 fold change = −ΔΔCT, fold change = 2−ΔΔCT). The primers used to amplify both human and mouse genes for this study are provided in supplemental Table S1. Pearson correlation analysis for gene expression data were performed using Microsoft Excel (data analysis tool pack add-in). The squares of these values (R2) are shown in Fig. 9.
FIGURE 7.
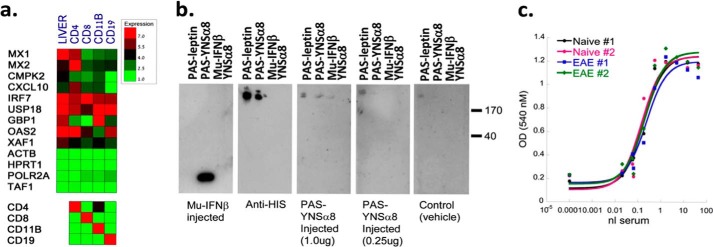
No observed loss of IFN-I responsiveness after repeated PAS-YNSα8 injections. a, 24 h after the last of repeated IFN (or vehicle) injections of EAE-induced mice (21 DPI), tissues were extracted, and the indicated splenocyte cell lineages were FACS-purified and processed for qPCR gene expression analysis for 10 IFN-I response genes and a panel of four reference genes (upper box). Samples were also tested for gene expression of immune cell markers as quality control to verify the FACS sort purity (lower box). The given values are averages from two to three mice for each group tested. b, Western blots were performed to test for immunogenicity of IFN-injected mice to PAS-leptin (as a control), PAS-YNSα8, Mu-IFNβ, and YNSα8. Panels from left to right show Western blots developed with the following sera. First panel, a mouse after repeat Mu-IFNβ injections; second panel, a commercial anti-His6 antibody; third panel, a mouse after repeat injections with 1 μg; fourth panel, 0.25 μg PAS-YNSα8 (active IFN); fifth panel, serum from a naïve mouse. c, EAE-induced mice after repeated injections of PAS-YNSα8 (0.25 μg q.a.d., from 0 to 20 DPI) had serum collected 24 h after the final IFN injection. Likewise, serum was also collected 24 h after naïve HyBNAR mice were administered with a single dose of PAS-YNSα8 for comparative purposes. To indirectly measure IFN serum levels, serial dilutions of blood samples were assayed by vesicular stomatitis virus antiviral assay using human WISH cells (see Fig. 1). All blood samples demonstrated a similar EC50 biological response conferred by 0.5 nl of serum, indicating that repeat PAS-YNSα8 treatment did not lead to a reduction in active IFN levels in the mouse compared with the naïve mouse controls. Two representative plots are shown for each treatment type.
FIGURE 9.

Increased expression of PD-L1 directly correlates with improved EAE clinical response. EAE-induced mice were treated for 21 or 27 days with vehicle, mouse IFNβ, or YNSα8 using the same injection regiments described in Fig. 4. Twenty-four hours after the last IFN injections spleens and livers were collected. CD4+, CD8+, CD11+, and CD19+ cells were freshly isolated by FACS analyses, and along with the liver samples all samples were processed for RNA generation. Samples were then tested for gene expression for MX1, MX2, PD-L1 (CD274), and PD1 (PDCD1) as described in Fig. 5. a, gene expression (log2 signal expressed as −ΔΔCT) for spleen-derived CD4+ve cells were plotted against a percentage of pre-EAE induction mouse weight for individual mice. Each point represents the average of duplicate measurements taken for an individual mouse. b, summary of a multivariable correlation analysis including EAE clinical score, percentage of change in mouse body weight and expression of the genes MX1, MX2, CD274, and PDCD1 for four different spleen-derived cell types, as well as from liver homogenate. R2 values are presented.
Mouse Stocks, Maintenance, and Ethics Statement
Stocks of C57BL/6 mice were purchased from Harlan Laboratories. Mice were maintained on site in the Weizmann Institute of Science animal facilities. All mouse experiments were performed strictly according to the Weizmann Institute of Science ethics committee guidelines and permissions.
Mouse Luminescence Studies
Life imaging and luciferase activity assays on tissue homogenates were performed as described in Ref. 24. In brief, d-luciferin was injected 10 min prior to luminescence measurements for each time point. The mice were anesthetized, and images were recorded in an IVIS Spectrum optical imaging device (Caliper Life Sciences) with quantification software (Living Image 4.1) provided by the same supplier. For tissue analyses, organs were extracted, and luciferin was added to each well immediately prior to measurements.
Pharmacokinetic Studies
Following intraperitoneal IFN injection, mouse blood samples were drawn at the indicated time points (see Fig. 2c) or after 24 h (see Fig. 7), and the EC50 values of antiviral activity relative to an IFNα2 standard were determined in WISH cells. pharmacokinetics (PKs) were calculated by fitting the EC50 values to a single exponential decay.
FIGURE 2.

Extended pharmacodynamic and pharmacokinetic lifespan of PAS-YNSα8. a, transgenic mice expressing the luciferase reporter gene under the control of the IFN-responsive MX2 promoter (MX2-LUC) were interbred with HyBNAR mice and injected intraperitoneally with 1 μg (IFN component) of either PASylated or non-PASylated YNSα8. At different time points, mice were injected with luciferin and anesthetized, and in vivo luminosity was measured by an image-capturing device (IVIS spectrum). Representative time course images from a mouse injected with PAS-YNSα8 (I, left panels) or a mouse injected with YNSα8 (II, right panels) are shown. b, quantification of in vivo luciferase signal from triplicate mice for each injection group. The data were fitted to a double exponential to model the biodistribution and elimination of the cytokine evoking the luciferase signal. c, mice were injected with 0.25 or 1 μg of PASylated YNSα8 (active IFN), and serum was collected 6, 24, and 48 h after injection. For control, 1.0 μg of human IFNβ was similarly injected, with serum collected at 6 and 24 h. The serum was then functionally quantified for human IFN by an anti-viral assay in human WISH cells.
Human MX1 Transcript Quantification
The data were extracted from a microarray study performed by Fernald et al. (29). The individual microarray signals were first normalized by subtraction of the average signal for each spot measurement against the average value of signals given by five reference genes (HMBS, HPRT1, POLR2A, TAF1, and TAF2), thus yielding the “Δ signal.” To determine relative change in expression of MX1, the signal for the IFNβ-treated patients was further subtracted against the average MX1 signal determined from four untreated control patients, yielding the “ΔΔ signal.” Fold change in gene expression was hence determined by the equation 2−(ΔΔ signal).
Experimental Autoimmune Encephalomyelitis Studies
EAE was induced in 9–11-week-old wild type and homozygous HyBNAR (C57BL/6) female mice (Harlan Laboratories Israel/Weizmann Institute animal facilities) by injecting a peptide comprising residues 35–55 of mouse myelin oligodendrocyte glycoprotein (MOG35–55; PolyPeptide Laboratories, Strasbourg, France). Mice were injected subcutaneously above the lumbar spinal cord with 100 μl of emulsion containing 200 μg/mouse of the encephalitogenic peptide in complete Freund's adjuvant (BD-Difco) enriched with 250 μg/mouse of heat-inactivated Mycobacterium tuberculosis (BD-Difco) at 0 days post-induction (DPI). Pertussis toxin (Enzo Life Sciences) at a dose of 300 ng per mouse was injected intraperitoneally immediately after the encephalitogenic injection, as well as at 2 DPI. IFN treatments commenced by intraperitoneal injection 3–4 h before EAE induction. EAE disease was scored using a five-point grading with 0 for no clinical disease; 1, tail weakness; 2, paraparesis (incomplete paralysis of one or two hindlimbs); 3, paraplegia (complete paralysis of one or two hindlimbs); 4, paraplegia with forelimb weakness or paralysis; 5, moribund or dead animals. The mice were examined daily. Graphics and statistical analyses were performed using KaleidaGraph version 4.1 or Microsoft Excel (data analysis tool pack add-in).
Western Blot Analysis
0.5 μg (excluding PAS component) of each indicated protein was run by SDS-PAGE and transferred to PVDF membranes (Millipore). Subsequently, the membranes were incubated with sera collected from mice (1:1000 diluted in TBST) or with a positive control anti-His6 antibody (1:8000; Qiagen). After washing, each membrane was incubated with the appropriate HRP-conjugated secondary antibody conjugate and stained by ECL.
