

Background: VEGF-calcineurin-nuclear factor of activated T cells (NFAT) signaling regulates endothelial cell homeostasis.
Results: Novel NFAT targets were identified by genome-wide screening and epigenome analysis.
Conclusion: Inhibition of the NFAT targets led to blockade of VEGF-mediated functional angiogenesis.
Significance: VEGF-NFAT genome-wide occupancy may be useful for the development of target therapies for VEGF-mediated vascular diseases.
Keywords: Angiogenesis, Endothelial Cell, Gene Transcription, Genomics, Microarray, NFAT Transcription Factor, Signaling, Vascular Biology, Vascular Endothelial Growth Factor (VEGF), ChIP-seq
Abstract
VEGF is a key regulator of endothelial cell migration, proliferation, and inflammation, which leads to activation of several signaling cascades, including the calcineurin-nuclear factor of activated T cells (NFAT) pathway. NFAT is not only important for immune responses but also for cardiovascular development and the pathogenesis of Down syndrome. By using Down syndrome model mice and clinical patient samples, we showed recently that the VEGF-calcineurin-NFAT signaling axis regulates tumor angiogenesis and tumor metastasis. However, the connection between genome-wide views of NFAT-mediated gene regulation and downstream gene function in the endothelium has not been studied extensively. Here we performed comprehensive mapping of genome-wide NFATc1 binding in VEGF-stimulated primary cultured endothelial cells and elucidated the functional consequences of VEGF-NFATc1-mediated phenotypic changes. A comparison of the NFATc1 ChIP sequence profile and epigenetic histone marks revealed that predominant NFATc1-occupied peaks overlapped with promoter-associated histone marks. Moreover, we identified two novel NFATc1 regulated genes, CXCR7 and RND1. CXCR7 knockdown abrogated SDF-1- and VEGF-mediated cell migration and tube formation. siRNA treatment of RND1 impaired vascular barrier function, caused RhoA hyperactivation, and further stimulated VEGF-mediated vascular outgrowth from aortic rings. Taken together, these findings suggest that dynamic NFATc1 binding to target genes is critical for VEGF-mediated endothelial cell activation. CXCR7 and RND1 are NFATc1 target genes with multiple functions, including regulation of cell migration, tube formation, and barrier formation in endothelial cells.
Introduction
The endothelium is a highly malleable cell layer that constantly senses and responds to changes in the extracellular microenvironment. Many extracellular mediators modulate gene transcription, resulting in changes in cell migration, proliferation, leukocyte adhesion, and coagulation. Tight control of these processes is essential for maintaining homeostasis. Endothelial cell activation, if excessive, oversustained, or misplaced spatially and temporally, may lead to vascular diseases such as pathological angiogenesis, inflammation, and atherosclerosis. Therefore, an understanding of the molecular pathways regulating endothelial activation may provide novel insights into therapeutic targets (1).
Endothelial cell activation is associated with small G protein-mediated signaling involving RhoA activation, which contributes to vascular permeability and endothelial cell motility. Rnd1, a Rho GTPase, regulates actin filament disassembly in response to various extracellular stimuli (2). Overexpression of Rnd1 resulted in a “rounded” cell shape in fibroblasts, and these morphological changes were due to inhibition of RhoA activation because of the anchoring p190 RhoGAP to the plasma membrane (3). In addition, activated endothelial cells dynamically sense attractive/repulsive chemokines via several G protein-coupled receptors. Specific chemokines, such as stromal derived factor (SDF) 1, CXCL1, and thrombin, regulate endothelial cell integrity and permeability (4, 5). Two receptors for SDF-1, CXCR4 and CXCR7, have been identified (6, 7). In particular, CXCR7 has been demonstrated recently to be highly expressed on lung and breast cancer cells and tumor vascular endothelia (8, 9). Expression of CXRC7 correlates with tumor malignancy and metastasis.
VEGF is a well known endothelial cell-specific mitogen and chemotactic agent involved in microvascular permeability and endothelial cell survival. VEGF receptors have been shown to activate several signaling pathways, including PKC, PI3K, AKT, MAPK, and Ca2+-calcineurin (10, 11). Calcineurin signaling activates the nuclear factor of activated T cells (NFAT)2 transcription factors. NFAT was first identified from the extracts of activated T lymphocytes, but recent studies have demonstrated that NFAT not only functions in the immune response in leukocytes but also regulates bone homeostasis, cardiac development, tumor progression, and pathogenesis of Down syndrome (12). In endothelial cells, many putative NFAT target genes have been described, including tissue factor, IL-8, E-selectin, Cox-2, and ATP2A3 (11, 13, 14). The balance of NFAT activity is important for proper endothelial activation. We demonstrated recently that Down syndrome critical region (DSCR) 1, an NFAT primary target, is critical for the formation of an autoinhibitory loop via DSCR-1 suppression of the NFAT activator calcineurin (11). In contrast, Egr-3, another NFAT target, positively transmits VEGF-mediated angiogenesis and proinflammatory signals (15). Overexpression of DSCR-1 completely attenuates NFAT activation, resulting in the down-regulation of VEGF-mediated angiogenesis and tumor growth (11). Null mutation of DSCR-1 leads to NFAT hyperactivation, which also causes primary tumor growth suppression via endothelial cell destabilization (16).
Although NFAT is increasingly recognized as an important transcription factor for endothelial cell activation, the mechanisms underlying VEGF-induced NFATc1 binding on the target locus are not well understood. In this report, we performed ChIP with deep sequencing (ChIP-seq) to determine the genome-wide occupancy of NFATc1 in endothelial cells and compared these results with the respective gene expression profiles to understand NFAT function on a genome-wide scale. By combining this with ChIP-seq data examining epigenetic markers, we demonstrated that NFATc1 binds preferentially to H3K4me3-positive active promoter regions. Moreover, we identified RND1 and CXCR7 as novel NFAT direct binding targets. Multiple VEGF-mediated functions are transduced via NFAT activation of these endothelial cells.
EXPERIMENTAL PROCEDURES
Cell Culture
Human umbilical vein endothelial cells (HUVEC) and human dermal microvascular endothelial cells were purchased from Lonza and cultured in EGM-2 MV medium (Lonza). We used three independent lots of HUVEC, and human dermal microvascular endothelial cells were a mixture from multiple donors. HEK-293 (ATCC, catalog no. CRL-1573), COS7 cells (ATCC, catalog no. CRL-1651), and human primary skin fibroblasts (Lonza) were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin.
Immunohistochemistry
HUVEC were cultured in EGM-2 MV medium, serum-starved for ∼18 h, and then pretreated with 1 μm cyclosporin A (CsA) or 10 μm MG-132 (Calbiochem) for 30 min before stimulation. Cells were treated with 50 ng/ml VEGF for the indicated times, fixed with 4% paraformaldehyde, and permeabilized with ice-cold methanol and 0.1% Triton X-100 in PBS. Samples were incubated with our generated anti-NFATc1 antibody, anti-CXCR7 (GeneTex), or anti-PECAM1 (BD Biosciences), washed with PBS, and then treated with Alexa Fluor 488- or Alexa Fluor 594-conjugated secondary antibodies (Invitrogen). Slides were mounted by ProLong Gold with antifade reagent with DAPI (Invitrogen). Images were captured with CoolsnapHQ charge-coupled device camera (Roper Scientific) at four independent objectives and quantified by examination with Metamorph software (Molecular Devices).
DNA Microarray
HUVEC were serum-starved overnight, pretreated with 1 μm CsA for 30 min, and then stimulated with 50 ng/ml VEGF (Peprotec) for 1 h. RNA was extracted with TRIzol (Invitrogen). Preparation of cRNA and hybridization of probe arrays were performed according to the instructions of the manufacturer (Affymetrix). Data were analyzed according to the Minimum information about a microarray experiment (MIAME) rule.
Western Blot Analyses
Subconfluent HUVEC were serum-starved for ∼18 h and then pretreated with 1 μm CsA or 10 μm MG-132 for 30 min before VEGF stimulation. 1 h after 50 ng/ml VEGF was added, cells were harvested, and these extracts were divided into nuclear and cytosol fractions as described previously (17, 18). 15 μg of each protein was loaded in 10% SDS-PAGE and immunoblotted with anti-NFATc1 antibody. The PVDF membrane was stripped and reprobed with anti-Lamin A (Santa Cruz Biotechnology) and Hsp 90 (Cell Signaling Technology) as nuclear and cytosol internal control markers, respectively.
