

Background: Prions can be transmitted by blood transfusion, but their origin and distribution in blood are unknown.
Results: Prions were detected in plasma extracellular vesicles from preclinical and clinically sick mice.
Conclusion: Prions associate with blood-circulating extracellular vesicles.
Significance: These findings provide information about prion distribution in blood and set the groundwork for novel prion removal and disease diagnosis technologies.
Keywords: Blood, Cell Culture, Exosome, Extracellular Vesicles, Mouse
Abstract
The development of variant Creutzfeldt-Jakob disease (vCJD) in three recipients of non-leukoreduced red blood cells from asymptomatic donors who subsequently developed the disease has confirmed existing concerns about the possible spread of transmissible spongiform encephalopathies (TSEs) via blood products. In addition, the presence of disease-associated misfolded prion protein (PrPTSE), generally associated with infectivity, has been demonstrated in the blood of vCJD patients. However, its origin and distribution in this biological fluid are still unknown. Various studies have identified cellular prion protein (PrPC) among the protein cargo in human blood-circulating extracellular vesicles released from endothelial cells and platelets, and exosomes isolated from the conditioned media of TSE-infected cells have caused the disease when injected into experimental mice. In this study, we demonstrate the detection of PrPTSE in extracellular vesicles isolated from plasma samples collected during the preclinical and clinical phases of the disease from mice infected with mouse-adapted vCJD and confirm the presence of the exosomal marker Hsp70 in these preparations.
Introduction
Human transmissible spongiform encephalopathies (TSEs)2 are fatal neurodegenerative disorders that can develop as sporadic, genetic, or infectious diseases. In the United Kingdom and France, which have reported the highest number of variant Creutzfeldt-Jakob disease (vCJD) cases due to exposure to bovine spongiform encephalopathy (BSE)-contaminated products (1), uncertainty exists in regard to the number of infected people (2, 3), raising concerns about the safety of blood- and plasma-derived products.
Transmission of vCJD through transfusion of non-leukoreduced red blood cells (RBCs) has been shown in four individuals, three of whom developed clinical disease and one who died from non-TSE-related causes, but in which the presence of disease-associated misfolded prion protein (PrPTSE) in lymphoreticular tissues was indicative of preclinical or subclinical disease (4). In addition, one hemophilic patient might have been infected through treatment with plasma-derived products (5). Importantly, PrPTSE has been detected in whole blood of vCJD patients (6–8). Moreover, a number of healthy people have been found to harbor this agent in lymphoreticular tissues (3, 9, 10); however, it is not known whether PrPTSE circulates in the blood of these individuals and whether they will develop the disease later in life. Recently, infectivity was reported in the plasma of two out of four patients affected by sporadic CJD (sCJD) (11). Although similar levels of PrPTSE have been detected in spleens, tonsils, and lymph nodes of vCJD and sCJD patients by Western blotting (12), the identification of this protein in the blood of individuals afflicted with the latter by various methods, including PrPTSE capture coupled to direct immunodetection of surface-bound material (6), capillary electrophoresis (13), and protein misfolding cyclic amplification (PMCA) coupled to surrounding optical fiber immunoassay (12), has been highly elusive for decades, with only two cases recently reported (8). Nevertheless, extensive epidemiological data continue to provide no evidence of human-to-human transmission of sCJD through blood transfusion (14).
Multiple studies showed that TSE infectivity is present in the blood of experimental rodents, sheep with natural and experimental scrapie and experimental BSE, and deer affected with chronic wasting disease (CWD) (15–20). PrPTSE has been successfully detected in the following: in whole blood of experimentally infected mice and hamsters, in sheep with natural scrapie and experimental BSE, and in white-tailed deer afflicted with CWD (21–23); in buffy coats of hamsters and sheep experimentally infected with scrapie (24–26), as well as in plasma of mice and hamsters with scrapie; and in sheep and white-tailed deer naturally and experimentally infected with scrapie and CWD, respectively (27–30). However, it is still not fully understood in what blood component TSE infectivity and/or PrPTSE reside and whether and how blood contributes to the spread of the disease from the periphery to the brain.
Exosomes are extracellular membrane vesicles of 40 to 200 nm in diameter, secreted by most cell types upon fusion of multivesicular bodies with the plasma membrane. Originally, they were considered to be a cellular mechanism to eliminate unwanted proteins during reticulocyte maturation (31–33). Further studies have shown the presence of nucleic acid and protein cargo inside exosomes (34–36). These extracellular vesicles (EVs) are believed to participate in intercellular communication processes and to execute functions regulating the immune response, angiogenesis, coagulation, inflammation, and programmed cell death (37). Importantly, it has been shown that viruses like HIV-1 can take over the cellular machinery for exosome secretion and use it as another mechanism for viral release (38, 39). This “Trojan exosome” hypothesis, posited by Gould et al. (39), is currently being evaluated in protein-misfolding neurodegenerative diseases as a possible mechanism for the spread of misfolded proteins (40–45). Cellular prion protein (PrPC) and misfolded PrPTSE have been identified in exosomes from various TSE models (46–55) and intracerebral inoculation of exosomes obtained from TSE-infected cell cultures has caused clinical disease in mice (47, 49). Little is known about the distribution of PrPTSE in blood. PrPC has been detected in exosomes isolated from platelets and annexin V-positive EVs released from apoptotic endothelial cells (48, 56). These findings raise the possibility that endothelial and blood cells other than platelets may be capable of releasing PrPC and possibly PrPTSE in association with exosomes and other types of EVs, contributing to the transmission of TSEs through blood-derived products (48, 56–58). Because of their availability in biological fluids, their stability, and their ability to carry specific cargo, exosomes are ideal targets for detection of biomarkers for the diagnosis of various diseases (34, 59–61). Detection of PrPTSE in EVs obtained from blood or other body fluids (urine, saliva, nasal secretions, and tears) will enable the design of minimally invasive or noninvasive diagnostic tests for TSEs. In this study, EVs, containing exosomes, were isolated from the conditioned media of cell cultures infected with mouse-adapted vCJD (Mo-vCJD) (16) or Fukuoka-1, a mouse-adapted isolate from a Gerstmann-Sträussler-Scheinker disease patient (62, 63), and from plasma collected periodically during the preclinical and clinical phases from Mo-vCJD-infected mice. They were used to seed serial automated protein misfolding cyclic amplification (saPMCA) reactions followed by PrPTSE detection by Western blotting.
Our findings represent the first evidence of the presence of PrPTSE in EVs obtained from plasma samples from preclinical and clinically sick mice, and they provide proof-of-concept for the design of a novel microvesicle-based diagnostic test for prion diseases.
EXPERIMENTAL PROCEDURES
Ethics Statement
The Institutional Animal Care and Use Committee of the American National Red Cross reviewed and approved the animal protocols numbered 0807-023 and 1006-045. The American National Red Cross maintains a centralized animal care and use program registered by the United States Food and Drug Administration, ensured with the Office of Laboratory Animal Welfare, and accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International. Housing and care of animals are consistent with the Public Health Service Policy on Humane Care and Use of Laboratory Animals, Guide for the Care and Use of Laboratory Animals, the Animal Welfare Act, and other applicable state and local regulations.
Human blood was collected into citrate/phosphate dextrose upon informed written consent under protocol number 1998-18, approved by the Institutional Review Board of the American National Red Cross.
Animals
Mouse Inoculations
Wild type (WT), C57BL/6 (C57BL), and FVB/NCr (FVB) mice (Charles River Laboratories, Andover, MA) were intracerebrally injected with 30 μl of a 10−2 diluted or a 10-fold serially diluted (10−1 to 10−7) Mo-vCJD-infected brain homogenate, respectively. Control mice received a similar injection of physiological saline. The inoculum titer was determined by the method of Reed and Muench (64) based on the survival rate of FVB mice inoculated with 10−1 to 10−8 dilutions of Mo-vCJD. FVB mice in the experimental groups were euthanized when they developed clinical signs of TSE. C57BL mice were euthanized periodically during the incubation period (6, 9, 12, 15, and 20 weeks post inoculation (wpi)) and at clinical onset (∼23 wpi). Negative controls were euthanized at the end of the experiment, together with the last mouse from the experimental group. TSE in mice was confirmed by detecting PrPTSE in brain extracts by Western blotting and/or immunohistochemistry as described elsewhere (65).