FACS Analysis
Brain-infiltrating leukocytes: Minced brain and spinal cord tissue were processed in C-tubes (Miltenyi Biotec). Brain lymphocytes were isolated from the interface of a Percoll density gradient. The isolated lymphocytes were washed with cold PBS and resuspended in PBS containing 1% BSA and 0.1% NaN3 for direct cell surface staining. Single-cell suspensions were stained with antibodies for 30 min on ice. mAbs against CD8, CD4, CD11b, CD45, CD11c, CD205, and GR1 were from BD Biosciences or Biolegend. Nonspecific binding to cell surface Fc receptors was blocked with unlabeled FcγRII/FcγRIII-specific antibody (clone 2.4G2). Stained cells were acquired on a ten-LSRII cytometer and were analyzed with Kaluza software. Spleens were minced and cells were dissociated (BD-Falcon 40 μm cell strainer) before staining with indicated antibodies and live sorted using a SORP FACSAriaII device. Sorted cells were processed for qPCR as described above.
Immunohistochemistry
Animals were anesthetized and perfused transcardially with Dulbecco's PBS. Spines were dissected, fixed with paraformaldehyde (4% (w/v) for 3 days), and decalcified (6% (w/v) trichloroacetic acid; 5 days) as described (30). The lumbar and thorax SC segments were paraffin-embedded and sectioned coronally (4 μm) with a microtome (Leica, Nussloch, Germany). Sections from the fourth lumbar vertebra (L4) region were chosen for staining. Paraffin sections were deparaffinized; antigen-retrieved in 10 mm sodium citrate, pH 6, or with 10 mm Tris-HCl, 1 mm EDTA, pH 9, in the case of CD3 staining; next preincubated in PBS containing 20% (v/v) normal horse serum (Vector Laboratories, Burlingame CA) and 0.2% (v/v) Triton X-100 for 1 h; and then incubated overnight with rabbit anti-IBA1 (Wako Chemicals) or rat anti-myelin basic protein (Abcam, Cambridge, UK) primary antibodies. Then the sections were incubated with species-specific highly cross-absorbed Cy2- or Cy3-conjugated antibodies for 50 min (Jackson ImmunoResearch). Sections were counterstained with Hoechst 33258 (Molecular Probes, Eugene, OR) for nuclear labeling. Stained sections were examined in a fluorescence microscope (E600; Nikon, Tokyo, Japan) equipped with Plan Fluor objective connected to a CCD camera (DMX1200F; Nikon). Digital images were analyzed by Image-Pro Plus 4.5 software (Media Cybernetics, Bethesda, MD).
RESULTS
Molecular Design of PASylated YNSα8: A Long-lived Human Type I IFN Superagonist
Introduction of three side chain substitutions into IFNα2b at the IFNAR1 binding interface via protein engineering, i.e. H57Y, E58N, and Q61S (YNS), results in a 60-fold higher affinity to the receptor. Exchange of the five C-terminal amino acids to those from human IFNα8 increases binding to IFNAR2 by 15-fold (23). Combination of both sets of mutations led to the variant YNSα8, which exhibits significantly higher potency than IFN-β in cell culture systems (22–24). To enhance the plasma half-life of this molecule, we appended to the N terminus a PAS polypeptide comprising 600 residues of Pro, Ala, and Ser, which forms a natively disordered, uncharged biopolymer with large hydrodynamic volume, i.e. with biophysical properties very similar to the chemical polymer PEG (25). The resulting long-lived superagonist has been named PAS-YNSα8 (Fig. 1a).
FIGURE 1.

Development and evaluation of PAS-YNSα8. a, PAS-YNSα8 was depicted using PyMOL, also showing IFNAR1, IFNAR2, and the PAS tag (here with 200 residues in one exemplary random coil conformation) as cartoons in green, magenta, and gray, respectively, using two orientations rotated by 180°. Mutations H57Y, E58N, and Q61S (YNS) and E159K, S160R, and R162K (α8-tail) are depicted on the IFN surface facing IFNAR1 and IFNAR2, respectively. b, analysis of PAS-YNSα8, purified from the periplasmic cell fraction of E. coli, by Coomassie-stained 12% SDS-PAGE under reducing (lane 1) and nonreducing (lane 2) conditions. Lane M, protein size marker. c, ESI-MS analysis of PAS-YNSα8, confirming monodisperse composition and the calculated average molecular mass of 70,271.8 Da (ExPASy ProtParam tool). x-axis: mass (Da); y-axis: counts (×10,000). d, representative real time SPR analysis of PAS-YNSα8 binding to the soluble human IFNAR2-Fc chimera immobilized at ΔRU = 200–250 on a Xantec CMDP sensor chip measured on a BIAcore 2000 instrument and fitted to a 1:1 Langmuir model. The resulting kinetic and affinity parameters are listed in Table 1. e and f, anti-viral (e) and antiproliferative (f) dose-response curves in human WISH cells for different IFN subtypes. g, transcript expression levels of representative IFN-I response genes after stimulation with 1 pm of different human IFN-Is. The cells were exposed to the different IFNs for 24 or 6 h (followed by 18 h in IFN-free medium) before harvest and gene transcript analysis.
PAS-YNSα8 was produced in E. coli in a soluble state via periplasmic secretion and purified to homogeneity by means of immobilized metal ion affinity chromatography, cation exchange chromatography, and size exclusion chromatography. The correct formation of the two disulfide bridges was verified by SDS-PAGE under nonreducing conditions. In this assay, PAS-YNSα8 showed a generally slower electrophoretic mobility caused by low binding of SDS to the hydrophilic PAS polymer, giving rise to an apparent size of ∼140 kDa in comparison with a calculated mass of 70.3 kDa (Fig. 1b). The correct mass and monodisperse composition of the recombinant protein preparation was confirmed by ESI-MS both for PAS-YNSα8 (Fig. 1c) and for the PASylated original IFNα2b.
PASylated YNSα8 Retains High Biological Activity
Binding of PAS-YNSα8 to the extracellular domain of IFNAR2 was measured by SPR real time (BIAcore) analysis, and the results are shown in Fig. 1d and Table 1. The observed affinity of 0.43 nm to the receptor was similar to that previously measured for YNSα8 (0.4 nm) (23), indicating no loss of binding activity to IFNAR2. PAS-YNSα8 was then subjected to two in vitro biological assays using human WISH cells (22). In an antiproliferative dose-response assay (Fig. 1f) PAS-YNSα8 induced a 50% growth inhibition at 7.7 pm, essentially the same value as for YNSα8 (6.6 pm). Notably, this corresponds to approximate 4- and 360-fold higher potency than that for human IFNβ and IFNα2, respectively (Fig. 1f and Table 1). Unlike antiproliferative activity, anti-viral potency is generally similar, whether using low or high affinity IFN-Is (31). This is shown in Fig. 1e, where IFN-induced protection from challenge with vesicular stomatitis virus took place for all IFNs tested with EC50 between 0.4 and 0.7 pm. These data support that the PAS tag has no effect on the biological activity of IFNs in vitro (Table 1).
TABLE 1.
In vitro and biochemical characteristics of type I IFNs used in this study
| IFN | Anti-proliferationa |
Antiviral activitya |
BIAcore (SPR)b |
|||
|---|---|---|---|---|---|---|
| EC50 | Ratioc | EC50 | Ratioc | KD | Ratioc | |
| pm | pm | nm | ||||
| IFNα2 | 3600d | 1× | 0.6 | 1× | 2.2d | 1× |
| IFNβ | 30.6 | 120× | 0.42 | 1.4× | 0.4d | 5.5× |
| YNS | 23d | 160× | 0.2d | 3× | 2.0d | 1.1× |
| YNSα8 | 6.6 | 540× | 0.46 | 1.3× | 0.4d | 5.5× |
| PAS-YNSα8 | 7.7 | 470× | 0.50 | 1.2× | 0.435 | 5.0× |
a Antiviral and antiproliferative potencies were determined in WISH cells as described. The experimental error (σ) for these biological assays was 35%. Therefore, a confidence level of 2× S.E. would suggest that differences smaller than 2-fold between interferons are within the experimental error.
b KD values against IFNAR2 were determined by SPR using either the ratio of ka/kd or mass action over five different concentrations of the analyte.
c Ratios are relative to wild type IFNα2.