Plasmid Construction and Luciferase Reporter Assay
The 5′ flanking region (−2193/+83) of the CXCR7 gene, the 5′ flanking region (−890/+103), and the distal region (−16066/−18577) of the RND1 gene were amplified by PCR with KOD polymerase (Toyobo) from the human genomic DNA templates. Subsequent PCR fragments were inserted into the pGL3-basic vector (Promega). Primers for cloning or point mutation were as follows: CXCR7 promoter, GGGGTACCGTGTGGCATCGATTCATTGG (forward) and CCGCTCGAGTGAGCTCTGCTGGCTGCA (reverse); CXCR7 promoter mutation 1, GAAAGAAGGCTGGGGTAACCCAAGAGTACA (forward) and TGTACTCTTGGGTTACCCCAGCCTTCTTTC (reverse); CXCR7 promoter mutation 2, TTTGGCTGACGTAATCCCCCCGTGGGGT (forward) and ACCCCACGGGGGGATTACGTCAGCCAAA (reverse); CXCR7 promoter mutation 3, GAGGAATTAACAAGGATTACCCAGGCTT (forward) and AAGCCTGGGTAATCCTTGTTAATTCCTC (reverse); RND1 promoter, GCAACAAGAGCGAAACTCCATCTC (forward) and GGTTGCAGTGTCCGCGGGACTT (reverse); and RND1 enhancer, CTCGAGCTTCCTGCACGAGATCCAAGAATCC (forward) and ACGCGTGGCTCGCTCAGAAAAGTTTCCAAGA (reverse).
HUVEC or COS7 cells were transiently transfected with plasmid DNA using FuGENE HD reagents (Promega), and luciferase activity was detected by the Dual-Luciferase assay kit (Promega) as described previously (17). All data were normalized by Renilla luciferase luminescence derived from the cotransfected pRL-SV40 vector (Promega).
ChIP-qPCR and ChIP Sequence Analysis
HUVEC were cross-linked with 1% formaldehyde and sonicated. Antibodies against NFATc1, histone H3 lysine 4 trimethyl (H3K4me3) (provided by H. Kimura), and acetylated histone H4 (H4Ac) (Upstate) were added and immunoprecipitated with protein A/G beads (Invitrogen). Prepared DNA was processed for ChIP-qPCR or ChIP sequence analysis. Real-time qPCRs were performed with the following primer pairs: CXCR7 promoter, AGGCTAGAGGCTCCTTTCTGCAGTG (forward) and CCCTTAGTGCTGAGCACTTTGCAAC (reverse); RND1 promoter, CTCTTTCTCTTAAAGCTGCACCGTT (forward) and TGCTTCCAGTACCCTTTCCA (reverse); RND1 enhancer, CTCGAGCTTCCTGCACGAGATCCAAGAATCC (forward) and ACGCGTGGCTCGCTCAGAAAAGTTTCCAAGA (reverse); EGR3 promoter, GGATAGGATCCCGAACGCTGG (forward) and TGCTGGGGAACCCGGAAGGC (reverse); and DSCR-1 promoter, GGTGTTGACGTCACCTCTTTCCAGT (forward) and TGAGTCAAGTCCTGCATGCT (reverse). All protocols for Illumina/Solexa sequence preparation, sequencing, and quality control were provided by Illumina.
Cell Migration Assay
HUVEC were treated with siRNAs for 24 h, incubated with EBM-2 (Lonza) plus 0.5% FBS for 18 h, stimulated by 50 ng/ml VEGF for 1 h, and labeled with PKH26 red fluorescent dye (Sigma-Aldrich). Migration assays were carried using the BD Biocoat angiogenesis system (BD Biosciences). Labeled cells were seeded on the upper chamber (105 cells/250 μl of EBM-2 plus 5 ng/ml VEGF) and incubated with 750 μl of EBM-2 plus 5 ng/ml VEGF in the presence or absence of 100 ng/ml SDF-1 (R&D Systems) in the lower chamber. After 8 or 24 h, migrated cells were visualized under a fluorescent microscope (Nikon) and quantified using a fluorescence detection cell image analyzer (Kurabo).
siRNA Treatment and Scratch Migration Assay
HUVEC were treated with siRNA for 24 h and incubated with EBM-2 plus 0.5% FBS for 18 h. After 50 ng/ml VEGF stimulation for 1 h, the confluent cell layer was scratched by a small tip (1-mm diameter). Resulting cell plates were incubated with 100 ng/ml SDF-1 for 24 h. Cells that migrated into the scratched area were counted under a phase-contrast microscope (Nikon) as described previously (19).
Flow Cytometry Analysis
siRNA-treated HUVEC were harvested by 1 mm EDTA (pH 8.0) and incubated with phycoerythrin (PE)-conjugated anti-CXCR7 or anti-CXCR4 antibody (BioLegend) for 30 min on ice. Samples were washed three times, resuspended with PBS, and then analyzed immediately by a flow cytometer (Merck Millipore).
Permeability Assay
siRNA-transfected HUVEC were seeded on transwell inserts with 8-μm pores (BD Biosciences) and then cultured in EGM-2 MV. After reaching confluence in dishes, HUVEC were serum-starved for ∼18 h and treated with 50 ng/ml hVEGF plus 1% FITC-dextran for 1 h. The lower chamber aliquots were collected and measured with an absorption spectrometer at A490.
Aortic Ring Assay
48-well plates were covered with growth factor-reduced Matrigel (BD Biosciences) (100 μl/well) and incubated for 30 min at 37 °C, 5% CO2. Thoracic aortas were excised from C57/BL6j male mice at 6–8 weeks of age and transferred into a dish with sterilized 1× PBS. After removing the fibroadipose tissue, arteries were sectioned into 1-mm-long cross sections, rinsed five times with EBM-2, and placed on the Matrigel-coated wells. Artery rings were covered with an additional 100 μl of Matrigel and incubated in 1.5 ml of EBM-2 plus 0.5% FBS for 24 h. Aortic rings were transfected with siRNA using Lipofectamine RNAi max (Invitrogen) and then cultured with EBM-2 containing 50 ng/ml mouse VEGF (PeproTech) and 2% FBS. Culture media containing siRNA and VEGF were replaced every 2 days. After 10 days, vessels sprouting from the aortas were stained with 1 μg/ml Calcein-AM (Invitrogen) and observed using a fluorescent microscope. Total tube length was calculated by image analyzer software (Kurabo). The siRNA sequences against mouse Rnd1 was GAACAGAAAUCCUAGACUATT or CGGUAUUGCUGUGCUUUGATT, and that against mouse Cxcr7 was GAUGGUACGCCGUGUUGUATT or GGAAGAUCAUCUUCUCCUATT.
RhoA Pulldown Assay
siRNA-transfected HUVEC were serum-starved for ∼18 h, stimulated with 50 ng/ml VEGF, and harvested immediately with lysis/binding/washing buffer from the Active Rho pull-down and detection kit (Thermo Scientific). Pulldown using purified GST-Rhotekin and immunoblot analysis for RhoA were performed according to the instructions of the manufacturer (Thermo Scientific).
Coculture Angiogenesis Assay
The coculture system of HUVEC and fibroblasts was purchased from Kurabo. Cells were incubated in medium containing VEGF (10 ng/ml). siRNA treatment was performed twice, on days 1 and 7. Eleven days later, cells were fixed and immunostained following the instructions of the manufacturer (Kurabo). Vessel trees and branches were calculated by an image analyzer (Kurabo).
Statistics
Data are shown as mean ± S.D. p values were calculated by using two-tailed unpaired Student's t test.