Blood Collection and Processing
Mouse blood was collected into citrate/phosphate dextrose by cardiac puncture under isoflurane (Patterson Veterinary, Devens, MA) anesthesia. Blood samples were usually processed separately, but on some occasions samples from a few mice were pooled together. Samples were spun at 2,300 × g for 10 min, at room temperature in a FA45-24-11 rotor (Eppendorf, Hauppauge, NY). Plasma was collected, aliquoted, and stored at −80 °C.
Substrate Preparation for saPMCA
Mice were euthanized by CO2 inhalation and perfused with 40 ml of 5 mmol/liter EDTA in PBS by intra-cardiac injection. Brains were collected and frozen at −80 °C until further processing. Brains were homogenized in appropriate volumes of conversion buffer (1% Triton X-100 in PBS with 1× Complete protease inhibitor mixture (Roche Applied Science)) to prepare a 10% (w/v) WT-mouse brain homogenate (MoBH) that was then split into 1-ml aliquots and stored at −80 °C.
Human Samples
Human blood was separated into RBCs, buffy coat, and platelet-rich plasma by centrifugation at 800 × g for 30 min. Platelet-rich plasma was centrifuged at 3,900 × g for 16 min. Platelet-poor plasma was collected, aliquoted, and stored at −80 °C. All centrifugations were performed at room temperature in an A-4-44 rotor (Eppendorf).
Cell Culture
Murine spleen-derived stromal cell culture persistently infected with Mo-vCJD (Mo-vCJD/SP-SC), murine bone marrow-derived stromal cell culture persistently infected with Fukuoka-1 (OF1-BMS), and appropriate uninfected control cell cultures (NB/SP-SC and BMS, respectively) were developed in our laboratory and have been described previously (66, 67). Cells were plated in 25-cm2 flasks with 5 ml of Opti-MEM media (Invitrogen) not supplemented with fetal bovine serum and allowed to grow to confluence at 37 °C and 5% CO2 for 2–3 days for EV preparation.
EV Isolation
EV Isolation from Conditioned Media by ExoQuickTM-TC, Optimized for Culture Media and Urine Samples (System Biosciences, Mountain View, CA)
The medium from each cell culture was collected when cells reached confluence and was centrifuged at 3,000 × g for 15 min to remove cells and cell debris. Supernatants were collected, split in 5-ml aliquots, frozen on dry ice, and stored at −80 °C until further processing. Supernatant aliquots of 5 ml were thawed at room temperature, mixed with 1 ml of ExoQuickTM-TC, and incubated at 4 °C overnight. The mixture was centrifuged at 1,500 × g for 30 min, and the EV pellets were collected and stored at −80 °C until analysis. All centrifugations were done at 4 °C in an A-4-44 rotor.
EV Isolation from Plasma by ExoQuickTM, Optimized for Serum or Ascites (System Biosciences)
Plasma samples were thawed at 37 °C and spun for 15 min at 3,000 × g. The resulting supernatant was mixed with ExoQuickTM (63 μl of ExoQuick per 250 μl of plasma) and incubated overnight at 4 °C. Samples were centrifuged at 1,500 × g for 30 min and EV pellets were collected and stored at −80 °C until analysis. All centrifugations were performed at 4 °C in a FA45-24-11 rotor.
Exosome Purification Using a 30% Sucrose Cushion
EV pellets obtained after ExoQuickTM were processed through a 30% sucrose cushion as indicated in the text, following a protocol adapted from Thery et al. (68). The ExoQuickTM pellet was thawed at room temperature, resuspended in 4 ml of PBS, and layered on top of 0.7 ml of 30% sucrose prepared in a Tris/D2O solution (30 g of sucrose, 2.4 g of Tris base, D2O up to 100 ml). After an initial ultracentrifugation at 110,000 × g for 75 min, three fractions were collected as follows: a top (“top”) layer of ∼0.2 ml, the “middle” layer containing the sucrose layer and the PBS/sucrose interface, and the pellet (“bottom”). The pellet was resuspended in PBS, and all three fractions were brought up to 5 ml with PBS. Samples were subjected to a second ultracentrifugation at 110,000 × g for 70 min. The three resulting pellets were collected and stored at −80 °C. All centrifugations were performed at 4 °C in an SW55 Ti rotor.
saPMCA Amplification
To determine the presence of PrPTSE, EV preparations from different sources (source of PrPTSE) were resuspended into 10% WT-MoBHs (source of PrPC) that were prepared as indicated above. The volume of 10% WT-MoBH used to resuspend the EV pellets is specified for each individual experiment under “Results.” Furthermore, samples were aliquoted into 0.2-ml PCR tubes containing zirconia/silica beads of 1 mm in diameter (Biospec Products Inc., Bartlesville, OK). Samples were amplified by 48 cycles (one round) of incubation at 37 °C, followed by a 20-s pulse of sonication at power 4 in a Q700MPX microplate horn sonicator (QSonica, Newtown, CT) (69, 70). After each round, sample aliquots were mixed 1:1 or 1:3, as indicated below for each experiment, with 10% WT-MoBH to perform the next round of saPMCA. The number of rounds varied for each experiment and is indicated in the appropriate sections.
Proteinase K (PK) Digestion
Aliquots of 9 μl of samples subjected to saPMCA were treated with 20 μg/ml PK (Novagen®, Darmstadt, Germany) for 1 h at 37 °C with agitation at 450 rpm. PK treatment was stopped by sample denaturation in NuPAGE LDS sample buffer (Invitrogen) at 100 °C for 10 min.
Western Blotting
Proteins were diluted in NuPAGE LDS sample buffer (Invitrogen) and separated by SDS-PAGE, electroblotted onto a nitrocellulose membrane (Invitrogen), and probed with various primary and secondary antibodies as indicated in the text. Immunoreactive bands were visualized using West Pico (Pierce). The primary antibodies used were as follows: mouse monoclonal anti-PrP antibody 6D11 (Covance®, Berkeley, CA), and rabbit monoclonal antibodies anti-Hsp70 (Epitomics, Burlingame, CA) and anti-GM130 (Abcam, Cambridge, MA). Secondary antibodies used were as follows: HRP-conjugated goat anti-mouse IgG (Kirkegaard & Perry, Gaithersburg, MD); HRP-conjugated rat anti-mouse IgG Mouse Trueblot® (Rockland, Gilbertsville, PA); and HRP-conjugated mouse anti-rabbit IgG rabbit Trueblot® (Rockland).
Methanol Precipitation
Where applicable, samples were mixed with 5 volumes of prechilled 100% methanol and incubated overnight at −20 °C. Samples were centrifuged at 25,000 × g for 30 min in a FA45-24-11 rotor and resuspended in 1% Sarkosyl.
EV Protein (EVP) Solubilization
EV proteins were solubilized by resuspending extracellular vesicle pellets in EVP lysis buffer consisting of 100 mmol/liter NaCl, 10 mmol/liter EDTA, 0.5% Triton X-100, 0.5% sodium deoxycholate, and 10 mmol/liter Tris, pH 7.4.