We next tested the ability of different IFN-Is, including PAS-YNSα8, to induce the expression of IFN response genes. WISH cells were stimulated with low dose IFN-Is (1 pm) for either 24 h without interruption or alternatively, for 6 h followed by washing and further incubation without IFN for the remaining 18 h. As expected, after 24 h of uninterrupted cytokine exposure, the amplitude of activation was found to be dependent on the type of IFN, with higher levels of gene induction measured for YNSα8 and IFNβ in comparison with the lower affinity IFNα2. The gene activation profile of PAS-YNSα8 was very similar to the ones of IFNβ and YNSα8, again indicating no loss of potency (Fig. 1g). For samples subjected to a 6-h pulse of IFN-Is, gene expression levels returned to nearly baseline at t = 24 h, demonstrating that the PASylated version of YNSα8 does not inherently induce prolonged IFN-I signaling in vitro (Fig. 1g). Thus, at least at the cellular signaling level, PAS-YNSα8 behaves in a manner very similar to that of its non-PASylated high affinity IFN counterpart.
PASylated YNSα8 Activates a Long Lasting IFN-I Response in Mice
HyBNAR transgenic mice that harbor humanized IFNAR1 and IFNAR2 receptors and that respond sensitively to human IFN-I injection (24) were used to evaluate the in vivo activity of PAS-YNSα8. These mice were further crossed with a transgenic mouse strain expressing the luciferase reporter under the control of the IFN-I-responsive MX2 promoter (24, 32) to follow the course of human IFN-I activation in real time. These double transgenic mice were injected intraperitoneally with 1 μg of YNSα8 or either 0.25 or 1.0 μg of PAS-YNSα8 (the described weight of PAS-YNSα8 relates only to the active IFN component of the chimeric protein). The luciferase signal was followed in live animals in the course of 1 week (three mice per group). Representative images for two mice injected with either 1 μg of PASylated or non-PASylated YNSα8 are shown in Fig. 2a.
A similar rapid rise in signal for both IFN-I variants was noted at t = 6 h, the first time point tested, indicating similar early stage bio-distribution. Thereafter, the pharmacodynamic response of PASylated and non-PASylated YNSα8 was dramatically different: mice injected with the non-PASylated IFN already exhibited a loss in activation signal at 12 h post-injection, further decreasing to nearly baseline at t = 24 h (II, right panels in Fig. 2a). In contrast, mice injected with PAS-YNSα8 exhibited prolonged activation of the luciferase signal, with a sustained biological response over >120 h (I, left panels in Fig. 2a). The live mouse luciferase images were quantified and fitted to a double exponential according to the Bateman function, i.e. one exponential representing increase of the signal during the distribution phase and the second exponential representing decay during elimination (Fig. 2b). The fitted pharmacodynamic half-life of the luciferase signal for non-PASylated YNSα8 was 3 h versus 30 h for PAS-YNSα8. Furthermore, injecting a 4-fold higher initial dose (1.0 μg instead of 0.25 μg of active component) of PASylated YNSα8 translated into a corresponding elevation of MX2-Luc levels during the entire period of the experiment, suggesting both that PAS-YNSα8 is stable in circulation and that its pharmacological activity is linearly dose-dependent.
To test whether our pharmacodynamic (PD) readouts correlate directly with PK properties of the PASylated IFN superagonist, we injected mice intraperitoneally with either 0.25 or 1.0 μg of PAS-YNSα8, and serum was collected 6, 24, and 48 h thereafter. The concentration of active IFN was then quantitatively determined by measuring its antiviral potency in the serum, showing a decay half-life of ∼24 h (Fig. 2c). In sharp contrast, when we injected 1 μg of human IFNβ intraperitoneally into mice, although high levels of IFN activity were detected in the serum at the 6-h time point, no detectable signal remained at 24 h (Fig. 2c). Thus, both our PK and PD studies indicate a similarly extended biological lifespan upon PASylation of our IFN superagonist without observable loss of bioactivity in relation to its non-PASylated counterpart.
To examine the effects of PASylation on tissue distribution of IFN, PAS-YNSα8 and a panel of other IFNs were injected intraperitoneally into HyBNAR/MX2-Luc double transgenic mice, and after 6 h a panel of organs was extracted for analysis. MX2-driven luciferase signals were shown to be most highly up-regulated in the liver but gave strong activation in all tissues tested with exception of the brain (Fig. 3a). Importantly, the activation profile did not differ between Mu-IFNβ and both the PASylated and non-PASylated forms of YNSα8, demonstrating that neither the tight binding of the superagonist to IFNARs nor the addition of the PAS tag results in an altered biodistribution or bioactivity at this early time interval postinjection.
FIGURE 3.
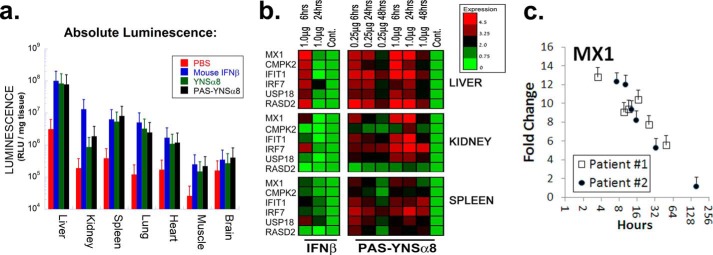
PAS-YNSα8 activates a robust yet distinct IFN-I signaling response in different tissues. a, mice were injected intraperitoneally with 1.0 μg of the indicated type I IFN or with PBS as control. Six hours after the mice were perfused with PBS, tissues were collected for homogenization and measured for luciferase activity. Absolute values of luciferase activity were standardized per unit wet weight for each tissue. Data for non-PASylated IFN-Is were taken from Ref. 24. b, left panels, wild type C57BL/6 mice were injected with 1 μg of Mu-IFNβ. Right panels, HyBNAR homozygous mice were injected with either 0.25 or 1 μg of PAS-YNSα8 (active IFN). Control mice were injected with vehicle (PBS). At the indicated time points, livers, kidneys, and spleens were harvested for RNA extraction and cDNA preparation. qPCR was then performed for a panel of IFN-I response genes as indicated at the left. The results representing log2 fold change (−ΔΔCt) are representative of two animals tested per time point. c, MX1 gene expression levels after single dose IFNβ injection into two human individuals (extracted from a microarray study published elsewhere (29)). Pharmacodynamics of IFNβ injection into humans shows a longer half-life in comparison with mice as expected from the rules of allometric scaling. The error bars represent S.E. from triplicate measurements.
Next, we tested the effects of unmodified IFN-I and PAS-YNSα8 on induction of IFN-I response genes. To this end, HyBNAR mice were injected intraperitoneally with 1 μg of Mu-IFNβ or either 0.25 or 1 μg of PAS-YNSα8 (active component). Then tissues were dissected, and activation of six IFN-I-controlled genes is analyzed by qPCR. The IFN-I response was found to be both tissue- and gene-specific (Fig. 3b). Whereas gene induction by mouse IFNβ lasted less than 24 h in all tissues and for all genes tested, a strong and prolonged induction was observed upon PAS-YNSα8 treatment even at the low dose of 0.25 μg. Interestingly, gene induction was remarkably tissue-specific, with the liver displaying the highest level of IFN-I induced gene activation as already seen in the in vivo luciferase study. Although all six genes were robustly activated in the liver (both by IFNβ and PAS-YNSα8), CMPK2 and RSDA2 were poorly induced in the kidney and spleen, indicating that the IFN-I activation profile is subject to tissue-specific epigenetic differences. Follow-up measurements of gene expression 24 and 48 h after PAS-YNSα8 injection demonstrated durable activation of these genes (Fig. 3b). Overall, this analysis of six IFN-I response genes in three tissues reveals a complex pharmacodynamic profile of gene activation, which is influenced by both gene and tissue specific parameters.