RESULTS
VEGF Induces Dynamic Calcineurin-mediated NFATc1 Activation in Primary Cultured Endothelial Cells
We reported previously that VEGF and thrombin activates the Ca2+-calcineurin-NFAT axis, which is a critical intercellular signaling pathway in primary cultured endothelial cells (11). To further study VEGF-mediated NFATc1 activation, we generated a mouse monoclonal antibody against the human NFATc1 antigen, amino acids 134–223, with high affinity (Fig. 1A). Our antibody detected all DNA binding domain-containing NFATc1 splicing isoforms, but not the closely related NFATc2, in a Western blot analysis (Fig. 1, B and C). To determine how quickly NFATc1 was activated after VEGF treatment, we carried out time course immunohistochemistry staining with the anti-NFATc1 antibody in HUVEC. As shown in Fig. 1, D and E, NFATc1 was localized both in the cytoplasm and in the nucleus without VEGF. However, VEGF treatment caused NFAT to translocate to the nucleus, with maximum NFAT nuclear localization at 10 min. NFATc1 remained in the nucleus at 1 h but returned to the cytoplasm by 4 h after VEGF treatment. Moreover, VEGF-mediated NFATc1 activation was dependent on calcineurin activity because CsA treatment largely abrogated VEGF-mediated NFATc1 nuclear translocation (Fig. 1E). To test the possibility of increased turnover/degradation of cytoplasmic NFATc1, we examined the amount of nuclear and cytoplasmic NFATc1 after VEGF stimulation to quantify changes in protein levels. To evaluate NFATc1 turnover, immunoblot analyses with anti-NFATc1 antibody were performed on nuclear and cytoplasmic fractions of HUVEC treated with or without the proteasome inhibitor MG-132. As a positive control, we also examined p65 nuclear localization. MG-132 treatment abrogated TNF-α-mediated p65 nuclear localization (data not shown) but did not interfere with VEGF-mediated NFATc1 nuclear localization (Fig. 1F). In contrast, CsA strongly attenuated translocation into the nucleus (Fig. 1F). This suggests that VEGF promptly activates the calcineurin pathway, which leads to NFATc1 nuclear localization in endothelial cells.
FIGURE 1.

VEGF-mediated immediate and dynamic NFATc1 activation in primary cultured endothelial cells. A, schematic of spliced isoforms of human NFATc1 and the antigen for our generated mouse monoclonal antibody. A.A., amino acid. B, ChIP-Western blot analysis of immunoprecipitates using the anti-NFATc1 antibody from HUVEC extracts. The right column indicates the NFATc1 isoforms, n = 5., M.W. molecular weight. C, human NFATc1 (pCMV-NFATc1), human NFATc2 (pCMV-NFATc2), or mock control (pCMV) were transfected in COS7 cells. Whole cell extracts were prepared and blotted with an anti-NFATc1 or NFATc2 antibody. Anti-β-actin antibody was used as a loading control, representative of four independent experiments. D, NFAT immunofluorescent staining. NFAT localization in the nucleus versus the cytoplasm was calculated in HUVEC treated with VEGF at the indicated time points. Data are mean ± S.D. of the nuclear localization levels obtained from three independent experiments. *, p < 0.01 compared with without VEGF. E, immunofluorescent staining of NFATc1 (left column) in mock- and VEGF-treated HUVEC with or without CsA and MG-132. Merged images with PECAM1 and DAPI are shown in the right column. Scale bars = 50 μm. F, Western blot analyses with anti-NFATc1 antibody. HUVEC were treated with PBS (mock) or VEGF for 1 h in the presence or absence of CsA and MG-132. Subsequently, nuclear and cytosolic fractions were subjected to 10% SDS-PAGE. LaminA and Hsp90 are shown as nuclear and cytosolic markers, respectively. n = 3.
A Genome-wide Approach Revealed Comprehensive VEGF-responsive Regions by NFATc1 Binding Site-enriched Analysis in Primary Cultured Endothelial Cells
We next wished to identify globally how VEGF-mediated NFATc1 nuclear localization regulates target gene expression. To that end, we performed a cDNA microarray analysis using VEGF-stimulated HUVEC in the presence or absence of CsA or the overexpression of constitutively active, nuclearly localized NFATc1 (CA-NFATc1). A comparison of microarray data from endothelial cells with adenovirally overexpressed CA-NFATc1 or endothelial cells treated with VEGF identified 416 genes that were more than 2-fold up-regulated by VEGF treatment, whereas 1678 genes were induced by CA-NFATc1. Among these, 193 genes were commonly induced (Fig. 2A), and the representative genes are shown in supplemental Table 1. Among the genes that were down-regulated, only nine genes were reduced more than 2-fold by VEGF for 1 h. 196 genes were reduced by overexpression of CA-NFATc1 (Fig. 2A). Collectively, these data suggest that VEGF signaling via NFATc1 favors gene up-regulation rather than down-regulation.
FIGURE 2.

Global survey for NFATc1 targets in activated endothelial cells via VEGF. A, Venn diagram showing the overlap of up- or down-regulated genes by VEGF and adenovirally overexpressed NFATc1. B, distribution of NFATc1-binding regions in VEGF-treated HUVEC. TSS, transcription start site. C, determination of the sequence recognized by NFATc1 in HUVEC. The MEME method was used for identification of enriched sequences. The size of character correlates with the rate of enrichment. p values and E values indicating probability and similarity were calculated by TOMTOM. D, correlation between VEGF-induced gene expression and NFATc1 bindings in HUVEC was determined by gene set enrichment analysis (GSEA) in the top panel. Bottom panel, heat map from the average difference of gene expression. The enriched signal (p < 10−30) around known genes is indicated by the gray bar from ChIP-seqs with NFATc1, H3K4me3, and H4Ac. E, schematic of NFAT-mediated turning on of the emergent epigenetic VEGF switch. The H3K4me3 histone mark is shown in green. H4Ac is shown in pink. F, combined data from DNA microarray and NFATc1 ChIP-seq analysis in HUVEC. In the DNA microarray, cells were treated with VEGF in the presence or absence of CsA or treated with Ad-CA-NFATc1. Representative NFATc1-regulated genes with gene ID and ontology are shown in the right panel. G, genome browser view around the Dscr-1, Egr-3, Vcam-1, and Bmp-2 loci. ChIP signals in the presence or absence of VEGF are shown with NFATc1 (red), H3K4me3 (yellow), and H4Ac (blue). Insulator CTCF binding is shown in black.
Subsequently, to globally survey where nuclear localized NFATc1 is bound to the genome, we performed ChIP with an NFATc1 antibody, and then the precipitated genome was sequenced comprehensively (ChIP-seq). To test the accuracy of the ChIP assay with the NFATc1 antibody, we used the previously reported NFATc1-regulated Dscr-1 and Egr-3 promoter regions as positive controls and the non-endothelial MyoD1 promoter region as a negative control. The NFATc1-mediated immunoprecipitated DNA and non-immunoprecipitated whole genome control (input DNA) were used to prepare libraries for deep sequencing and analyzed using massively parallel sequencing. The genome-wide NFATc1 binding regions were calculated by QuEST (see Ref. 20 for details). 4119 regions were identified as NFATc1-associated areas with VEGF treatment from the ChIP-seq. To reconstruct the NFATc1-associated binding regions, we clustered the regions into three sections on the basis of the distance from the transcription start site in the respective genes. As shown in Fig. 2B, half of the NFATc1-bound peaks were located at the proximal promoter region within 1 kb from the transcription start site and the 5′-UTR. 17% of NFATc1-bound peaks were categorized in the gene body, exon, intron, and 3′-UTR. The remaining 33% of the NFATc1 binding sites were at intergenes.
Next, we identified the commonly recognized motif from the whole NFATc1-mediated ChIP genome sequences. As shown in Fig. 2C, the NFATc1 consensus motif, AGGAAA, was identified to be the highest enriched binding element, with an E value of 1.3× e−194. In addition to NFAT, CCAAT/enhancer binding protein (C/EBP)- and CREB1-recognized sequences were also found as the second and third most coenriched elements. NFATc1 activation predominantly induced the target genes (Fig. 2A). Therefore, to determine whether NFATc1 binding correlated with the active chromatin state, we added ChIP-seq analyses using antibodies against H3K4me3 as an active proximal promoter mark and H4Ac as an enhancer mark using HUVEC with or without VEGF treatment. Combination of the enrichment scores for NFATc1 and H3K4me3 indicated that basal H3K4me3 methylation was independent of VEGF and that, in the absence of VEGF, some NFATc1 was bound to these transcriptionally active promoter regions. In contrast, H4Ac-enriched genes were correlated significantly with VEGF-mediated gene expression because H4Ac marks were minimally present in untreated HUVEC but increased more than 3-fold after VEGF stimulation (Fig. 2D). These findings suggest that subsets of NFATc1 are constitutively harbored in the H3K4me3-positive region. After VEGF stimulation, more abundant NFATc1 was bound on H4Ac-positive enhancer as well as H3K4me3-positive promoter regions, which allowed maximum activation of VEGF-regulated genes (Fig. 2E).