Nanoparticle Tracking Analysis (NTA)
The EV pellet obtained from the plasma of a healthy FVB mouse after purification with ExoQuickTM was resuspended in 200 μl of PBS, and the suspension was diluted 1,000-fold prior to being analyzed. Alternatively, the EV pellet obtained as above, was further purified through a 30% sucrose cushion as described under “Exosome Purification Using a 30% Sucrose Cushion.” The three fractions collected (top, middle, and bottom) were resuspended in 700 μl of PBS prior to being analyzed. The particle hydrodynamic diameter of the EVs was measured for 90 s using a NanoSight LM10 system (Malvern, Worcestershire, UK). The Nanoparticle Tracking Analysis 2.3 analytical software version was used for capturing and analyzing the data. Particle number per ml and standard deviation were calculated from three measurements from the same sample.
RESULTS
saPMCA Detection of PrPTSE in EVs Isolated from Conditioned Media Collected from TSE-infected Cells
We first investigated the presence of PrPTSE in EVs isolated from conditioned media collected from Mo-vCJD/SP-SC and OF1-BMS and corresponding uninfected cells (NB/SP-SC and BMS, respectively) used as control. EVs were isolated from 5 ml of conditioned media with ExoQuickTM-TC as described under “Experimental Procedures” (EV Isolation from Conditioned Media by ExoQuickTM-TC, Optimized for Culture Media and Urine Samples (System Biosciences, Mountain View, CA)). EV pellets from spleen-derived and bone marrow-derived cells were resuspended in 250 and 200 μl, respectively, of 10% WT-MoBH. Samples were amplified by saPMCA through 48 cycles (one round). After each round, aliquots of 30 μl were mixed 1:1 with 10% WT-MoBH to perform the next round of saPMCA. Additionally, aliquots of 9 μl of amplified material were taken from each sample and processed for the detection of PrPTSE by PK digestion followed by Western blotting as under “Experimental Procedures.”
EVs from 5 ml of conditioned media contained sufficient amounts of PrPTSE to trigger PrPC conversion in saPMCA reactions, validating the applicability of this approach to detect PrPTSE in these preparations (Fig. 1). One round of saPMCA led to PrPTSE detection in samples generated from Mo-vCJD/SP-SC cells (Fig. 1A), whereas two rounds were required to detect this protein in samples purified from OF1-BMS cells (Fig. 1C). The difference in number of rounds required for each cell model, which differ in their tissue of origin, might be due to variations in PrPTSE levels in the EV preparations and/or in the number of EVs secreted by the cells. Alternatively, it may reflect differences in the conversion efficiency of the two strains, Mo-vCJD and Fukuoka-1.
FIGURE 1.
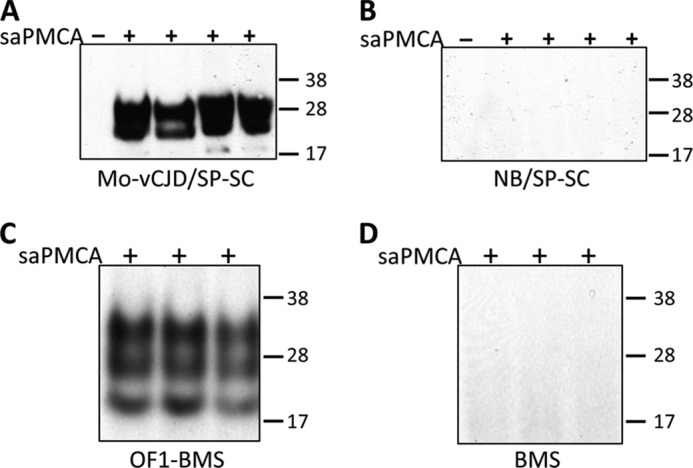
Detection by saPMCA of PrPTSE in EV preparations isolated from the conditioned media of chronically infected cells. EVs were purified with ExoQuickTM-TC (System Biosciences) from 5 ml of conditioned media collected from Mo-vCJD-infected (A) and -uninfected (B) spleen-derived stromal cells (Mo-vCJD/SP-SC and NB/SP-SC, respectively), and from Fukuoka-1 (FU)-infected (C) and -uninfected (D) bone marrow-derived stromal cells (OF1-BMS and BMS, respectively). EV pellets were mixed with 250 and 200 μl, respectively, of 10% wild-type mouse brain homogenate (used as source of PrPC). Aliquots of 9 μl were taken from each sample and used as controls not subjected to saPMCA (−). The remaining BMS and SP-SC samples were split into 3 and 4 aliquots, respectively, and amplified by saPMCA (+). PrPTSE was specifically detected in EV samples from Mo-vCJD/SP-SC cells (A) and from OF1-BMS cells (C) after one and two rounds of saPMCA, respectively, but not in samples from uninfected NB/SP-SC (B) and BMS (D) cells. All samples were treated with 20 μg/ml of proteinase K, resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance). Molecular mass is shown on the right.
Detection of PrPTSE in Uninfected Human and Mouse Plasma Samples Supplemented with EVs from Conditioned Media of TSE-infected Cells
Earlier studies have shown that some blood components impair PMCA amplification (24, 25). This inhibition can be overcome by performing several rounds of saPMCA. Under these conditions, inhibitors are diluted after each round, at the same time that new PrPTSE is generated. This approach enabled PrPTSE detection as follows: in buffy coat samples from 263K-infected hamsters during the preclinical and clinical phases of prion disease (24, 25); in plasma preparations from preclinical and clinically sick scrapie-affected sheep, CWD-infected white-tailed deer (28), and from clinically ill mice infected with mouse-adapted BSE (71); in blood leukocytes from clinically sick VRQ/VRQ scrapie-affected sheep (26); and in whole blood from clinical mice infected with scrapie (21). To evaluate the effect of plasma on the detection of EV-associated PrPTSE by saPMCA, EVs isolated from conditioned media of infected cells as described above were added to 250 μl of human plasma obtained from a healthy donor. Human plasma was used because of large volumes available to ensure experimental consistency during the study. EVs were re-isolated from EV-supplemented plasma samples with ExoQuickTM as described under “Experimental Procedures” (“EV Isolation from Plasma by ExoQuickTM, Optimized for Serum or Ascites (System Biosciences)”). Re-isolated EVs were thawed and mixed with 250 μl of 10% WT-MoBH. In parallel, EV samples that were not added to plasma were mixed with 250 μl of 10% WT-MoBH and used as positive controls for saPMCA. Four aliquots of 58 μl were prepared from control and experimental samples and amplified by saPMCA as described under “Experimental Procedures.” Samples were diluted 1:1 between saPMCA rounds.
Although PrPTSE was readily demonstrated in EVs from the conditioned media of Mo-vCJD/SP-SC cells after one round of saPMCA (Fig. 2A), a high unspecific signal background prevented detection of PrPTSE in plasma samples supplemented with EVs isolated from the conditioned media of infected cells (Fig. 2B). Nevertheless, dilution and amplification of these samples for three additional rounds of saPMCA resulted in the reliable detection of PrPTSE (Fig. 2A). Similar findings were obtained when EVs, prepared from the conditioned media of Fukuoka-1-infected OF1-BMS cells, were added to plasma collected from uninfected wild-type FVB mice, re-isolated, and used in saPMCA (Fig. 3).
FIGURE 2.