Single-dose intramuscular injection of IFNβ into humans also results in the activation of IFN response genes, as well as of response markers such as the metabolite neopterin. These responses can be measured by monitoring either their transcriptional or translational induction (18, 29, 33). In one of these studies, transcription microarray data were made available from two volunteers injected intramuscularly with single dose IFNβ, as well as from noninjected control patients (29). From these data, we determined the expression levels of the IFN-I response gene MX1 (Fig. 3c), revealing elevated activation over 3 days, in line with claims of others (18). Thus, IFNβ remains in circulation in humans ∼10 times longer than in mice, which is in accordance with the rules of allometric PK scaling (34). Consequently, IFNβ in humans has a half-life similar to that measured by us for PAS-YNSα8 in mice. This finding is of importance when planning the IFN treatment regime for a mouse disease model, which will be discussed next.
Therapeutic IFN Response in a Mouse Model of MS: EAE Induction
Induction of EAE with a MOG35–55 peptide is an extensively used mouse model to emulate chronic relapsing MS in humans (35, 36). Because IFNβ treatment is a major therapeutic option offered to MS patients, we were particularly interested in learning how well HyBNAR mice respond to human IFN therapy and how this response compares with that with PAS-YNSα8. We adjusted the system in a manner to induce a relatively severe form of EAE and then tested different Mu-IFNβ injection regimes beginning at the first day after disease induction: twice daily, once daily, and once every second day. The first injection was given at 0 DPI, and the last injection was at 16 DPI. In this setting, only the twice daily injection of IFNβ led to reduced EAE clinical severity (Fig. 4), which is in line with the known PK and PD data of this IFN. After cessation of therapy, the mice rapidly worsened to a clinical disease state similar to that of untreated EAE controls (Fig. 4).
FIGURE 4.

Clinical response of MOG peptide-induced EAE to mouse IFNβ. EAE was induced in C57BL/6 female mice, and different groups were treated with alternative regiments of mouse IFNβ therapy (9–10 mice per group). Mouse IFNβ (Mu-IFNβ) treatments commenced immediately prior to MOG injections (0 DPI) with the dosing regiments: 1 μg of IFNβ injected twice daily (b.i.d.), a single 2-μg dose injected once a day (q.d.), or a 2-μg dose injected once every 2 days (q.a.d.). At 11 DPI, all injection dosages were halved but without change in treatment periodicity. The last day of interferon treatment was 16 DPI. Two indexes to measure clinical disease severity are displayed. a, direct clinical measurement of EAE phenotype in a 5-point scale with increased disease symptoms correlating with higher score value. b, mice were weighed the day before EAE induction, and the change (as a percentage) in weight was recorded. c, nonparametric one-way analysis of variance was performed to determine pairwise significance for EAE vehicle control to that of IFN-treated groups measured until the last day of IFN injection (16 DPI). Three different statistical tests measuring cumulative EAE score, maximum EAE score, and cumulative mouse weight are given. A significant reduction in clinical severity and protection from weight loss was noted only for the Mu-IFNβ twice daily (b.i.d.) injected group.
Superior Efficacy of PAS-YNSα8 to Treat EAE
We next set out to perform a head to head comparison by treating mice with Hu-IFNβ, Mu-IFNβ, and PAS-YNSα8. Whereas IFNβ was injected twice daily at 1.0 μg/dose, the PASylated superagonist was injected only once every 2 days and at the much lower dosage of 0.25 μg (equivalent of IFN component). Injections took place from days 0 to 18, after which mice were observed without continued therapy for a further 3 days (Fig. 5). Whereas a relative severe EAE disease progression was similarly measured for both vehicle-injected WT and HyBNAR mice, all forms of IFN showed therapeutic efficacy to different degrees. Both Mu-IFNβ and Hu-IFNβ led to a less severe clinical score (Fig. 5a) and protection from inflammation-induced cachexia (Fig. 5b). In contrast, PAS-YNSα8 exerted a much stronger suppression of clinical symptoms and weight loss relative to both human and mouse IFNβ therapies during the course of the experiment, despite being administered at one-sixteenth of the cumulated protein dose, with merely one-quarter the frequency of injection (Fig. 5, a and b). A statistical comparison of cumulative EAE scores revealed that the clinical efficacy of PAS-YNSα8 in the treatment of the EAE mice was clearly significant (Fig. 5c). Similar to the previous experiment, after cessation of therapy, a worsening of disease soon followed. Our findings are consistent with previous reports on exacerbated clinical symptoms in both EAE and in MS patients after discontinuation of IFNβ therapy (37, 38). In summary, we demonstrate superior clinical response using our engineered long-lived human IFN variant compared with that of IFNβ in an EAE model using the transgenic HyBNAR mouse strain.
FIGURE 5.

Robust clinical protection of MOG peptide-induced EAE by PAS-YNSα8. EAE was induced in either wild type or in HyBNAR mice, split into groups of n = 5–8, each treated with a different regime of IFN therapy. Wild type and HyBNAR mice were injected with Mu-IFNβ or Hu-IFNβ, respectively, at a dose of 1 μg/injection, 2 injections/day (b.i.d.), whereas PAS-YNSα8 mice (0.25 μg of active IFN per injection) were injected once every 2 days (q.a.d.). a and b, average EAE disease scores (a) and percentage of original mouse weight (b). c, nonparametric one-way analysis of variance was performed from mice until the last day of IFN therapy as described in Fig. 4. d, box plots of EAE clinical score and percentage of weight loss summarizing the accumulated findings from a number of independent EAE experiments (taking the 19 DPI time point from each experiment). The mice were treated with different doses of PAS-YNSα8 (once every second day) or Mu- or Hu-IFNβ used for intraexperimental comparison (1 μg twice daily) for the duration of the experiment. The boxes represent the 25–75% data points, with lines and black squares representing the means and geometric means, respectively. Statistical significance: * p < 0.05; ** p < 0.01.
Next, we sought to test whether raising the dose of PAS-YNSα8 could further protect mice from EAE clinical symptoms. Indeed, clinical data using IFNβ to treat human MS indicate that increased drug dosage improves disease protection (7). Despite these findings, we found that maximum therapeutic efficacy was already observed with the lower dose of 0.25 μg of PAS-YNSα8, without added benefit by higher dosing (Fig. 5d).
Differences in the degree of EAE clinical severity and protection provided by IFN therapy were observed between the different experiments despite the identical reagents used. Despite these variations in absolute clinical scores, it was consistently observed that PAS-YNSα8 outperformed any other IFN-I used in this study (Figs. 5, a and d, and 6).
FIGURE 6.

Low affinity PAS-IFNα2 does not provide sustained clinical protection in MOG peptide-induced EAE. HyBNAR mice were injected with high affinity PAS-YNSα8 or alternatively low affinity PAS-IFNα2 (1 μg of active IFN injected once every 2 days for both 0 → 24 DPI) or with PBS vehicle control (eight mice per experimental group). Low affinity PAS-IFNα2 exerts a delay in disease progression, but this is not sustained over the course of drug treatment. a, average EAE disease score. b, average percentage of initial weight. c, statistical analyses (cumulative for 0 → 20 DPI) demonstrate that the improvement in clinical symptoms for the PAS-IFNα2-treated group is transient. NS, not significant.
To test the importance of using a high affinity receptor binding interferon in the treatment of EAE, we performed a similar experiment by injecting the low affinity (original) PASylated IFNα2 (25). Although PAS-IFNα2 might provide transient delay in the onset of clinical symptoms, full disease was evident by 21 DPI, despite continued drug injection (Fig. 6). These results support that high affinity IFN-Is are required for effective EAE disease protection, in agreement with clinical findings for human MS (14).