Binding of NFATc1 to DNA might not always necessarily correlate with changes in gene expression. To determine the association between genome-wide NFATc1 binding and target gene expression, we compared the results of ChIP-seq with those of DNA microarrays of VEGF- or CA-NFATc1-overexpressed HUVEC in the presence or absence of CsA. We selected microarray gene set probes that exhibited significant expression, as defined by a >100-fold average difference and more than 2-fold up-regulation by VEGF for 1 h, which were reduced more than 30% by CsA treatment. Alternatively, the genes that were reduced more than 2-fold by VEGF but oppositely induced by CsA were selected. 48 total genes met these criteria (Fig. 2F, left panel). Interestingly, more than half of these genes were directly bound by NFATc1 at the proximal promoter region. Moreover, 20 total genes were significantly up-regulated via CA-NFATc1 overexpression (Fig. 2F, right panel). In these clusters, we found Vcam-1, Dscr-1 short isoform (Dscr-1s), and Egr-3 genes, which we reported previously as transactivated by NFAT (11, 13, 15). Representative NFAT binding sites with epigenetic information are shown in Fig. 2G.
Dynamic NFATc1 Binding on the Promoter Directs VEGF-mediated CXCR7 Induction in Primary Cultured Endothelial Cells
Among the NFATc1 positively regulated gene clusters, we decided to further focus our analysis on Cxcr7 and Rnd1 gene expression because the identification of these genes as NFAT-responsive is novel and because they are candidates for VEGF-mediated angiogenic modulators. Cxcr7 mRNA was most highly (4.1-fold) induced at 1 h after VEGF treatment, and this induction was abrogated by the pretreatment with CsA (Fig. 3A). NFAT-mediated CXCR7 transactivation was also validated by adenoviral CA-NFATc1 overexpression in HUVEC (Fig. 3B). Moreover, as shown in Fig. 3C, NFATc1 bound on the H3K4me3-positive proximal promoter region where histone acetylation is associated with open chromatin. Both H4Ac and H3K4me3 binding levels were increased after the VEGF treatment. To determine whether NFATc1 binding is involved in changes in histone modifications, we performed comparative ChIP-qPCR with anti-H3K4me3 and anti-H4Ac antibodies. After VEGF stimulation, H3K4me3 and H4Ac binding was increased 4.1-and 2.8-fold, respectively, on the Cxcr7 promoter region but not MyoD1 as a negative control (Fig. 3D). Importantly, CsA treatment completely abrogated VEGF-stimulated H4Ac binding but not H3K4me3 binding to the Cxcr7 promoter region (Fig. 3D). Therefore, CXCR7 expression in endothelial cells is regulated by increased NFAT binding connected to histone acetylation. Moreover, we found three NFAT consensus elements around the positive region from NFATc1-mediated ChIP-seq data (Fig. 3C), but there were no other NFATc1-enriched binding regions within the insulator CCCTC-binding factor (CTCF)-divided Cxcr7 locus. The significant but transient Cxcr7 gene induction correlated directly with significantly increased but transient NFATc1 binding on the Cxcr7 promoter (Fig. 3E).
FIGURE 3.

NFATc1 binds and transactivates CXCR7 in VEGF-treated endothelial cells. A, real-time qPCR analysis of CXCR7 expression in VEGF alone (red) or VEGF plus CsA (blue) treated HUVEC for the indicated time points (*, p < 0.01 compared with VEGF plus CsA) (n = 3). B, CXCR7 expression in HUVEC infected by Ad-NFATc1 or Ad-lacZ for 24 h (*, p < 0.001 compared with the control) (n = 4). C, genome browser view around the CXCR7 gene. ChIP-signals in the presence or absence of VEGF are shown with NFATc1 (red), H3K4me3 (yellow), and H4Ac (purple). The number below each ChIP-seq indicates the threshold window on the integrated genome browser (IGB) software. D, HUVEC were treated with VEGF for 1 h. ChIP was performed with antibodies to H3K4me3 (green) and H4Ac (pink). Subsequent qPCR using primers specific to the Cxcr7 (left panel) and MyoD1 (right panel) target loci are as shown under “Experimental Procedures.” Fold enrichment was compared with non-immunoprecipitated chromatin (percent input). Data are expressed as mean ± S.E. obtained from three independent experiments. *, p < 0.05, compared with to without VEGF; #, p < 0.05 compared with to without CsA (n = 3). E, ChIP-qPCR analysis with NFATc1 antibody on the CXCR7 locus in HUVEC treated with VEGF at the indicated time points (*, p < 0.001 compared with 0 min of VEGF treatment) (n = 3). F, HUVEC were transfected with CXCR7-luc or CXCR7 (NFAT mut)-luc and then serum-starved, preincubated with 1 μm CsA or mock control, and treated with VEGF for 4 h. Data are mean ± S.D. of luciferase light units obtained from six independent experiments. *, p < 0.001 compared with to without VEGF; **, p < 0.001 compared with to without CsA. G, CXCR7-luc and the NFATc1 expression plasmid (pCMV-NFATc1) were transiently cotransfected into COS7 cells. Two days later, cells were assayed for reporter activity. Data are mean ± S.D. of relative expression compared with the mock control (pCMV). *, p < 0.001 compared with the control (n = 6).
To evaluate the transcriptional mechanism underlying NFATc1-mediated CXCR7 expression, we cloned the 5′ flanking region (−2193/+83) of the Cxcr7 gene and inserted it into the luciferase reporter vector, CXCR7-luc. VEGF stimulation of HUVEC containing this vector induced significant promoter activity. This promoter activity was completely abrogated by CsA treatment. Moreover, the NFAT point-mutated promoter failed to respond to VEGF (Fig. 3F). Cotransfection with the NFATc1 expression plasmid and Cxcr7 promoter, but not the NFAT mutated Cxcr7 promoter, resulted in the more than 3-fold Cxcr7 promoter activation compared with the mock control (Fig. 3G). Taken together, the increase in Cxcr7 mRNA and the promoter activity inductions via VEGF signaling indicate that NFATc1 directly regulates Cxcr7 expression.
NFATc-mediated CXCR7 Expression Leads to Endothelial Migration and Tube Formation
siRNA to NFATc1 significantly abrogated NFATc1 mRNA and protein expression (13) (data not shown), which almost completely abrogated VEGF-induced membrane expression of CXCR7 (Fig. 4A). CXCR7 is a chemokine receptor and has been identified recently as a receptor for CXCL12/SDF-1 ligand (6, 8). CXCR7 and the classic SDF-1 receptor CXCR4 are both expressed by endothelial cells (7). SDF-1 is expressed by various cells, including endothelial cells, but is more highly expressed in perivascular tissues from the endothelium (Fig. 4B). To verify CXCR4 and 7 regulation in endothelial cells, we compared VEGF-mediated induced patterns in each mRNA. CXCR4 is constitutively expressed by endothelial cells. In sharp contrast to Cxcr7, Cxcr4 mRNA was up-regulated weakly and slowly after 24 h via VEGF, which did not correlate directly to immediate NFAT activation (Fig. 4, C and D). To further distinguish between CXCR4 and CXCR7, we generated specific siRNA for each (Fig. 4E) and performed endothelial cell migration and tube formation assays. Mild induction of CXCR4 mRNA via VEGF did not lead to the up-regulation of membrane-expressed CXCR4 levels within 24 h. Therefore, siRNA against CXCR4 markedly reduced the basal expression of CXCR4, but this reduction was not changed in the presence of VEGF (Fig. 4F). In contrast, siRNA to CXCR7 resulted in >90% inhibition of VEGF-induced CXCR7 mRNA expression (Fig. 4G). VEGF treatment increased membrane expression of CXCR7, which was abolished by two independent siRNAs (si-CXCR7 oligo1 and 2, Fig. 4H and data not shown).
FIGURE 4.

Differences between CXCR4 and 7 via the VEGF response in the endothelium. A, quantification of membrane-expressed CXCR7 levels. Each number shows the median cell counts indicated in the region (n = 3). Black, si-control without VEGF; light blue, si-NFATc1 without VEGF; orange, si-control with VEGF; blue line, si-NFATc1 with VEGF. B, cell culture media from confluent HUVEC, human dermal microvascular endothelial cells (HMVEC), HEK293, human primary smooth muscle cells (SMC), and human fibroblast were harvested, and SDF-1 levels were measured using ELISA (*, p < 0.05 compared with HUVEC) (n = 5). C and D, real-time qPCR analysis of CXCR4 expression in VEGF alone (C) or VEGF plus CsA (D) treated HUVEC at the indicated time points. Data are mean ± S.D. from four independent experiments. *, p < 0.05 compared with 0 min (C) or without VEGF for 24 h (D). E, real-time qPCR analysis of Cxcr4 and 7 expression in HUVEC transfected with each specific siRNA. Data are mean ± S.D. of the relative expression to cyclophilin A from three independent experiments (n = 3). *, p < 0.001 compared with si-control. F, quantification of membrane-expressed CXCR4 using flow cytometry. Each number shows the median cell counts indicated in the region (n = 3). Black, si-control without VEGF; red, si-control with VEGF; pink, si-CXCR4 with VEGF. G, real-time qPCR analysis of CXCR7 expression in HUVEC transfected with siRNAs. *, p < 0.001 compared with si-control with VEGF (n = 3). H, quantification of membrane-expressed CXCR7 using flow cytometry. The results show one representative of three independent experiments. Each number shows the median cell counts indicated in the region (n = 3). Black, si-control without VEGF; red, si-control with VEGF; blue, si-CXCR7 with VEGF.