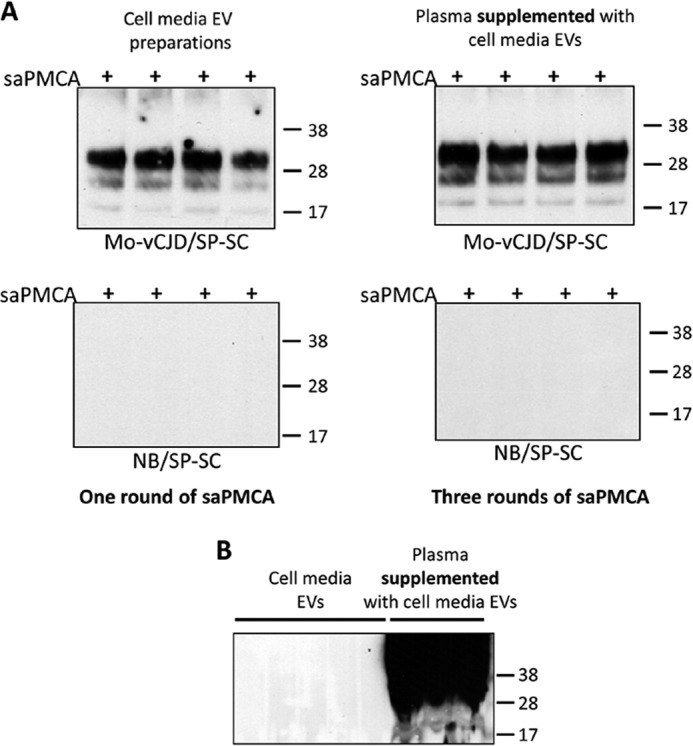
Detection by saPMCA of PrPTSE in EV preparations isolated from cell-conditioned media of chronically infected cells and added to normal human plasma. A, EV samples were obtained from 5 ml of conditioned media from Mo-vCJD-infected and -uninfected spleen-derived stromal cells (Mo-vCJD/SP-SC and NB/SP-SC, respectively) using ExoQuickTM-TC (System Biosciences). EV pellets were either mixed with 250 μl of 10% wild-type mouse brain homogenate (WT-MoBH) (A, top and bottom left panels, and B) or added to 250 μl of human plasma. Samples added to plasma were subjected to repeated EV extraction with ExoQuickTM, and the pellets were mixed with 1,250 μl of WT-MoBH (A, top and bottom right panels, and B). Each sample was split into 4 aliquots and subjected to cycles of saPMCA (+). The presence of heavy background signal in EV preparations from plasma prevented PrPTSE detection after one round of saPMCA (B). Dilution of samples in three additional rounds allowed reliable detection of PrPTSE (A, top right panel). Sample aliquots of 9 μl were treated with 20 μg/ml proteinase K, resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance). Molecular mass is shown on the right.
FIGURE 3.

Detection by saPMCA of PrPTSE in extracellular vesicles EVs isolated from cell-conditioned media of OF1-BMS cells that were added to normal FVB mouse plasma. EV samples were obtained from 5 ml of conditioned media from Fukuoka-infected bone marrow-derived stromal cells (OF1-BMS) using ExoQuickTM-TC (System Biosciences). Samples were either mixed with 250 μl of 10% wild-type mouse brain homogenate (WT-MoBH) (A, lanes 2 and 3, and B, lanes 3–6) or added to 250 μl of FVB mouse plasma. Samples added to plasma were subjected to repeated EV extraction with ExoQuickTM, and the pellets were mixed with 150 μl (A, lanes 4 and 5, and B, lanes 7 and 8), 250 μl (A, lanes 6–9, and B, lanes 9–12), and 1,250 μl of WT-MoBH (A, lanes 10 and 11, and B, lanes 13 and 14). Each sample was split into aliquots prior to saPMCA (+). The presence of heavy background signal in EVs re-isolated from plasma prevented PrPTSE by Western blotting detection after one round of saPMCA. Dilution of samples in three and four additional rounds allowed reliable detection of PrPTSE (A and B, respectively). Samples were treated with 20 μg/ml of proteinase K (PK), resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance). −PK, WT-MoBH not treated with PK. Molecular mass (M) is shown on the left. Samples were diluted 1:5 between saPMCA rounds.
Presence of IgG in Plasma EV Preparations Disables Detection of PrPTSE after the Initial Rounds of saPMCA
IgG is one of the most abundant proteins in plasma, and its presence in samples is a recognized problem in Western blotting applications due to reaction with secondary antibodies. We observed high background signal when performing Western blotting analysis of samples after initial rounds of saPMCA that were seeded with plasma EV preparations. Therefore, we attempted to overcome this problem by using the mouse Trueblot® HRP-conjugated anti-mouse IgG (Rockland) secondary antibody, which does not detect the reduced and SDS-denatured forms of IgG, instead of the commonly used secondary HRP-conjugated anti-mouse IgG (Kirkegaard & Perry) antibody. EVs were isolated with ExoQuickTM from 250 μl of plasma samples from two terminally ill FVB mice infected with Mo-vCJD. EV pellets were resuspended in 250 μl of 10% WT-MoBH and amplified by saPMCA. Selected saPMCA-amplified plasma EV samples corresponding to rounds one and two were treated with PK, subjected to electrophoresis, and immunoblotted with either antibody (Fig. 4A). The antibody from Kirkegaard & Perry revealed the presence of IgG and other reactive bands, in all samples from the two mice after one round of saPMCA. The intensity of the signal significantly decreased after two rounds demonstrating that cycles of dilution/conversion efficiently eliminated unspecific proteins, including IgG, from the samples. Bands corresponding to IgG were not detected with the antibody from Rockland after rounds one and two. To demonstrate the appropriate binding to the primary antibody 6D11, which is specific to PrP, antibodies from both Kirkegaard & Perry and Rockland were used to reveal PrPTSE in Western blotting of samples subjected to five rounds of saPMCA. Three bands corresponding to di-, mono-, and unglycosylated PrPTSE were observed with either antibody in samples treated with PK; however, the intensity of the signal obtained with the antibody from Rockland was significantly lower, and longer exposure times were necessary to obtain results comparable with those generated with the antibody from Kirkegaard & Perry (Fig. 4B).
FIGURE 4.

Presence of IgG in plasma-derived EV preparations. Western blotting analysis of the initial rounds of saPMCA showed high background that prevented the detection of PrPTSE. A, presence of IgG was evaluated by assessing sample reactivity with two secondary antibodies: HRP-conjugated goat anti-mouse IgG (Kirkegaard & Perry antibody (Ab)) and mouse Trueblot® HRP-conjugated rat anti-mouse IgG (Rockland antibody), which does not detect the reduced SDS-denatured forms of IgG. saPMCA-amplified plasma EVs from terminally ill FVB/NCr mice infected with Mo-vCJD after rounds one and two were digested with proteinase K and separated on SDS-PAGE prior to immunoblotting with either antibody (A, left and right panels, respectively). Immunoblotting with the antibody from Kirkegaard & Perry revealed the presence of IgG in all aliquots from both mice after one round of saPMCA. The intensity of the signal significantly decreased after two rounds of saPMCA, demonstrating that cycles of dilution/conversion effectively eliminate IgG in plasma EV-seeded samples. No signal was detected with mouse Trueblot® antibody. B, same plasma EV preparations were probed with the PrP-specific antibody 6D11 (Covance) as the primary antibody was followed by secondary antibodies from either Kirkegaard & Perry or Rockland. Molecular mass is shown on the right. Note the difference in the intensity signals between the antibodies used from Kirkegaard & Perry and Rockland, with the antibody from Kirkegaard & Perry giving significantly better signal readout. Lanes 1–3 represent sample triplicates.
Isolation and Characterization of Plasma EVs, PrPTSE Associates with Plasma EVs
To investigate association of PrPTSE with plasma EVs, we took advantage of the flotation properties of nanovesicles on a 30% sucrose cushion. Ultracentrifugation of EVs in the presence of 30% sucrose eliminates nonspecifically associated proteins, or protein aggregates, which are sedimented by centrifugation but do not float on a sucrose gradient and allow isolation of highly purified exosomes (68). Two 500-μl aliquots of pooled plasma from clinically sick FVB mice infected with 10−1 and 10−2 dilutions of Mo-vCJD were processed with ExoQuickTM to isolate EVs. The pellet from 1 aliquot was stored at −80 °C until analysis and was labeled as “infected non-sucrose.” The pellet from the second aliquot was processed through a 30% sucrose cushion as described under “Experimental Procedures.” Three fractions were collected after ultracentrifugation as follows: a thin infected top layer of ∼0.2 ml of white material of lipidic appearance; the infected middle layer containing the PBS/sucrose interface and the sucrose layer; and the infected bottom pellet, which was not visible. All three fractions were brought up to 5 ml of PBS and subjected to a second ultracentrifugation. Plasma samples from uninfected FVB mice were processed similarly and used as negative controls (“uninfected non-sucrose” and “uninfected top, middle, and bottom”). Infected and uninfected “non-sucrose” fractions were resuspended in 250 μl of 10% WT-MoBH; infected and uninfected “sucrose” fractions were resuspended in 125 μl of 10% WT-MoBH. All samples were subjected to various rounds of saPMCA.