PAS-YNSα8 Retains a Similar Level of Signaling over the Course of the EAE Experiment
Repeated injections of Hu-IFN-Is or of the PASylated proteins into mice may potentially result in a reduced response over time, either through an immune response that neutralizes their effect or through receptor down-regulation. To test the IFN-I response over the course of the EAE treatment, gene induction was monitored at the end of the experiment (21 DPI), which was 1 day after the last PAS-YNSα8 injection. Livers and spleens were collected and the latter were dissociated, stained with different antibodies against surface markers (CD4, CD8, CD11B, and CD19), and subjected to FACS to separate the different immune cell populations. These samples were then used to generate cDNA for qPCR analysis. Gene expression levels for nine IFN-I response genes and four reference genes were analyzed and compared with those for untreated EAE mice (Fig. 7a). Even after 21 days of repeated injections, a strong IFN-I gene response was detected for the different cell types. Consistent with our earlier study of IFN-I gene activation in different tissues (Fig. 3b), a conspicuous cell-specific variation in induction of different IFN-I response genes was noted (Fig. 7a). IRF7 and USP18 were up-regulated in all examined cell types, confirming that the IFN superagonist is activating a gene response for all the cell types examined. Nevertheless, all other genes exhibited tissue/cell type-specific activation profiles. qPCR was next performed to verify the level of purity of the sorted cells, and a one to one concordance between cell type and the gene encoding the surface marker used for its purification was demonstrated. Exceptionally, the CD11B+-sorted cells encoded both CD4 and CD11B gene products, in line with findings that subpopulations of dendritic cells and macrophages co-express both markers (39, 40).
Next, the immunogenicity of repeated PAS-YNSα8 treatment was evaluated. Serum from mice that had received repeated injections of IFN was extracted at 21 DPI and directly tested for the presence of anti-drug antibodies. Western blot analysis revealed that mice repeatedly dosed with PAS-YNSα8 did not generate antibodies, despite the human origin of the engineered IFN that was administered (Fig. 7b). Surprisingly, however, some mice treated with Mu-IFNβ therapy did raise antibodies to the murine cytokine. Mu-IFNβ is naturally glycosylated, whereas the recombinant Mu-IFN produced in E. coli is not. This may explain why these mice generated an immune response to the injected cytokine. To directly assess the concentration of active nonbound interferon in the serum, the antiviral potency of the serum on human WISH cells was determined and found to be unchanged between mice after 21 days of treatment in comparison with naïve controls (representative plots are shown in Fig. 7c), demonstrating the absence of drug neutralizing activity. Together, our findings demonstrate that the level of PAS-YNSα8 response was not altered during the duration of the experiment, exerting sustained activation of IFN response genes, with no neutralizing anti-drug antibodies being generated.
PAS-YNSα8 Curtails Myeloid Cell Lineage in the Central Nervous System of EAE-induced Mice
Brain and spinal cord infiltrating leukocytes were isolated from individual EAE-induced HyBNAR mice and examined by FACS for changes in immune phenotype following treatment with PAS-YNSα8. Superagonist treatment resulted in an almost 2-fold reduction of infiltrating (CD11B+/CD45Hi) macrophages, yet without observing a significant change in the proportion of resident (CD11B+/CD45Low) microglia (Fig. 8a). The decrease in infiltrating macrophages may be accompanied by a reduction in MHCII+ cells, an indicator of activation of antigen presenting cells, although this latter observation lacked statistical significance (Fig. 8a). In contrast, no differences in the relative frequencies in (CD205+/CD206−) dendritic cells and T-cells (CD4+ or CD8+) were found between experimental groups. We looked into this further by performing a qualitative histological study with cross-sections of spinal cord at 19 DPI from EAE HyBNAR mice treated with either vehicle (PBS), Hu-IFNβ, or PAS-YNSα8. Hematoxylin and eosin staining revealed regions of leukocyte infiltration as indicated by clusters of darkly stained nuclei found in the EAE-vehicle control and, to a lesser degree, in the IFNβ-treated sample (Fig. 8b, top panels, white arrows).
FIGURE 8.

PAS-YNSα8 curtails myeloid cell lineage in the EAE CNS. a, infiltrating leukocytes were prepared from spinal cords of EAE-induced HyBNAR mice that were treated with or without PAS-YNSα8 (four animals per group, 18 DPI) and subjected to FACS analysis (± S.E.). Black columns, vehicle-treated EAE mice; gray columns, PAS-YNSα8-treated EAE mice. DCs, dendritic cells. Of the studied populations, only the macrophage lineage was shown to be decreased with statistical significance (p < 0.02, Student's t test; one tail analysis). b, qualitative histological study. Cross-sections of spinal cord from EAE-induced HyBNAR mice 19 DPI (L6 region), treated with vehicle, Hu-IFNβ, or PAS-YNSα8, were stained with hematoxilin and eosin (top panels, H&E), antibodies directed against IBA1 (middle panels), and antibodies directed against myelin basic protein (MBP) (bottom panels). c, quantification of IBA1-positive cells (IBA+) taken from L6 medial region of spinal cord. The average number of cells counted (± S.E.) corresponds to a field view of 10,000 μm2. Measurements were taken from duplicate mice per treatment group. Three slides were quantified per mouse. Significance values correspond to one-way analysis of variance (Tukey post hoc test). Mice from independent EAE experiments were used for the described FACS and immunohistochemistry studies.
We then stained for IBA1 immunoreactivity, a marker of microglia and infiltrating macrophages. A marked up-regulation of IBA1 immunopositive cells was detected for the EAE-induced vehicle control but appeared markedly reduced for both IFNβ- and PAS-YNSα8-treated mice (Fig. 8b, middle panels). Finally, using a myelin basic protein antibody, the degree of demyelination (labeled by asterisks) in the IFN-treated EAE-induced mice was reduced in comparison with those with vector control (Fig. 8b, bottom panels).
We next performed semiquantification of the IBA+ immunoreactive myeloid cells. A significant decrease in IBA-positive cells was detected both for IFNβ and PAS-YNSα8-treated spinal cord sections in comparison with vehicle-treated EAE mice (Fig. 8c). This result concurs with our FACS findings of a decrease in CD11B+/CD45HI myeloid cells. In contrast, consistent with our FACS data, we found no obvious difference in the staining of CD3+ T-cells or CD45R+ B cells as a result of IFN therapy by immunohistochemistry analysis (data not shown). Thus, the FACS and immunohistochemical studies both support that IFN therapy leads to decrease in the number of myeloid cells in the CNS of EAE-induced mice. Based on the FACS data, we suspect that IFN therapy acts to suppress the infiltration of (CD11B+/CD45Hi) macrophages into the CNS.
Increased PD-L1 (CD274) Transcript Expression Correlates with Improved EAE Clinical Outcome
IFN-I therapeutic response in the treatment of MS or in EAE must by default take place by the activation or repression of effector IFN response genes that modulate disease status. It has recently been reported that a novel regulatory T-cell population expressing high levels of PD-L1 (CD274) plays a central role in the IFN-mediated therapeutic activity upon treatment of EAE and MS (41). We have identified this gene as one that is differentially up-regulated in human WISH cells by IFNβ but not by IFNα (31) and thus hypothesized that CD274 may act as a mediator of the IFN effect in this disease. In comparison, we also tested for a relationship between the expression of MX1, as well as MX2, and the disease status. Indeed, MX1 in particular has often been used as a reference marker to test for correlation between gene expression and response to IFNβ therapy in MS. However, although associations have been found, it is of limited predictive value (42–47).
Consequently, FACS-sorted CD4 spleen cells from EAE-induced mice that were either untreated or treated with Mu-IFNβ or with PAS-YNSα8 were tested for a relationship between gene expression levels for CD274, MX1, and MX2 and disease severity. The findings are represented in scatter plots with each data point representing a value determined from a single mouse (Fig. 9a). Here, the degree of clinical severity is represented as change in mouse weight in comparison with pre-EAE induction for each animal based on a strong correlation between weight loss and disease scoring (Pearson correlation coefficient r = −0.85; R2 = 0.72; Fig. 9b). Weight loss gives a stronger correlative relationship to gene expression than that for measurements of EAE disease score (Fig. 9b), the latter which depends upon the human evaluator. In CD4+ cells, PD-L1 gene expression levels strongly correlate with EAE disease severity, significantly more than found for either MX1 or MX2 (Fig. 9, a and b). Whereas MX1 and MX2 both demonstrated a trend toward higher gene expression with more potent IFN treatment, the correlation with disease progression was poor. The high quality of gene expression measurements in CD4+ cells was confirmed by the excellent mutual correlation of co-expression of MX1 and MX2 for the different mouse samples (Fig. 9b).