Under these conditions, si-NFATc1 treatment significantly attenuated cell migration compared with si-control. CXCR4 specific knockdown resulted in ∼70% reduction of SDF-1-mediated cell migration. Importantly, CXCR7-specific knockdown reduced cell migration toward SDF-1 more prominently (∼90%). Combined knockdown of CXCR4 and CXCR7 caused complete abrogation of the response to SDF-1 in endothelial cells (Fig. 5A). Similarly, cell scratch assays also demonstrated that, in the absence of CXCR7 (si-CXCR7), HUVEC sensitivity to SDF-1 was not increased by VEGF (Fig. 5B). These data suggest that both CXCR4 and CXCR7 are important for SDF-1-mediated motility in endothelial cells but that NFATc1-mediated CXCR7 is regulated more tightly by VEGF stimulation.
FIGURE 5.

The NFATc1-CXCR7 axis is required for SDF-1-mediated cell migration and angiogenic sprouting in the endothelium. A, SDF-1-mediated cell migration of HUVEC with NFATc1, CXCR4, and CXCR7 knockdown. Right panel, mean ± S.D. migration level derived from triplicate experiments. *, p < 0.01; **, p < 0.01 compared with si-control in the presence of SDF-1 for 8 and 24 h, respectively; #, p < 0.05 compared with si-CXCR4 in the presence of SDF-1 for 24 h (n = 3). B, SDF-1-mediated endothelial cell migration in si-CXCR4 and si-CXCR7 HUVEC. HUVEC were scratched and incubated with EBM-2 plus 100 ng/ml SDF-1 or 100 ng/ml SDF-1 plus 50 ng/ml VEGF. The scratched field is shown by a broken red line. After 24 h, migrated cells were counted under a phase-contrast microscope. Right panel, mean ± S.E. from four optical independent images. #, p < 0.05 compared with si-control with SDF-1 alone; *, p < 0.05 compared with si-control with SDF-1+VEGF; N.S., not significant (n = 4). C, confluent HUVEC were denudated and recultured with basal conditioning (EBM-2 plus 0.5% FBS) medium. One day later, cells were treated in the presence or absence of VEGF for 2 h. Subsequently, cells were fixed and immunostained with anti-NFATc1 (green) and -CXCR7 (red) antibodies. The dashed rectangle indicates the representative growth frontier region (n = 4). Scale bars = 50 μm. D, HUVEC and fibroblasts were cocultured and treated with si-control or si-NFATc1 in the presence of VEGF-containing media. siRNAs were added twice, on days 1 and 7. Eleven days later, cells were fixed and visualized with anti-PECAM1 (green) or anti-CXCR7 (red). Merged images with DAPI are shown on the right. Images are representative of eight independent experiments. Vascular trees and branches were calculated with a cell imager (Kurabo) and are mean ± S.E. *, p < 0.05 compared with si-control. Scale bars = 50 μm. E, real-time qPCR analysis of mCxcr7 expression in mouse endothelial cells. Data are mean ± S.D. derived from three independent experiments. *, p < 0.001 compared with si-control. F, aortic ring assays in si-control or si-Cxcr7 treatment. The outgrowth of neovessels from the aorta was observed under a phase-contrast microscope, and tube length was calculated using a cell image analyzer. The mean ± S.D. was derived from six independent experiments. *, p < 0.05 compared with the si-control with VEGF.
Next, to study the functional relevance of the VEGF-mediated NFATc1-CXCR7 axis, we evaluated angiogenesis activity in vitro. VEGF stimulated regrowth of the endothelial cell frontier region after denudation, where nuclear localization of NFATc1 and concurrent CXCR7 expression were apparent. Without VEGF treatment, NFATc1 nuclear localization and CXCR7 expression were minimal (Fig. 5C). Moreover, coculture of endothelial cells and SDF-1-rich fibroblasts enhanced vascular tree and branch formation. However, NFATc1 knockdown markedly reduced this coculture effect, despite VEGF stimulation (Fig. 5D). Finally, to evaluate NFATc1-CXCR7 activity under the physiological vasculature, we designed siRNA to mouse cxcr7 and then performed aortic ring assays. Cxcr7 siRNA reduced cxcr7 mRNA expression after VEGF treatment by more than 90% (Fig. 5E). VEGF induced neovessel sprouting from the aortic ring treated with control culture medium (Fig. 5F). In contrast, aortic rings in which Cxcr7 was reduced by siRNA failed to induce vessel sprouting from the aorta, even in the presence of VEGF (Fig. 5F). Taken together, these findings suggest that NFATc1 activation via VEGF signaling in endothelial cells causes membrane expression of CXCR7, which promotes SDF-1-expressed fibroblast or SDF-1-expressed, aorta-mediated endothelial cell migration and tube formation.
Rnd1 Is a Target of the VEGF-NFATc1 Signaling Axis
Rnd1 mRNA was highly (5.8-fold) induced at 1 h by VEGF treatment, and this was strongly attenuated by pretreatment with CsA (Fig. 6A). NFAT-mediated Rnd1 transactivation was also validated by adenoviral CA-NFATc1 expression in HUVEC (Fig. 6B). Importantly, NFATc1 bound on the H3K4me3-positive proximal promoter and the H4Ac-positive 19 kb upstream distal enhancer regions (Fig. 6C). These two NFATc1 binding regions were predicted to belong to the Rnd1 regulatory locus because of the location within two CTCF divisions (Fig. 6C). To determine whether NFATc1 binding on the Rnd1 locus involves changes in histone modifications, we performed ChIP-qPCR with antibodies to H3K4me3 and H4Ac. VEGF stimulation significantly increased the H3K4me3 and H4Ac modification rate. CsA treatment attenuated VEGF-mediated histone acetylation on the Rnd1 enhancer region (Fig. 6D). We found two separated NFAT consensus elements around the proximal Rnd1 promoter region and multiple (eight) NFAT elements in the distal enhancer. Transient RND1 expression correlated with profound but transient NFATc1 binding on the Rnd1 gene locus (Fig. 6E). To evaluate the transcriptional regulation underlying NFATc1-regulated RND1 expression, we isolated the proximal Rnd1 promoter (−890/+103 bp) region and distal (−16,066/−18,577 bp) region and inserted these regions into a luciferase reporter vector, Rnd1-luc. We failed to detect VEGF-mediated promoter activation when only the proximal promoter was transiently transfected to HUVEC. However, combination of the proximal promoter and the distal enhancer from the Rnd1 locus showed significant reporter up-regulation after VEGF treatment, which was reduced significantly by CsA treatment (Fig. 6F). Moreover, cotransfection of Rnd1-luc with the distal enhancer and NFATc1 expression plasmids resulted in significant (∼8.2-fold) up-regulation of the luciferase activity compared with the mock control (Fig. 6G). Collectively, Rnd1 mRNA and the VEGF-stimulated promoter activity suggest that transient NFATc1 binding at the Rnd1 distal enhancer region induces RND1 expression.
FIGURE 6.