Samples were evaluated for the presence of PrPTSE by Western blotting with the anti-PrP primary antibody 6D11 and mouse Trueblot® secondary antibody (Fig. 5). This allowed PrPTSE detection in plasma EVs at early rounds of saPMCA without IgG interference. Under these conditions, PrPTSE was specifically detected in “non-sucrose” plasma exosome fractions from infected mice after just one round of saPMCA (Fig. 5A, infected non-sucrose). A second round of saPMCA showed the presence of PrPTSE in the pellet fraction obtained after centrifugation through a sucrose cushion (Fig. 5B, lanes 3 and 4). In addition, PrPTSE was detected in the “top” layer (Fig. 5B, lanes 1 and 2), which represents a fraction of co-purified PrPTSE that is likely to be associated with lipids, very low density and low density lipoproteins (72), and/or because the ExoQuickTM protocol does not include a filtration step, probably with membrane fragments. Most importantly, PrPTSE was detected in the “middle” fraction (Fig. 5B, lanes 5 and 6), containing plasma exosomes. No PrPTSE was detected in corresponding fractions from plasma EV preparations from uninfected mice (Fig. 5, A, lanes 5–8, and B, lanes 7–12). Samples were diluted 1:1 between rounds.
FIGURE 5.
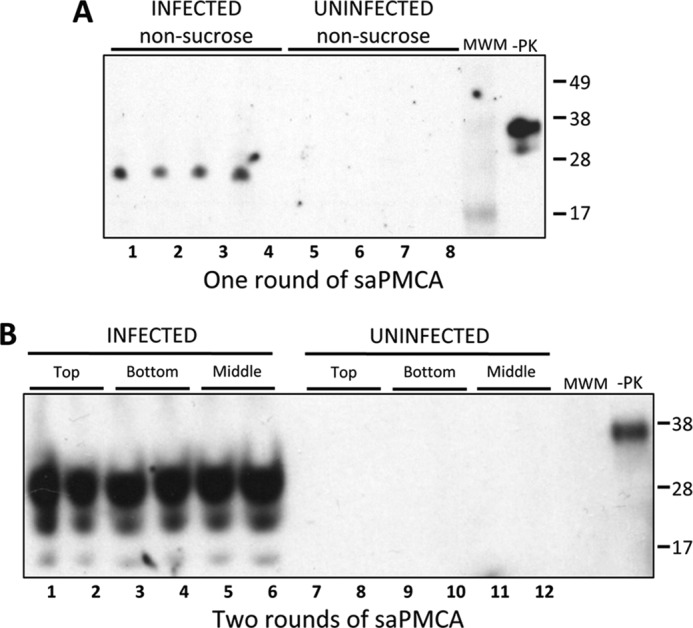
Detection by saPMCA of PrPTSE in EV preparations from plasma samples of Mo-vCJD-infected mice. EV samples were prepared using ExoQuickTM (System Biosciences) from 500 μl of plasma from clinically sick or uninfected control mice as described in the text. A, EV pellets were mixed into 250 μl of 10% wild-type mouse brain homogenate (WT-MoBH), split into 4 aliquots, and subjected to one round of saPMCA (non-sucrose). B, EV pellets were used to purify exosomes by ultracentrifugation on a 30% sucrose cushion. Three fractions were collected (top, middle (containing exosomes), and bottom) and subjected to a second round of ultracentrifugation. Pellets were mixed into 125 μl of 10% WT-MoBH, split into 2 aliquots, and subjected to two rounds of saPMCA. Aliquots of 9 μl were collected from each sample, treated with 20 μg/ml proteinase K, resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance) and mouse Trueblot® (Rockland) secondary antibody. −PK, WT-MoBH not treated with PK. Molecular mass is shown on the right. Samples were diluted 1:1 between rounds.
Exosomal Marker Hsp70 Associates with Plasma EVs
To characterize EV preparations, we analyzed the presence of the exosomal marker Hsp70 and the absence of the Golgi marker GM130. EVs were isolated from 500 μl of plasma from a healthy FVB mouse using ExoQuickTM. The EV pellet was subjected to ultracentrifugation through a 30% sucrose cushion and repeated ultracentrifugation as described above. The three pellets, representing sucrose top, middle, and bottom fractions, were resuspended in EVP lysis buffer to solubilize membrane proteins. Proteins were concentrated by methanol precipitation and resuspended in 1% Sarkosyl in PBS. For comparison, EVs were also isolated from the conditioned media of NB/SP-SC cells with ExoQuickTM-TC, resuspended in EVP lysis buffer, methanol-precipitated, and resuspended in 1% Sarkosyl in PBS (“cell EVs”). This preparation was used as positive control for Hsp70. Additionally, NB/SP-SC cells were lysed in EVP lysis buffer (“NB/SP-SC cell lysate”) and used as control for GM130. The same amount of total protein (12 μg) was loaded per sample. Western blotting analysis of samples obtained from plasma, conditioned media, and cell lysates revealed the presence of Hsp70 in the conditioned media sample (cell EVs) and sucrose middle plasma fraction (Fig. 6A, lanes 1 and 4, respectively) but not in sucrose top and bottom plasma fractions obtained after ultracentrifugation (Fig. 6A, lanes 3 and 5), thus confirming that samples prepared from plasma using ExoQuickTM contained exosomes. As expected, the Golgi marker GM130 was exclusively detected in cell lysates (Fig. 6B, lane 1) but was absent in samples prepared from conditioned media and from sucrose top, middle, and bottom fractions (Fig. 6B, lanes 2–5). The specificity of the immunoreactive bands was confirmed by the absence of signal while probing the samples with the secondary rabbit Trueblot® antibody only (data not shown).
FIGURE 6.