We expanded our study to assess the expression of MX1, MX2, PD-L1, and its receptor PDCD1 (PD-1) in different leukocyte subsets including CD8+, CD11B+, and CD19+ cells, as well as liver (which we assume to not directly relate to EAE disease state). The analyses are summarized in the form of Pearson correlations squared (Fig. 9b). Although for all but CD19+ cells, positive correlations were found between gene expression and the disease state, the most predictive of these were found for the CD4+, as well as the CD11B+ lineages, with CD274 giving the most strongly predictive values. In summary, our findings indicate that at least for appropriately isolated leukocyte populations, CD274 transcript expression may serve as a marker to predict successful IFN therapy in a mouse EAE model.
DISCUSSION
In this study, we have combined two technologies, a tight receptor-binding type I interferon variant and the novel PASylation technology to generate a long-lived IFN superagonist for potential pharmaceutical application. The PAS amino acid sequence is unstructured and hydrophilic, although uncharged, conferring a large hydrodynamic volume in aqueous solution with the capacity to greatly increase the in vivo half-life of a fused protein by retarding kidney filtration. The PAS tag used in this study comprises 600 residues of Pro, Ala, and Ser with a total mass of ∼50 kDa, which adopts an expanded random coil conformation with an effective size of more than 600 kDa under physiological conditions as previously demonstrated by size exclusion chromatography (25). PASylation thus provides a simple alternative to PEGylation technologies, which suffer from intrinsic technical complexities such as the necessity of chemical in vitro coupling, high cost of goods, inherent polydispersity, and lack of biodegradability. In the case of the IFN superagonist YNSα8, the PAS tag conferred an increased pharmacodynamic half-life of ∼10-fold in mice after intraperitoneal injection (Fig. 2), without inducing a measurable change in biodistribution or biological potency relative to the non-PASylated cytokine (Fig. 3a). If translated to humans, this could conceivably shift a thrice weekly IFNβ injection regime to a monthly injection of PAS-YNSα8 with potentially enhanced clinical efficacy.
Testing the physiological effects of injected human IFNs in animal models is inherently problematic, because interspecies variations in receptor sequence result in suboptimal activation of human IFNs when injected into mice (24). The traditional solution to this approach is to directly perform preclinical studies of human IFNs in primates. To exemplify this, another engineered human IFN-I variant has recently been tested for efficacy in treatment of HIV, utilizing the rhesus macaque SIV model (48). The inherent ethical considerations of working with primates and the extremely high cost of their maintenance make this a problematic experimental option. Here, we adopted an alternative approach using a transgenic mouse strain expressing humanized IFNAR receptors, which respond sensitively to human IFN (24). The HyBNAR mice, which were generated in the purebred C57BL/6 genetic background, are amenable to MOG35–55 peptide-induced EAE, a commonly used mouse model to emulate human MS.
In a head to head comparison, PAS-YNSα8 outperformed both Hu-IFNβ and Mu-IFNβ in ameliorating EAE disease symptoms in the HyBNAR mouse model, despite being injected with a 4-fold less frequency and an overall 16-fold lower injection dose (Fig. 5). Administration of low affinity PASylated IFNα2 was not efficacious in treating EAE (Fig. 6), confirming that the tight receptor binding activity of the YNSα8 variant built on the backbone of this IFN converts it into an effective drug for the treatment of this indication. This mirrors clinical trials showing a lack of efficacy for low affinity IFNα in the treatment of MS (14).
Twenty years of clinical data confirm that IFNβ therapy is efficacious in the treatment of MS, albeit with a significant proportion of patients (∼35–50%) responding poorly to the drug (49). Currently, there is no accepted biomarker to identify a priori good responders to IFNβ treatment in advance of therapy. This problem extends to other drugs where we still lack reliable personalized medicinal markers to tailor a suitable therapy to treat individual MS patients (50). One major problem for MS research on finding associations between clinical and genomic/transcriptomic data is that there remains a lack of consensus as to what clinical features of MS are suitable to quantify disease severity (51, 52). A further complication is that neuroimmunological studies suggest that MS may not be a homogeneous disease but rather comprises a collection of disease states with related but distinct pathologies (53, 54). The mouse EAE model provides an alternative to study transcriptomic markers that might provide predictive power in assessing IFN therapy in MS. In this model, disease onset is timed and controlled, scoring of disease status is relatively straightforward, and the mice used are inbred; thus differential states of disease severity cannot be attributed to genotypic variation. Using this experimentally controlled EAE model, a clear signal of increased expression of CD274 in CD4+ and CD11B+ spleen-derived cells correlated directly with improved clinical outcome, suggesting its potential role as biomarker. In accordance with our original hypothesis, we further consider that CD274 may not only be acting as a marker of IFN response to in EAE and MS but is by itself an effector molecule that modulates the immune phenotype. This notion is strengthened by a recent study showing that a novel suppressive regulatory T-cell population expressing the transcription factor FoxA1 is instrumental in driving the IFN therapeutic response in EAE and MS (41). These FoxA1-positive T-reg cells were also shown to co-express high levels of CD274 (PD-L1). The number of these FoxA1+ PD-L1 cells was significantly reduced in IFNAR1 knock-out mice (41), indicating that the IFN-I signaling axis plays a major role in delineation of this novel T-Reg cell population. Our gene expression study with CD274 shown herein complements these findings.
In our spinal cord histopathological studies, we observed decreased myeloid cell numbers, perhaps resulting from a decrease of macrophage infiltration into the CNS, a phenotype consistent with studies by others testing the EAE phenotype with genetic deletions of IFNAR1 (55, 56) or IFNβ (57, 58). In particular, a study using cell lineage specific IFNAR1 knock-out mice has identified the myeloid cells as critical for disseminating the IFN-therapeutic potential in EAE (56). How these myeloid cells interact with infiltrating CD4+ subsets in particular will be a subject of future studies, complementing important findings made in this field (41, 59).
CONCLUSIONS
We have used the novel PASylation technology to generate a long-lived human type I IFN superagonist. PASylation of our IFN variant resulted in no loss of receptor binding affinity whatsoever and when injected into a mouse multiple sclerosis model demonstrated superior efficacy to that of IFNβ, without inducing an observable immunogenic footprint. In addition to its increased efficacy in relation to IFNβ, PAS-YNSα8 can potentially act as a replacement therapy for patients defined a priori as good responders to IFNβ treatment but who have developed neutralizing antibodies to the injected interferon. Our preclinical findings provide supportive evidence to vindicate the advance of PAS-YNSα8 to human clinical trials.
Using the HyBNAR transgenic mouse model, we have found a strong linear correlation between increased PD-L1 transcript levels in spleen-derived CD4+ and CD11+ cells and a decrease in EAE disease severity. Thus, we present a model claiming that PD-L1 acts as a predictive marker to determine efficacious IFN therapy in EAE. We extrapolate that this may also hold true for human MS, which should be a matter worth of future study.
Supplementary Material
Acknowledgments
We are grateful for the generosity and help from a number of people, especially Dr. Sandrine Pouly (Merck-Serono) for important discussions relating to the HyBNAR PK and PD studies, Drs. Slava Kalchenko and Yuri Kuznetsov for advice regarding IVIS Spectrum use, and members of the Veterinary Services for help, handling, and maintenance of our mice colonies. Mouse and human IFNβ were both supplied by Merck-Serono.
This work was supported by a research grant from Merck-KGaA (to G. S.) and by the Leading Edge Cluster m4 Grant 16EX1022V funded by the Bundesministerium für Bildung und Forschung (to A. S.). A. Z. and P. S. were employed by Merck-Serono. A. S. is a member of XL-protein GmbH.

This article contains supplemental Table S1.
- MS
- multiple sclerosis
- PAS
- proline, alanine, and serine
- EAE
- autoimmune encephalomyelitis
- qPCR
- quantitative PCR
- PK
- pharmacokinetic
- MOG
- myelin oligodendrocyte glycoprotein
- DPI
- days post-induction
- PD
- pharmacodynamic
- SPR
- surface plasmon resonance
- IFNAR
- type I interferon receptor
- HyBNAR
- HyBbrid IFNAR.