NFATc1 binds and transactivates RND1 in VEGF-treated endothelial cells. A, real-time qPCR analysis of RND1 expression in HUVEC treated with VEGF alone (red) or treated with VEGF plus CsA (blue) at the indicated time points *, p < 0.01 compared with VEGF plus CsA (n = 3). B, RND1 expression in HUVEC infected with Ad-NFATc1 or Ad-lacZ for 24 h (*, p < 0.001 compared with Ad-lacZ) (n = 3). C, genome browser view around the Rnd1 gene. ChIP signals in the presence or absence of VEGF are shown with NFATc1 (red), H3K4me3 (yellow), and H4Ac (purple). Insulator CTCF bindings are shown in blue. The number below each ChIP-seq indicates the threshold window on the IGB software. D, HUVEC were treated with VEGF for 1 h. ChIP was performed with antibodies to H3K4me3 (green) and H4Ac (pink). Subsequently, qPCR was performed using primers specific to the Rnd1 target locus. The fold enrichment was compared with non-immunoprecipitated chromatin (percent input). Data are mean ± S.E. from three independent experiments. *, p < 0.05 compared with to without VEGF; #, p < 0.05 compared with to without CsA (n = 3). E, ChIP-qPCR with NFATc1 antibody on the Rnd1 locus in HUVEC treated with VEGF at the indicated time points. *, p < 0.001 compared with 0 min (n = 3). F, HUVEC were transfected with Rnd1-luc or Rnd1 (NFAT mut)-luc, serum-starved, preincubated with 1 μm CsA or mock control, and treated with VEGF for 4 h. Data are mean ± S.D. of luciferase light units obtained from six independent experiments. *, p < 0.001 compared with to without VEGF; **, p < 0.01 compared with to without CsA. G, RND1-luc and the NFATc1 expression plasmid (pCMV-NFATc1) were transiently cotransfected into COS7 cells. Two days later, cell lysates were assayed for reporter activity. Data are mean ± S.D. of relative expression compared with the mock control (pCMV). *, p < 0.001 compared with the control (n = 6).
NFATc1-mediated Rnd1 Expression Regulates Endothelial Cell Integrity and Neovessel Formation
RND1 activates p190RhoGAP, an inhibitor of Rho family member activity, and is, therefore, an inhibitor of Rho signaling (3). We hypothesized that RND1 induction modulates RhoA activity following the normalization of hyperpermeability. First, we wished to evaluate the correlation between RhoA and Rnd1 activity in endothelial cells. Transfection of two independent siRNAs to Rnd1 (si-Rnd1) reduced VEGF-mediated Rnd1 mRNA up-regulation by 91 and 81% (Fig. 7A). VEGF treatment caused a transient increase of RhoA activation (1 min) in the control. Importantly, inhibition of NFATc1 by si-NFATc1 caused basal and VEGF-mediated RhoA hyperactivation at both acute (1 min) and sustained (60 min) time points (Fig. 7B). Moreover, knockdown of the NFATc1-direct target, Rnd1, similarly resulted in RhoA hyperactivation (Fig. 7B). Subsequently, to test vessel integrity in cultured endothelia, we performed HUVEC permeability assays in vitro using FITC-labeled dextran and a modified Boyden chamber in which permeability through the endothelial monolayer is measured. HUVEC permeability was increased with si-NFATc1 compared with si-control treatment in the presence of VEGF, resulting in 138-fold higher penetration of dextran. Moreover, two independent si-Rnd1 treatments showed similarly increased higher permeability in the presence of VEGF (Fig. 7C). Collectively, this suggests that NFATc1 and the direct downstream target, Rnd1, regulate RhoA activity and endothelial cell integrity.
FIGURE 7.

VEGF-mediated Rnd1 induction blocks RhoA hyperactivation. A, real-time qPCR analysis of RND1 expression in HUVEC transfected with siRNAs. *, p < 0.001 compared with si-control with VEGF for 1 h (n = 4). B, pulldown assay using GST-rhotekin in whole cell extracts derived from siRNA-treated HUVEC. GDP and GTPγS are the negative and positive control, respectively (n = 5). C, FITC-dextran permeability assay in a Boyden chamber using VEGF-treated HUVEC. Data are mean ± S.D. of FITC concentrations derived from three independent experiments. *, p < 0.001 compared with si-control. D, VEGF-1-mediated cell migration of HUVEC with Rnd1 knockdown. Right panel, mean ± S.D. migration level derived from triplicate experiments. *, p < 0.05; #, p < 0.01 compared with the absence of VEGF and si-control in the presence of VEGF for 24 h, respectively (n = 3). E, real-time qPCR analysis of mRnd1 expression in mouse endothelial cells. Data are mean ± S.D. derived from three independent experiments. *, p < 0.001 compared with the si-control. F, aortic ring assays in si-control or si-Rnd1 treatment. The outgrowth of neovessels from the aorta was observed under a phase-contrast microscope, and tube length was calculated using a cell image analyzer. The mean ± S.D. were derived from six independent experiments. *, p < 0.05 compared with the si-control plus VEGF.
RhoA activation is directly linked to vessel migration and extension (21). Therefore, to survey the functional outcome of VEGF-mediated Rnd1 induction in neovessel formation, we carried out cell migration assays using a modified Boyden chamber. Endothelial cell migration was increased more than 40-fold in the presence of VEGF for 24 h (Fig. 7D). Importantly, knockdown of Rnd1 augmented VEGF-mediated cell migration (Fig. 7D). To further characterize VEGF-mediated Rnd1 function, we carried out mouse aortic ring assays in the presence of si-Rnd1. As shown in Fig. 7E, two independent siRNA to mouse Rnd1 resulted in more than 80% reduction of VEGF-stimulated Rnd1 mRNA up-regulation. These siRNAs increased neovessel formation from aortic rings in response to VEGF. Total tube length and the vessel-invaded areas were increased significantly compared with the control in the presence of VEGF (Fig. 7F). Taken together, these findings suggest that VEGF-mediated NFAT activation leads to Rnd1 expression as a direct downstream target, which, in turn, modulates VEGF-mediated RhoA activation.
DISCUSSION
Recent technological advancements now allow transcription factor binding regions to be defined and the histone code to be unlocked. In this report, we examined VEGF-stimulated NFATc1 binding on a genome-wide scale in human primary cultured endothelial cells and then comprehensively mapped and compared these signals with epigenetic histone marks and whole-genome expression arrays. VEGF treatment of endothelial cells caused NFAT nuclear localization, which led to expression of proangiogenic and proinflammatory genes. At the early time point, around one-third of VEGF stimulated genes were also stimulated by NFAT (Fig. 2A), suggesting that the calcineurin-NFAT pathway is an important route for VEGF-mediated signal transduction in endothelial cells.
ChIP-seq with NFATc1 in the presence of VEGF demonstrated that many of the NFATc1 binding elements were located within proximal promoters, the majority of which were commonly located with proximal H3K4me3, active promoter histone marks (22) (Fig. 2D). These NFATc1 occupancy patterns were unique, considering our previous reports of GATA2- and STAT6-mediated ChIP-seqs in endothelial cells (20, 23). GATA2 and STAT6 predominantly occupied distal enhancer regions. These differences might be on the basis of the functional difference of the transcription factors. NFAT is crucial for turning on the VEGF switch, but GATA2 and STAT6 function in endothelial specificity and chronic inflammation, at least in part, via changing the chromatin structure. Generally, compared with methylation, histone acetylation is thought to be more dynamically and accurately correlated to target gene expression levels (24). VEGF-stimulated NFAT activation correlated significantly with H4Ac histone modification (Fig. 2D). Surprisingly, the H3K4me3 mark was also increased moderately by VEGF treatment in our data. Although the mechanism and the functional consequences are still unknown, H3K4me3 levels do not always correlate with gene expression levels. For example, increased H3K4me3 marks in the Rnd1 promoter were important, but not important enough, to activate the Rnd1 core promoter-luc without accompaniment of histone acetylation (Fig. 6F). Genes obtaining bivalent H3K4me3 and H3K27me3 marks in ES cells have been demonstrated previously to be ready for expression, but transcription had not begun (25). Our data, taken together with previous studies, suggest that small amounts of NFAT might already be present in the transcriptionally permissive core promoter and that, after VEGF stimulation, abundant responsive NFAT can bind on H3K4me3-positive as well as H4Ac-positive regions with dynamic histone modifications (Fig. 2E). Our model proposes that gene expression in response to VEGF is amplified rapidly via this sequence. Chromatin is already in an expression-permissive state because of the presence of H3K4me3 methyl marks, and small amounts of NFAT are localized to the promoter region prior to VEGF stimulation. After VEGF treatment, more NFAT translocates to the promoter region, accompanied by a significant increase in H4Ac marks and some increase in H3K4me3, which further promote open chromatin. Together, increased NFAT binding and chromatin modifications promote gene expression.