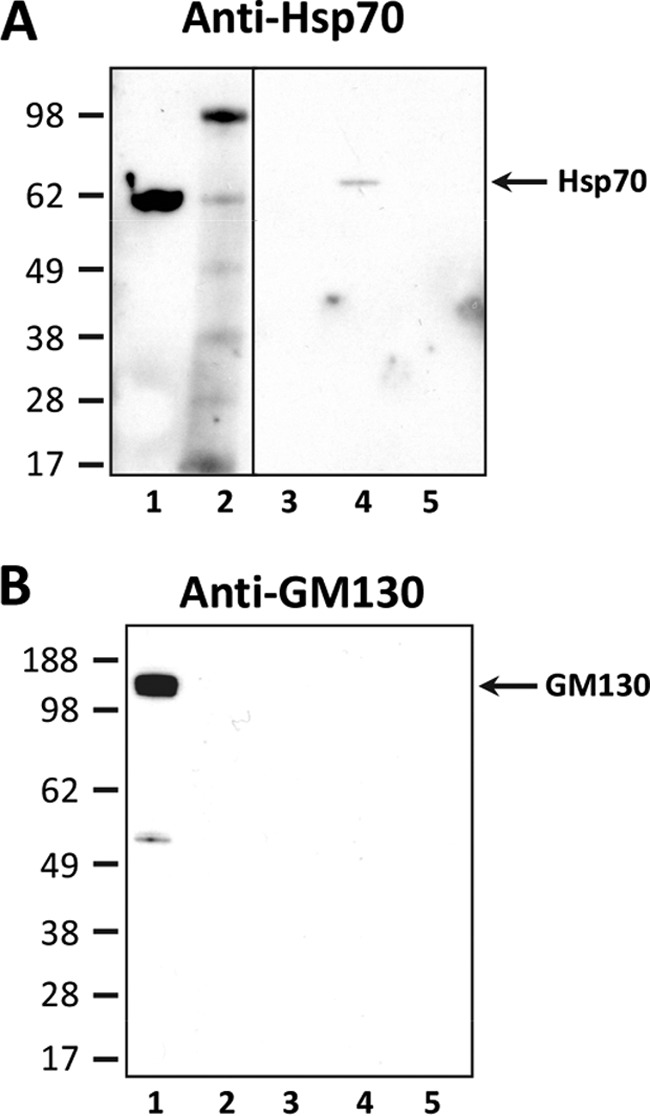
Demonstration of the presence of the exosomal marker Hsp70 in EV preparations from normal FVB mouse plasma. EVs were isolated from 500 μl of plasma from uninfected FVB mice with ExoQuickTM (System Biosciences) and purified as described in the text. Proteins from the top, middle, and bottom fractions were concentrated by methanol precipitation and resuspended in 1% Sarkosyl in PBS. The exosomal marker Hsp70 (A) and the Golgi marker GM130 (B) were detected with corresponding rabbit primary antibodies and the rabbit Trueblot® (Rockland) secondary antibodies. A, lane 1, cell EVs: non-sucrose EVs from uninfected NB/SP-SC conditioned media (positive control); lane 2, molecular mass marker; lanes 3–5, sucrose top, middle (containing EVs), and bottom fractions, respectively. B, lane 1, NB/SP-SC cell lysate (positive control); lane 2, cell EVs: non-sucrose-cleaned EVs from NB/SP-SC-conditioned media; lanes 3–5, sucrose top, middle (containing EVs), and bottom fractions, respectively. Twelve micrograms of total protein were loaded per well in A and B. Molecular mass is shown on the left.
Detection of Particle Hydrodynamic Diameter in EV Preparations Purified by ExoQuickTM
To identify the particle hydrodynamic diameter of EVs, samples prepared from the plasma of healthy FVB mice were subjected to NTA. The vesicle hydrodynamic diameter ranged from 50 to 300 nm, with an average diameter of 114 nm, in a sample with a vesicle count of 5.46 × 108 particles/ml (Fig. 7A). Established EV hydrodynamic diameter was within the range reported for human plasma EVs (73). Next, we evaluated the presence of EVs in the three fractions collected following centrifugation through a 30% sucrose cushion. To this end, EVs were isolated from the plasma of healthy FVB mice with ExoQuickTM and further purified through a 30% sucrose cushion as described above. The top, middle, and bottom fractions were resuspended in PBS and subjected to NTA. We identified the presence of EVs in all fractions analyzed, albeit with differences in particle concentration and hydrodynamic diameter. The top fraction was characterized by the presence of a single population of EVs ranging from 38 to 350 nm and an average hydrodynamic diameter of 103 nm (Fig. 7B). The middle fraction contained EVs with a hydrodynamic diameter range of 25 to 450 nm and a major particle population of 102 nm. Two other minor peaks were detected in this preparation showing average hydrodynamic diameters of 124 and 145 nm (Fig. 7C). The bottom fraction contained EVs of 40 to 500 nm in diameter. Various peaks could be identified in this fraction with average hydrodynamic diameters of 87, 114, 139, and 170 nm (Fig. 7D). The particle concentration also varied between fractions, with the bottom fraction containing the lowest number of particles, 7.2 ± 0.21 × 108 particles/ml, followed by the top fraction with 11.6 ± 0.21 × 108 particles/ml, and the middle fraction with 17.3 ± 1.33 × 108 particles/ml.
FIGURE 7.

Analysis of plasma-derived EVs. A, EVs were purified from 250 μl of uninfected mouse plasma, resuspended in 200 μl of PBS, and further diluted 1,000 times in PBS. B–D, EVs were isolated from 500 μl of uninfected mouse plasma. EVs were further purified through a 30% sucrose cushion. Top (B), middle (C), and bottom (D) fractions were collected and resuspended in 700 μl of PBS. All samples were analyzed in a NanoSight LM-10 instrument. The particle hydrodynamic diameter distribution is represented relative to the particle concentration. A, NTA analysis of the total EVs isolated from plasma shows a main particle population of 114 nm, which is consistent with the exosome hydrodynamic diameter range of 40 to 200 nm. B, top fraction was characterized by the presence of a single population of EVs with an average hydrodynamic diameter of 103 nm. C, middle fraction contained EVs with a major particle population of 102 nm in diameter and two minor peaks with average hydrodynamic diameters of 124 and 145 nm. D, bottom fraction was characterized by the presence of various peaks of 87, 114, 139, and 170 nm in diameter.
Detection of PrPTSE in Plasma EVs Isolated from Symptomatic Mo-vCJD-infected FVB Mice
After demonstrating the efficiency of ExoQuickTM to isolate PrPTSE-containing EVs and the potential of saPMCA to detect PrPTSE in plasma EV preparations (Fig. 5), we aimed to confirm the presence of PrPTSE in plasma preparations from a larger number of samples collected from terminally sick mice infected with Mo-vCJD.
EVs were isolated from 200 and 250 μl of plasma from Mo-vCJD-infected FVB mice and appropriate uninfected controls, using ExoQuickTM as described under “Experimental Procedures.” Samples were resuspended in an equal volume of 10% WT-MoBH, split into 4 aliquots, and subjected to multiple rounds of saPMCA. Samples were diluted 3-fold between saPMCA rounds. Results of PrPTSE detection, starting from round five, in which the first positive samples were clearly identified, are summarized in Table 1 and Fig. 8. PrPTSE was detected in all plasma EV pellets from infected mice in which TSE had been biochemically confirmed. Samples from mouse 4, which received a 10−7 dilution of Mo-vCJD, were negative through all seven rounds. In agreement with this result, mouse 4 showed no clinical signs of TSE and tested negative for PrPTSE in the brain by Western blotting. The calculated LD50 value for the Mo-vCJD brain homogenate, used for inoculation, was estimated to be 10−6.3. The other three mice in the group infected with a 10−7 dilution exhibited clinical signs and tested positive for PrPTSE in the brain by Western blotting. Samples from uninfected control mice remained negative for all rounds of saPMCA (Fig. 8, FVB-I and FVB-II). Likewise, PrPTSE was specifically detected in plasma EVs isolated from FVB mice inoculated with Fukuoka-1; samples collected from noninfected mice remained negative for five rounds of saPMCA (data not shown).
TABLE 1.
Detection by saPMCA of PrPTSE in plasma-derived EV preparations isolated from clinically sick Mo-vCJD-infected FVB mice
The presence of PrPTSE was tested in 4 aliquots of EV preparations from individual or pooled plasma samples obtained from terminally ill Mo-vCJD-infected mice. All infected mice had tested positive (+) for PrPTSE in the brain by Western blotting, except mouse 4 (−). EVs were isolated from 200 μl of plasma by ExoQuickTM (System Biosciences). EV pellets were mixed with 200 μl of 10% wild-type mouse brain homogenate and split into 4 aliquots that were subjected to up to seven rounds of saPMCA. A summary of the data from rounds 5 to 7 is presented. Background interference prevented identification of PrPTSE-positive samples prior to round five. Presence of PrPTSE was evaluated by Western blotting using the PrP-specific antibody 6D11 (Covance). FVB-I and FVB-II indicate plasma samples from FVB control mice.
| Mouse ID/inoculum dilution | Round 5, aliquots positive/total | Round 6, aliquots positive/total | Round 7, aliquots positive/total | PrPTSE in brain |
|---|---|---|---|---|
| Mouse 1/10−7 | 4/4 | 4/4 | 4/4 | + |
| Mouse 2/10−7 | 4/4 | 4/4 | 3/4 | + |
| Mouse 3/10−7 | UDa | 4/4 | 4/4 | + |
| Mouse 4/10−7 | 0/4 | 0/4 | 0/4 | − |
| Mouse 5/10−6 | 3/4 | 3/4 | 3/4 | + |
| Plasma pool/10−5 | 4/4 | 4/4 | 4/4 | + |
| Mouse 6/10−5 | 4/4 | 4/4 | 4/4 | + |
| Mouse 7/10−4 | 3/4 | 3/4 | 3/4 | + |
| Mouse 8/10−4 | 4/4 | 4/4 | 4/4 | + |
| Mouse 9/10−3 | 4/4 | 4/4 | 4/4 | + |
| Mouse 10/10−3 | 4/4 | 4/4 | 4/4 | + |
| Plasma pool/10−1 and 10−2 | 16/16 | 16/16 | 16/16 | + |
| FVB-I/control | 0/4 | 0/4 | 0/4 | − |
| FVB-II/control | 0/4 | 0/4 | 0/4 | − |
a UD, PrPTSE presence undetermined due to unspecific background; + or −, the presence or absence of PrPTSE in the brain. Boldface indicates plasma EV samples that tested positive for PrPTSE in all 4 aliquots.