REFERENCES
- 1. Goverman J. (2009) Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 9, 393–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ebers G. C. (2008) Environmental factors and multiple sclerosis. Lancet Neurol. 7, 268–277 [DOI] [PubMed] [Google Scholar]
- 3. International Multiple Sclerosis Genetics Consortium, Wellcome Trust Case Control Consortium 2 (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. International Multiple Sclerosis Genetics Consortium (IMSGC) (2013) Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 45, 1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McFarland H. F., Martin R. (2007) Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol. 8, 913–919 [DOI] [PubMed] [Google Scholar]
- 6. IFNB Multiple Sclerosis Study Group (1993) Interferon β-1b is effective in relapsing-remitting multiple sclerosis: I. clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology 43, 655–661 [DOI] [PubMed] [Google Scholar]
- 7. Schwid S. R., Panitch H. S. (2007) Full results of the Evidence of Interferon Dose-Response-European North American Comparative Efficacy (EVIDENCE) study: a multicenter, randomized, assessor-blinded comparison of low-dose weekly versus high-dose, high-frequency interferon β-1a for relapsing multiple sclerosis. Clin. Ther. 29, 2031–2048 [DOI] [PubMed] [Google Scholar]
- 8. Mahurkar S., Suppiah V., O'Doherty C. (2014) Pharmacogenomics of interferon β and glatiramer acetate response: a review of the literature. Autoimmun. Rev. 13, 178–186 [DOI] [PubMed] [Google Scholar]
- 9. Pestka S., Krause C. D., Walter M. R. (2004) Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 202, 8–32 [DOI] [PubMed] [Google Scholar]
- 10. Platanias L. C. (2005) Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386 [DOI] [PubMed] [Google Scholar]
- 11. Borden E. C., Sen G. C., Uze G., Silverman R. H., Ransohoff R. M., Foster G. R., Stark G. R. (2007) Interferons at age 50: past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 6, 975–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Uzé G., Schreiber G., Piehler J., Pellegrini S. (2007) The receptor of the type I interferon family. Curr. Top. Microbiol. Immunol. 316, 71–95 [DOI] [PubMed] [Google Scholar]
- 13. Lavoie T. B., Kalie E., Crisafulli-Cabatu S., Abramovich R., DiGioia G., Moolchan K., Pestka S., Schreiber G. (2011) Binding and activity of all human α interferon subtypes. Cytokine 56, 282–289 [DOI] [PubMed] [Google Scholar]
- 14. AUSTIMS Research Group. (1989) Interferon-α and transfer factor in the treatment of multiple sclerosis: a double-blind, placebo-controlled trial. J. Neurol. Neurosurg. Psychiatry 52, 566–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heathcote E. J., Shiffman M. L., Cooksley W. G., Dusheiko G. M., Lee S. S., Balart L., Reindollar R., Reddy R. K., Wright T. L., Lin A., Hoffman J., De Pamphilis J. (2000) Peginterferon alfa-2a in patients with chronic hepatitis C and cirrhosis. N. Engl. J. Med. 343, 1673–1680 [DOI] [PubMed] [Google Scholar]
- 16. Dhalluin C., Ross A., Huber W., Gerber P., Brugger D., Gsell B., Senn H. (2005) Structural, kinetic, and thermodynamic analysis of the binding of the 40 kDa PEG-interferon-α2a and its individual positional isomers to the extracellular domain of the receptor IFNAR2. Bioconjug. Chem. 16, 518–527 [DOI] [PubMed] [Google Scholar]
- 17. Baker D. P., Lin E. Y., Lin K., Pellegrini M., Petter R. C., Chen L. L., Arduini R. M., Brickelmaier M., Wen D., Hess D. M., Chen L., Grant D., Whitty A., Gill A., Lindner D. J., Pepinsky R. B. (2006) N-terminally PEGylated human interferon-β-1a with improved pharmacokinetic properties and in vivo efficacy in a melanoma angiogenesis model. Bioconjug. Chem. 17, 179–188 [DOI] [PubMed] [Google Scholar]
- 18. Hu X., Miller L., Richman S., Hitchman S., Glick G., Liu S., Zhu Y., Crossman M., Nestorov I., Gronke R. S., Baker D. P., Rogge M., Subramanyam M., Davar G. (2012) A novel PEGylated interferon β-1a for multiple sclerosis: safety, Pharmacology, and biology. J. Clin. Pharmacol. 52, 798–808 [DOI] [PubMed] [Google Scholar]
- 19. Reuss R. (2013) PEGylated interferon β-1a in the treatment of multiple sclerosis: an update. Biol. Targets Ther. 7, 131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Calabresi P. A., Kieseier B. C., Arnold D. L., Balcer L. J., Boyko A., Pelletier J., Liu S., Zhu Y., Seddighzadeh A., Hung S., Deykin A., and ADVANCE Study Investigators (2014) Pegylated interferon β-1a for relapsing-remitting multiple sclerosis (ADVANCE): a randomised, phase 3, double-blind study. Lancet Neurol. 13, 657–665 [DOI] [PubMed] [Google Scholar]
- 21. Roisman L. C., Jaitin D. A., Baker D. P., Schreiber G. (2005) Mutational analysis of the IFNAR1 binding site on IFNα2 reveals the architecture of a weak ligand-receptor binding-site. J. Mol. Biol. 353, 271–281 [DOI] [PubMed] [Google Scholar]
- 22. Kalie E., Jaitin D. A., Abramovich R., Schreiber G. (2007) An interferon α2 mutant optimized by phage display for IFNAR1 binding confers specifically enhanced antitumor activities. J. Biol. Chem. 282, 11602–11611 [DOI] [PubMed] [Google Scholar]
- 23. Levin D., Harari D., Schreiber G. (2011) Stochastic receptor expression determines cell fate upon interferon treatment. Mol. Cell. Biol. 31, 3252–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harari D., Abramovich R., Zozulya A., Smith P., Pouly S., Köster M., Hauser H., Schreiber G. (2014) Bridging the species divide: transgenic mice humanized for type-I interferon response. PLoS One 9, e84259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schlapschy M., Binder U., Börger C., Theobald I., Wachinger K., Kisling S., Haller D., Skerra A. (2013) PASylation: a biological alternative to PEGylation for extending the plasma half-life of pharmaceutically active proteins. Protein Eng. Des. Sel. 26, 489–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meerman H. J., Georgiou G. (1994) Construction and characterization of a set of E. coli strains deficient in all known loci affecting the proteolytic stability of secreted recombinant proteins. Biotechnology (NY) 12, 1107–1110 [DOI] [PubMed] [Google Scholar]
- 27. Schlapschy M., Grimm S., Skerra A. (2006) A system for concomitant overexpression of four periplasmic folding catalysts to improve secretory protein production in Escherichia coli. Protein Eng. Des. Sel. 19, 385–390 [DOI] [PubMed] [Google Scholar]
- 28. Schiweck W., Skerra A. (1995) Fermenter production of an artificial Fab fragment, rationally designed for the antigen cystatin, and its optimized crystallization through constant domain shuffling. Proteins 23, 561–565 [DOI] [PubMed] [Google Scholar]
- 29. Fernald G. H., Knott S., Pachner A., Caillier S. J., Narayan K., Oksenberg J. R., Mousavi P., Baranzini S. E. (2007) Genome-wide network analysis reveals the global properties of IFN-β immediate transcriptional effects in humans. J. Immunol. 178, 5076–5085 [DOI] [PubMed] [Google Scholar]
- 30. Begum F., Zhu W., Namaka M. P., Frost E. E. (2010) A novel decalcification method for adult rodent bone for histological analysis of peripheral-central nervous system connections. J. Neurosci. Methods 187, 59–66 [DOI] [PubMed] [Google Scholar]
- 31. Jaitin D. A., Roisman L. C., Jaks E., Gavutis M., Piehler J., Van der Heyden J., Uze G., Schreiber G. (2006) Inquiring into the differential action of interferons (IFNs): an IFN-α2 mutant with enhanced affinity to IFNAR1 is functionally similar to IFN-β. Mol. Cell. Biol. 26, 1888–1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pulverer J. E., Rand U., Lienenklaus S., Kugel D., Zietara N., Kochs G., Naumann R., Weiss S., Staeheli P., Hauser H., Köster M. (2010) Temporal and spatial resolution of type I and III interferon responses in vivo. J. Virol. 84, 8626–8638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Williams G. J., Witt P. L. (1998) Comparative study of the pharmacodynamic and pharmacologic effects of Betaseron and AVONEX. J. Interferon Cytokine Res. 18, 967–975 [DOI] [PubMed] [Google Scholar]