NFAT nuclear localization and subsequent target gene activation are tightly regulated by calcineurin phosphatase, which is inhibited by the immunosuppressors CsA and FK506. When NFAT is phosphorylated by the priming kinase DYRK1A, NFAT is exported from the nucleus to the cytoplasm (26). In T cells, NFAT directly transactivates genes. Many of these genes function in inflammation or immune responses (27). The majority of NFAT-activated genes in endothelial cells are unique compared with T cells, except Egr-3 and Cox2. The differential activation mechanism between endothelial cells and T cells might be on the basis of the epigenetic microenvironment. Indeed, T cell-specific induced genes, TNF-α, colony-stimulating factor 1, IL-2, and IL-4 were fully covered with silencer histone markings and never enriched by active promoter markings in primary cultured endothelial cells. Specific regulation of these cell types was similarly observed in our previous study of IL-4-STAT6-mediated gene expression patterns in endothelial cells and T cells (23).
In HUVEC, ChIP-seq with NFATc1 antibody identified DNA enriched for the sequence AGGAAA, a well recognized consensus binding sequence. This is not random enrichment. The associated E value is 1.3× e−194 (Fig. 2C). HUVEC results were coenriched with C/EBP and CREB as second and third occupied sequences via ChIP-seq with NFATc1 (12). We and others have reported that C/EBP and CREB cobinding with NFAT functioned for maximum transactivation of DSCR-1 and Egr-3 in endothelial cells (15, 28). These data suggest that not only the specificity with agonist-receptor interaction but also the epigenetic microenvironment for NFAT would regulate the specific downstream target genes in endothelial cells.
We newly identified two novel functional NFATc1 endothelial cell targets, CXCR7 and RND1. CXCR7 is now classified as a chemokine receptor that is able to bind SDF-1 and CXCL11 (6). SDF-1 is a chemoattractant for vascular endothelial cells (29). Because we failed to detect SDF-1 as a VEGF-mediated inducible factor in endothelial cells, we thought it likely that SDF-1 expressed by perivascular cells activates CXCR7 in endothelial cells in a paracrine manner. Indeed, smooth muscle cells and fibroblasts showed much higher SDF-1 expression than endothelial cells, which may also potentiate VEGF-stimulated angiogenesis in aortic rings. Importantly, CXCR7 is increasingly reported as being expressed by malignant tumors and tumor endothelial cells (9). Moreover, we demonstrated that CXCR7 is a direct downstream target of the VEGF-NFAT axis in endothelial cells. Consistent with our data of CXCR7 as an NFATc1 target, similar phenotypes were observed between NFATc1 null mutation and CXCR7-deficient mice. Ventricular septal defects and heart valve malformation have been reported for each targeted knockout mouse (30, 31). It is well known that the interaction between SDF-1 and its unique receptor, CXCR4, is critical for tumor growth and metastasis (7). However, the recent identification of CXCR7 merits an addition to the short list of known SDF-1 receptors. Blockade of CXCR7, as well as CXCR4, in tumor- and VEGF-stimulated endothelial cells may lend itself to antitumor therapy.
We identified a second target of VEGF-NFAT signaling, Rnd1. Rnd1 inhibits RhoA activation to regulate actin filament disassembly, which leads to a rounded cell shape (2). VEGF-mediated Rnd1 expression was transient. We hypothesized that it functions as a modulator of RhoA activity to prevent hyperactivation, which could cause dysfunctional angiogenesis. In line with our theory, Rnd1 knockdown further exaggerated VEGF-promoted endothelial permeability and migration, at least following the hyperactivation of RhoA. RhoA signaling plays an essential role in VEGF-dependent in vivo angiogenesis and in the initial step of in vitro endothelial cord assembly. Tumor capillary endothelial cells, or targeted deletion of central cavernous malformation, led to hyperactivation of RhoA, which resulted in excessive but dysfunctional angiogenic sprouting (32, 33). Normalization of RhoA activity rescues functional angiogenesis. Therefore, VEGF regulation of Rnd1 to normalize RhoA activity would be important for angiogenesis. NFAT induction of Rnd1 to modulate VEGF-induced angiogenesis is similar to our recent findings related to the calcineurin modulator DSCR-1. Both overexpression and null mutation of DSCR-1 cause the attenuation of the VEGF-mediated angiogenesis because of blocking or excessive activation of NFAT, respectively (11, 16). Therefore, physiological and transient DSCR-1 and Rnd1 induction seem to be important for proper angiogenesis promotion. Taken together, we demonstrated that the VEGF-mediated calcineurin-NFAT signaling axis is one of the key angiogenesis regulatory pathways. NFAT directs a number of multifunctional downstream targets in endothelial cells, including feedback modulators, chemokine receptors, and transcription factors (Fig. 8).
FIGURE 8.
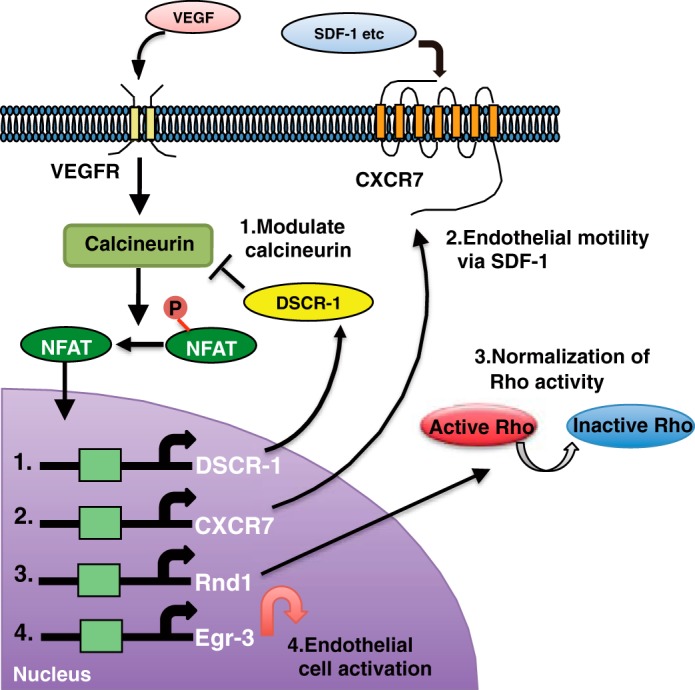
Schematic of multifunctional VEGF-calcineurin-NFAT signaling in endothelial cells. VEGF activates the calcineurin-NFAT signaling axis. Nuclear localized NFAT transactivates many angiogenesis-related genes. 1, DSCR-1 modulates calcineurin for feedback loop formation. 2, CXCR7 regulates SDF-1 mediated cell motility. 3, RND1 normalizes cell permeability following the modulation of RhoA activity. 4, Egr-3 mediates endothelial cell activation as a direct downstream transcription factor.
In summary, our findings reveal a mechanism by which VEGF-mediated NFATc1 widely binds the genome on epigenetically coordinated active DNA elements and regulates the expression of genes with functional importance in endothelial cells. Given the critical role of NFAT in endothelial cell activation and dysfunction, the discovery of the new transcriptional circuits via NFAT may provide a foundation for targeted therapies in VEGF-mediated vascular diseases.
Supplementary Material
Acknowledgments
We thank Drs. Naoki Mochizuki and Shigetomo Fukuhara (NCCRI, Japan) for discussions regarding NFATc1 downstream genes and Drs. Shogo Yamamoto and Hiroko Iwanari and Akashi Izumi (RCAST, The University of Tokyo) for technical assistance with ChIP-seqs, antibody generation, and microarrays.
This work was supported by the Leading-edge Research Promotion Fund from the Japan Society for the Promotion of Science (to T. M.) and by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology in Japan (to T. M.). This work was also supported by a project for developing innovation systems of MEXT in Japan (to T. M.).
The array and sequence data can be accessed through the NCBI Protein Database under NCBI accession numbers GSE49426 and GSE49429, respectively.

This article contains supplemental Table S1.
- NFAT
- nuclear factor of activated T cells
- ChIP-seq
- ChIP with deep sequencing
- HUVEC
- human umbilical vein endothelial cell(s)
- CsA
- cyclosporin A
- qPCR
- quantitative PCR
- H3K4me3
- histone H3 lysine 4 trimethyl
- H4Ac
- acetylated histone H4
- CA
- constitutionally active
- CREB
- cAMP response element-binding protein.