FIGURE 8.

Detection by saPMCA of PrPTSE in plasma EV preparations isolated from clinically sick Mo-vCJD-infected FVB mice. EVs were isolated with ExoQuickTM (System Biosciences) from 200 μl of plasma collected from terminally sick FVB mice intracerebrally injected with 10-fold diluted samples of a Mo-vCJD-infected brain homogenate (the inoculum dose is indicated below each mouse number). EV pellets were mixed with 200 μl of 10% wild-type mouse brain homogenate (10% WT-MoBH), split into 4 aliquots, and amplified by saPMCA. After each round, aliquots of 20 μl were taken from each sample, mixed with 40 μl of 10% WT-MoBH, and amplified in a new round of saPMCA; additionally, aliquots of 9 μl were collected from each sample, treated with 20 μg/ml of proteinase K (PK), resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance). The figure illustrates the presence/absence of PrPTSE signal in each of 4 sample aliquots from each mouse after six rounds of saPMCA. FVB-I and FVB-II, samples from uninfected wild-type mice used as negative control; −PK, WT-MoBH not treated with PK. Molecular mass is shown on the right.
Detection of PrPTSE in EVs Isolated from Presymptomatic and Symptomatic Mo-vCJD-infected C57BL Mice
Plasma EV pellets were obtained from groups of Mo-vCJD-infected C57BL mice that were euthanized at 6, 9, 12, 15, and 20 wpi and at clinical stage of TSE (∼23 wpi) and from uninfected control mice. Samples were evaluated for the presence of PrPTSE after several rounds of saPMCA as described above. The results of this experiment are summarized in Table 2 and Fig. 9. All mice had tested positive for PrPTSE in the brain by Western blotting (Fig. 10). Four rounds of saPMCA reliably revealed the presence of PrPTSE in all aliquots of plasma EVs obtained from mice euthanized at 20 wpi and during the clinical phase. Moreover, PrPTSE was demonstrated in at least 1 of 4 aliquots from mice from earlier disease stages starting from 6 wpi, except for mouse 1. One additional round of conversion increased the number of positive aliquots from animals 3, 5, and 12, euthanized at 6, 9, and 15 wpi. Mouse 1 again tested negative for PrPTSE. Aliquots from samples generated from uninfected mice remained negative for all five rounds of saPMCA. Conclusively, the presence of PrPTSE was demonstrated in plasma EVs from Mo-vCJD-infected mice during the preclinical phase, as early as 6 wpi. This finding is in agreement with the work of Tattum et al. (21) showing detection of PrPTSE, after four rounds of saPMCA, in whole blood collected at 60 days post-inoculation from scrapie-infected CD1 mice.
TABLE 2.
Detection by saPMCA of PrPTSE in plasma EV preparations isolated from Mo-vCJD-infected C57BL/6 (C57CL) mice, pre-symptomatic and clinical phase
The presence of PrPTSE was tested after four to five saPMCA rounds in 4 aliquots of EV preparations from individual plasma samples obtained at various post-inoculation times from Mo-vCJD-infected C57BL mice. All infected mice had tested positive for PrPTSE in the brain. Plasma samples from uninfected mice were used as negative controls (C57BL-I and C57BL-II). EVs were isolated from 200 or 250 μl of plasma by ExoQuickTM (System Biosciences). EV pellets were mixed with 200 or 250 μl of 10% wild-type mouse brain homogenate, respectively, and split into 4 aliquots that were subjected to saPMCA. Repeated rounds of saPMCA were performed. The presence of PrPTSE was evaluated by Western blotting using the PrP-specific antibody 6D11 (Covance). Boldface indicates plasma extracellular vesicle samples that tested positive for PrPTSE in all 4 aliquots.
| Blood collection time post-inoculation | Mouse ID | Round 4, aliquots positive/total | Round 5, aliquots positive/total |
|---|---|---|---|
| 6 weeks | 1 | 0/4 | 0/4 |
| 2 | 3/4 | 2/4 | |
| 3 | 2/4 | 3/4 | |
| 9 weeks | 4 | 4/4 | 4/4 |
| 5 | 1/4 | 4/4 | |
| 6 | 4/4 | 4/4 | |
| 12 weeks | 7 | 2/4 | 2/4 |
| 8 | 3/4 | 3/4 | |
| 9 | 4/4 | 4/4 | |
| 15 weeks | 10 | 4/4 | 4/4 |
| 11 | 4/4 | 4/4 | |
| 12 | 1/4 | 4/4 | |
| 20 weeks | 13 | 4/4 | 4/4 |
| 14 | 4/4 | 4/4 | |
| 15 | 4/4 | 4/4 | |
| Clinical phase (∼23 weeks) | 16 | 4/4 | 4/4 |
| 17 | 4/4 | 4/4 | |
| Uninfected controls | C57BL-I | 0/4 | 0/4 |
| C57BL-II | 0/4 | 0/4 |
FIGURE 9.

Detection by saPMCA of PrPTSE in plasma EV preparations isolated from presymptomatic and symptomatic Mo-vCJD-infected C57BL mice. EVs were isolated with ExoQuickTM (System Biosciences) from 200 and 250 μl of plasma collected from presymptomatic and symptomatic C57BL mice intracerebrally injected with a 1% Mo-vCJD-infected brain homogenate. EV pellets were mixed with 200 and 250 μl, respectively, of 10% wild-type mouse brain homogenate (WT-MoBH), split into 4 aliquots, and amplified by saPMCA. After each round, aliquots of 20 μl were taken from each sample, mixed with 40 μl of 10% WT-MoBH, and amplified in a new round of saPMCA; additionally, aliquots of 9 μl were collected from each sample, treated with 20 μg/ml of proteinase K (PK), resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance). The figure illustrates the presence/absence of PrPTSE signal in each of 4 sample aliquots from each mouse after five rounds of saPMCA. C57BL-I and C57BL-II, samples from uninfected wild-type mice used as negative control; −PK, WT-MoBH not treated with PK. Molecular mass is shown on the right.
FIGURE 10.
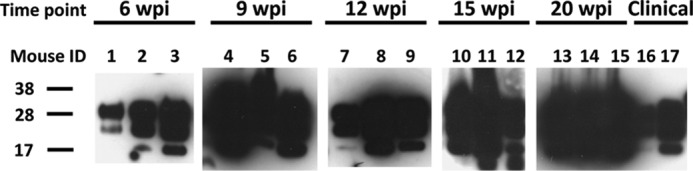
Detection of PrPTSE in brain tissue from presymptomatic and symptomatic Mo-vCJD-infected C57BL mice. 10% mouse brain homogenates were treated with 20 μg/ml of proteinase K, resolved by SDS-PAGE, and subjected to Western blotting using the PrP-specific antibody 6D11 (Covance). Molecular mass is shown on the left.