- 34. Mahmood I. (2005) Interspecies Pharmacokinetic Scaling: Principles and Application of Allometric Scaling, 1st Ed., Pine House Publishers, Rockville, MD [Google Scholar]
- 35. Mendel I., Kerlero de Rosbo N., Ben-Nun A. (1995) A myelin oligodendrocyte glycoprotein peptide induces typical chronic experimental autoimmune encephalomyelitis in H-2b mice: fine specificity and T cell receptor V β expression of encephalitogenic T cells. Eur. J. Immunol. 25, 1951–1959 [DOI] [PubMed] [Google Scholar]
- 36. Croxford A. L., Kurschus F. C., Waisman A. (2011) Mouse models for multiple sclerosis: historical facts and future implications. Biochim. Biophys. Acta 1812, 177–183 [DOI] [PubMed] [Google Scholar]
- 37. van der Meide P. H., de Labie M. C., Ruuls S. R., Groenestein R. J., Botman C. A., Olsson T., Dijkstra C. D. (1998) Discontinuation of treatment with IFN-β leads to exacerbation of experimental autoimmune encephalomyelitis in Lewis rats: rapid reversal of the antiproliferative activity of IFN-β and excessive expansion of autoreactive T cells as disease promoting mechanisms. J. Neuroimmunol. 84, 14–23 [DOI] [PubMed] [Google Scholar]
- 38. Meyniel C., Spelman T., Jokubaitis V. G., Trojano M., Izquierdo G., Grand'Maison F., Oreja-Guevara C., Boz C., Lugaresi A., Girard M., Grammond P., Iuliano G., Fiol M., Cabrera-Gomez J. A., Fernandez-Bolanos R., Giuliani G., Lechner-Scott J., Cristiano E., Herbert J., Petkovska-Boskova T., Bergamaschi R., van Pesch V., Moore F., Vella N., Slee M., Santiago V., Barnett M., Havrdova E., Young C., Sirbu C.-A., Tanner M., Rutherford M., Butzkueven H., and MSBasis Study Group (2012) Country, sex, EDSS change and therapy choice independently predict treatment discontinuation in multiple sclerosis and clinically isolated syndrome. PLoS One 7, e38661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vremec D., Pooley J., Hochrein H., Wu L., Shortman K. (2000) CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164, 2978–2986 [DOI] [PubMed] [Google Scholar]
- 40. Esashi E., Sekiguchi T., Ito H., Koyasu S., Miyajima A. (2003) Cutting edge: a possible role for CD4+ thymic macrophages as professional scavengers of apoptotic thymocytes. J. Immunol. 171, 2773–2777 [DOI] [PubMed] [Google Scholar]
- 41. Liu Y., Carlsson R., Comabella M., Wang J., Kosicki M., Carrion B., Hasan M., Wu X., Montalban X., Dziegiel M. H., Sellebjerg F., Sørensen P. S., Helin K., Issazadeh-Navikas S. (2014) FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat. Med. 20, 272–282 [DOI] [PubMed] [Google Scholar]
- 42. Pachner A. R., Bertolotto A., Deisenhammer F. (2003) Measurement of MxA mRNA or protein as a biomarker of IFNβ bioactivity: detection of antibody-mediated decreased bioactivity (ADB). Neurology 61, S24–S26 [DOI] [PubMed] [Google Scholar]
- 43. Sellebjerg F., Datta P., Larsen J., Rieneck K., Alsing I., Oturai A., Svejgaard A., Soelberg Sørensen P., Ryder L. P. (2008) Gene expression analysis of interferon-β treatment in multiple sclerosis. Mult. Scler. 14, 615–621 [DOI] [PubMed] [Google Scholar]
- 44. Hesse D., Sellebjerg F., Sorensen P. S. (2009) Absence of MxA induction by interferon β in patients with MS reflects complete loss of bioactivity. Neurology 73, 372–377 [DOI] [PubMed] [Google Scholar]
- 45. van der Voort L. F., Vennegoor A., Visser A., Knol D. L., Uitdehaag B. M., Barkhof F., Oudejans C. B., Polman C. H., Killestein J. (2010) Spontaneous MxA mRNA level predicts relapses in patients with recently diagnosed MS. Neurology 75, 1228–1233 [DOI] [PubMed] [Google Scholar]
- 46. Hundeshagen A., Hecker M., Paap B. K., Angerstein C., Kandulski O., Fatum C., Hartmann C., Koczan D., Thiesen H.-J., Zettl U. K. (2012) Elevated type I interferon-like activity in a subset of multiple sclerosis patients: molecular basis and clinical relevance. J. Neuroinflammation 9, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Serana F., Imberti L., Amato M. P., Comi G., Gasperini C., Ghezzi A., Martinelli V., Provinciali L., Rottoli M. R., Sotgiu S., Stecchi S., Vecchio M., Zaffaroni M., Cordioli C., Capra R. (2014) MxA mRNA quantification and disability progression in interferon β-treated multiple sclerosis patients. PLoS One 9, e94794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sandler N. G., Bosinger S. E., Estes J. D., Zhu R. T., Tharp G. K., Boritz E., Levin D., Wijeyesinghe S., Makamdop K. N., del Prete G. Q., Hill B. J., Timmer J. K., Reiss E., Yarden G., Darko S., Contijoch E., Todd J. P., Silvestri G., Nason M., Norgren R. B., Jr., Keele B. F., Rao S., Langer J. A., Lifson J. D., Schreiber G., Douek D. C. (2014) Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Río J., Nos C., Tintoré M., Téllez N., Galán I., Pelayo R., Comabella M., Montalban X. (2006) Defining the response to interferon-β in relapsing-remitting multiple sclerosis patients. Ann. Neurol. 59, 344–352 [DOI] [PubMed] [Google Scholar]
- 50. Baranzini S. E. (2013) Gene expression profiling in MS: a fulfilled promise? Mult. Scler. 19, 1813–1814 [DOI] [PubMed] [Google Scholar]
- 51. Lublin F. D., Reingold S. C. (1996) Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (U.S.A.) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46, 907–911 [DOI] [PubMed] [Google Scholar]
- 52. Goldman M. D., Motl R. W., Rudick R. A. (2010) Possible clinical outcome measures for clinical trials in patients with multiple sclerosis. Ther. Adv. Neurol. Disord. 3, 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lucchinetti C., Brück W., Parisi J., Scheithauer B., Rodriguez M., Lassmann H. (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707–717 [DOI] [PubMed] [Google Scholar]
- 54. Karussis D. (2014) The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J. Autoimmun. 48–49, 134–142 [DOI] [PubMed] [Google Scholar]
- 55. Guo B., Chang E. Y., Cheng G. (2008) The type I IFN induction pathway constrains Th17-mediated autoimmune inflammation in mice. J. Clin. Invest. 118, 1680–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prinz M., Schmidt H., Mildner A., Knobeloch K.-P., Hanisch U.-K., Raasch J., Merkler D., Detje C., Gutcher I., Mages J., Lang R., Martin R., Gold R., Becher B., Brück W., Kalinke U. (2008) Distinct and nonredundant in vivo functions of IFNAR on myeloid cells limit autoimmunity in the central nervous system. Immunity 28, 675–686 [DOI] [PubMed] [Google Scholar]
- 57. Teige I., Treschow A., Teige A., Mattsson R., Navikas V., Leanderson T., Holmdahl R., Issazadeh-Navikas S. (2003) IFN-β gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J. Immunol. 170, 4776–4784 [DOI] [PubMed] [Google Scholar]
- 58. Galligan C. L., Pennell L. M., Murooka T. T., Baig E., Majchrzak-Kita B., Rahbar R., Fish E. N. (2010) Interferon-β is a key regulator of proinflammatory events in experimental autoimmune encephalomyelitis. Mult. Scler. 16, 1458–1473 [DOI] [PubMed] [Google Scholar]
- 59. Axtell R. C., de Jong B. A., Boniface K., van der Voort L. F., Bhat R., De Sarno P., Naves R., Han M., Zhong F., Castellanos J. G., Mair R., Christakos A., Kolkowitz I., Katz L., Killestein J., Polman C. H., de Waal Malefyt R., Steinman L., Raman C. (2010) T helper type 1 and 17 cells determine efficacy of interferon-β in multiple sclerosis and experimental encephalomyelitis. Nat. Med. 16, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.