REFERENCES
- 1. Minami T., Aird W. C. (2005) Endothelial cell gene regulation. Trends Cardiovasc. Med. 15, 174–184 [DOI] [PubMed] [Google Scholar]
- 2. Nobes C. D., Lauritzen I., Mattei M. G., Paris S., Hall A., Chardin P. (1998) A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J. Cell Biol. 141, 187–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oinuma I., Kawada K., Tsukagoshi K., Negishi M. (2012) Rnd1 and Rnd3 targeting to lipid raft is required for p190 RhoGAP activation. Mol. Biol. Cell 23, 1593–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luo C., Pan H., Mines M., Watson K., Zhang J., Fan G. H. (2006) CXCL12 induces tyrosine phosphorylation of cortactin, which plays a role in CXC chemokine receptor 4-mediated extracellular signal-regulated kinase activation and chemotaxis. J. Biol. Chem. 281, 30081–30093 [DOI] [PubMed] [Google Scholar]
- 5. Minami T., Sugiyama A., Wu S. Q., Abid R., Kodama T., Aird W. C. (2004) Thrombin and phenotypic modulation of the endothelium. Arterioscler. Thromb. Vasc. Biol. 24, 41–53 [DOI] [PubMed] [Google Scholar]
- 6. Balabanian K., Lagane B., Infantino S., Chow K. Y., Harriague J., Moepps B., Arenzana-Seisdedos F., Thelen M., Bachelerie F. (2005) The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 280, 35760–35766 [DOI] [PubMed] [Google Scholar]
- 7. Lee B. C., Lee T. H., Avraham S., Avraham H. K. (2004) Involvement of the chemokine receptor CXCR4 and its ligand stromal cell-derived factor 1α in breast cancer cell migration through human brain microvascular endothelial cells. Mol. Cancer Res. 2, 327–338 [PubMed] [Google Scholar]
- 8. Burns J. M., Summers B. C., Wang Y., Melikian A., Berahovich R., Miao Z., Penfold M. E., Sunshine M. J., Littman D. R., Kuo C. J., Wei K., McMaster B. E., Wright K., Howard M. C., Schall T. J. (2006) A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 203, 2201–2213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miao Z., Luker K. E., Summers B. C., Berahovich R., Bhojani M. S., Rehemtulla A., Kleer C. G., Essner J. J., Nasevicius A., Luker G. D., Howard M. C., Schall T. J. (2007) CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. U.S.A. 104, 15735–15740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kroll J., Waltenberger J. (1997) The vascular endothelial growth factor receptor KDR activates multiple signal transduction pathways in porcine aortic endothelial cells. J. Biol. Chem. 272, 32521–32527 [DOI] [PubMed] [Google Scholar]
- 11. Minami T., Horiuchi K., Miura M., Abid M. R., Takabe W., Noguchi N., Kohro T., Ge X., Aburatani H., Hamakubo T., Kodama T., Aird W. C. (2004) Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J. Biol. Chem. 279, 50537–50554 [DOI] [PubMed] [Google Scholar]
- 12. Arron J. R., Winslow M. M., Polleri A., Chang C. P., Wu H., Gao X., Neilson J. R., Chen L., Heit J. J., Kim S. K., Yamasaki N., Miyakawa T., Francke U., Graef I. A., Crabtree G. R. (2006) NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600 [DOI] [PubMed] [Google Scholar]
- 13. Minami T., Miura M., Aird W. C., Kodama T. (2006) Thrombin-induced autoinhibitory factor, Down syndrome critical region-1, attenuates NFAT-dependent vascular cell adhesion molecule-1 expression and inflammation in the endothelium. J. Biol. Chem. 281, 20503–20520 [DOI] [PubMed] [Google Scholar]
- 14. Hadri L., Pavoine C., Lipskaia L., Yacoubi S., Lompré A. M. (2006) Transcription of the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase type 3 gene, ATP2A3, is regulated by the calcineurin/NFAT pathway in endothelial cells. Biochem. J. 394, 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suehiro J., Hamakubo T., Kodama T., Aird W. C., Minami T. (2010) Vascular endothelial growth factor activation of endothelial cells is mediated by early growth response-3. Blood 115, 2520–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ryeom S., Baek K. H., Rioth M. J., Lynch R. C., Zaslavsky A., Birsner A., Yoon S. S., McKeon F. (2008) Targeted deletion of the calcineurin inhibitor DSCR1 suppresses tumor growth. Cancer Cell 13, 420–431 [DOI] [PubMed] [Google Scholar]
- 17. Minami T., Aird W. C. (2001) Thrombin stimulation of the vascular cell adhesion molecule-1 promoter in endothelial cells is mediated by tandem nuclear factor-κ B and GATA motifs. J. Biol. Chem. 276, 47632–47641 [DOI] [PubMed] [Google Scholar]
- 18. Dignam J. D., Martin P. L., Shastry B. S., Roeder R. G. (1983) Eukaryotic gene transcription with purified components. Methods Enzymol. 101, 582–598 [DOI] [PubMed] [Google Scholar]
- 19. Song H., Suehiro J., Kanki Y., Kawai Y., Inoue K., Daida H., Yano K., Ohhashi T., Oettgen P., Aird W. C., Kodama T., Minami T. (2009) Critical role for GATA3 in mediating Tie2 expression and function in large vessel endothelial cells. J. Biol. Chem. 284, 29109–29124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kanki Y., Kohro T., Jiang S., Tsutsumi S., Mimura I., Suehiro J., Wada Y., Ohta Y., Ihara S., Iwanari H., Naito M., Hamakubo T., Aburatani H., Kodama T., Minami T. (2011) Epigenetically coordinated GATA2 binding is necessary for endothelium-specific endomucin expression. EMBO J. 30, 2582–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Nieuw Amerongen G. P., van Hinsbergh V. W. (2009) Role of ROCK I/II in vascular branching. Am. J. Physiol. Heart Circ. Physiol. 296, H903–H905 [DOI] [PubMed] [Google Scholar]
- 22. Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 23. Tozawa H., Kanki Y., Suehiro J., Tsutsumi S., Kohro T., Wada Y., Aburatani H., Aird W. C., Kodama T., Minami T. (2011) Genome-wide approaches reveal functional interleukin-4-inducible STAT6 binding to the vascular cell adhesion molecule 1 promoter. Mol. Cell Biol. 31, 2196–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Markowetz F., Mulder K. W., Airoldi E. M., Lemischka I. R., Troyanskaya O. G. (2010) Mapping dynamic histone acetylation patterns to gene expression in nanog-depleted murine embryonic stem cells. PLoS Comp. Biol. 6, e1001034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., Alvarez P., Brockman W., Kim T. K., Koche R. P., Lee W., Mendenhall E., O'Donovan A., Presser A., Russ C., Xie X., Meissner A., Wernig M., Jaenisch R., Nusbaum C., Lander E. S., Bernstein B. E. (2007) Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448, 553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Minami T. (2014) Calcineurin-NFAT activation and DSCR-1 auto-inhibitory loop: how is homoeostasis regulated? J. Biochem. 155, 217–226 [DOI] [PubMed] [Google Scholar]
- 27. Clipstone N. A., Crabtree G. R. (1992) Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357, 695–697 [DOI] [PubMed] [Google Scholar]
- 28. Oh M., Dey A., Gerard R. D., Hill J. A., Rothermel B. A. (2010) The CCAAT/enhancer binding protein beta (C/EBPβ) cooperates with NFAT to control expression of the calcineurin regulatory protein RCAN1–4. J. Biol. Chem. 285, 16623–16631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tachibana K., Hirota S., Iizasa H., Yoshida H., Kawabata K., Kataoka Y., Kitamura Y., Matsushima K., Yoshida N., Nishikawa S., Kishimoto T., Nagasawa T. (1998) The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393, 591–594 [DOI] [PubMed] [Google Scholar]
- 30. Sierro F., Biben C., Martínez-Muñoz L., Mellado M., Ransohoff R. M., Li M., Woehl B., Leung H., Groom J., Batten M., Harvey R. P., Martínez A. C., Mackay C. R., Mackay F. (2007) Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. U.S.A. 104, 14759–14764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Phoon C. K., Ji R. P., Aristizábal O., Worrad D. M., Zhou B., Baldwin H. S., Turnbull D. H. (2004) Embryonic heart failure in NFATc1−/− mice: novel mechanistic insights from in utero ultrasound biomicroscopy. Circ. Res. 95, 92–99 [DOI] [PubMed] [Google Scholar]
- 32. Ghosh K., Thodeti C. K., Dudley A. C., Mammoto A., Klagsbrun M., Ingber D. E. (2008) Tumor-derived endothelial cells exhibit aberrant Rho-mediated mechanosensing and abnormal angiogenesis in vitro. Proc. Natl. Acad. Sci. U.S.A. 105, 11305–11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whitehead K. J., Chan A. C., Navankasattusas S., Koh W., London N. R., Ling J., Mayo A. H., Drakos S. G., Jones C. A., Zhu W., Marchuk D. A., Davis G. E., Li D. Y. (2009) The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 15, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.