DISCUSSION
The finding of four human vCJD infections related to blood transfusion (4) highlights the need to develop a method that can reliably identify preclinical cases to minimize the risk of iatrogenic disease transmission via blood-derived products. Experimental detection of PrPTSE in animal cerebrospinal fluid (CSF) (29), whole blood (21, 22), plasma (27–30, 71), buffy coat (24–26), and urine (74) and in human CSF (12, 75), whole blood (6–8), and urine (76, 77) has been achieved by different methods that rely on the concentration or amplification techniques to bring PrPTSE levels to the detection threshold of biochemical assays. Although the presence of PrPTSE in blood exosomes has been suggested previously (48), biochemical detection has been complicated by the low levels of PrPTSE in blood and the concomitantly large volumes required for exosome isolation by standard methods. Traditionally, exosome isolation has been achieved by a series of differential centrifugation and filtration steps from large sample volumes (68). This approach has been combined with saPMCA to demonstrate the presence of PrPTSE in exosomes isolated from the conditioned media of cell cultures infected with M1000, a mouse-adapted sCJD strain (52). In the context of disease diagnosis using body fluids, limitations exist as to the sample volume available for evaluation. In recent years, new reagents have become available for isolating exosomes from small volumes expected in such clinical applications. Two of these reagents, ExoQuickTM-TC and ExoQuickTM, have been repeatedly shown to successfully isolate exosomes from conditioned media and biological fluids for various downstream applications (78–82). By combining extracellular vesicle isolation using ExoQuickTM with PrPTSE amplification by saPMCA, we were able to demonstrate the presence of PrPTSE in plasma samples collected from preclinical and sick mice infected with Mo-vCJD, and we showed for the first time its association with blood-circulating EVs. Biochemical characterization of EV preparations obtained from plasma and conditioned media showed the presence of the exosomal marker Hsp70 and the absence of the Golgi marker GM130, thus demonstrating the efficiency of ExoQuickTM reagents to purify EV-containing exosomes. Moreover, processing of EVs by ultracentrifugation through a 30% sucrose cushion, coupled with saPMCA amplification, allowed the detection of PrPTSE in EVs that were confirmed to carry the exosomal marker Hsp70, further indicating the possible association of PrPTSE with these vesicles. There are three types of EVs released by the cell that differ in their biogenesis and that can be classified as follows: (i) apoptotic vesicles (50–5000 nm in diameter) released by apoptotic cells; (ii) exosomes (40–200 nm) released upon fusion of multivesicular bodies with the plasma membrane, and (iii) microvesicles (50–1000 nm) that directly bud from the plasma membrane (37, 48, 83). Because some of these vesicles overlap in size, it is difficult to separate them by the current isolation methods. In particular ExoQuick isolates EVs ranging in size from 17 to 200 nm. Nevertheless, the demonstration of the exosomal marker Hsp70 exclusively in the middle fraction, where exosomes fractionate following centrifugation in a 30% sucrose cushion, confirms the presence of these vesicles in our preparations. The fact that PrPTSE was also identified after saPMCA in the top and bottom fraction where other EVs have been detected by NTA suggests that it can associate with other classes of EVs, although this conclusion requires further investigation because, as mentioned above, the top and bottom fraction may contain lipoprotein-associated PrPTSE and PrPTSE aggregates, respectively, not associated with EVs. Interestingly, on the bases of the accumulated evidence that PrPC is present at the exosomal membrane (46–50, 53, 58, 84–87), this protein is being used to characterize exosomal preparations (88). Additionally, we demonstrated that EV isolation can be scaled down to volumes compatible with clinical applications. Two hundred microliters of plasma provided enough material to allow PrPTSE detection by saPMCA in murine samples. The challenge will be to detect PrPTSE in similar volumes of human plasma.
Western blotting analysis of plasma EVs, isolated with ExoQuickTM, revealed the presence of heavy and light chains of IgG in our samples. These findings are in line with various studies that showed association of immunoglobulin light chains (89) and various types and subtypes of heavy chains with exosomes isolated from tumors and body fluids (89–92). The presence of IgG in EVs isolated using ExoQuickTM hindered the detection of PrPTSE in the initial rounds of saPMCA by Western blotting due to a high nonspecific background signal. Serial dilution/amplification significantly reduced IgG, effectively eliminating the signal interference and revealing the presence of misfolded PrPTSE. Detection of PrPTSE in plasma EVs after just one round of saPMCA was achieved with a secondary antibody that does not recognize heavy and light chains of IgG under reduced conditions. However, this antibody was only used in our confirmatory assays due to practical reasons, i.e. high cost and low reading signal in Western blotting. Additional methods to remove IgG from plasma sample preparations are currently under evaluation; we expect that these approaches will reduce the number of rounds required to detect PrPTSE. The number of amplification rounds needed to identify PrPTSE varied between animals infected with the same TSE agent and between cell cultures infected with Mo-vCJD or Fukuoka-1. These findings suggest that variability exists between individual mice as far as the quantities of PrPTSE present in plasma at the terminal stages of disease as well as differences in conversion efficiencies between TSE strains. However, we cannot exclude differences in the PrPTSE replication capacity of our cell models.
In summary, our experiments provide a valuable foundation for the development of new diagnostics and potential targets for TSE treatment. Moreover, our data provide evidence that a fraction of blood-circulating PrPTSE co-localizes with exosomes. This finding opens new avenues for further TSE research. Because exosomes are known to not only participate in cell to cell communication processes (34, 58, 59, 93), but to also cross the blood-brain barrier (94, 95), association of PrPTSE with exosomes may serve as a platform for TSE spread from the periphery to the CNS (47, 56, 58). Additionally, we are further characterizing PrPTSE-bearing microvesicles with the goal of identifying the cellular origin of this protein in blood. This information may advance the development of new strategies to ensure the safety of blood- and plasma-derived products.
Acknowledgments
We thank Dr. Paul Brown for providing a Fukuoka-infected mouse brain and Dr. Moira Bruce for providing a Mo-vCJD-infected mouse brain for the infectivity studies. We also thank Donna Sobieski for editorial assistance, Anton Cervenak for technical support, and the American National Red Cross vivarium staff for providing excellent animal care. We are grateful to Dr. Andrew Hill for critical reading of the manuscript and for helpful discussions.
This study was supported by research funding from the American National Red Cross (to L. C.) and the Fondation Alliance BioSecure (to P. S.).
- TSE
- transmissible spongiform encephalopathy
- vCJD
- variant Creutzfeldt-Jakob disease
- PrPTSE
- TSE-associated prion protein
- PrPC
- cellular prion protein
- EV
- extracellular vesicle
- BSE
- bovine spongiform encephalopathy
- MoBH
- mouse brain homogenate
- sCJD
- sporadic CJD
- PMCA
- protein misfolding cyclic amplification
- CWD
- chronic wasting disease
- saPMCA
- serial automated PMCA
- wpi
- weeks post-inoculation
- EVP
- EV protein
- NTA
- nanoparticle tracking analysis
- FVB
- FVB/NCr
- C57BL
- C57BL/6
- WT
- wild-type.
REFERENCES
- 1. Brandel J. P., Heath C. A., Head M. W., Levavasseur E., Knight R., Laplanche J. L., Langeveld J. P., Ironside J. W., Hauw J. J., Mackenzie J., Alpérovitch A., Will R. G., Haïk S. (2009) Variant Creutzfeldt-Jakob disease in France and the United Kingdom: evidence for the same agent strain. Ann. Neurol. 65, 249–256 [DOI] [PubMed] [Google Scholar]
- 2. Hilton D. A., Ghani A. C., Conyers L., Edwards P., McCardle L., Ritchie D., Penney M., Hegazy D., Ironside J. W. (2004) Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J. Pathol. 203, 733–739 [DOI] [PubMed] [Google Scholar]
- 3. Gill O. N., Spencer Y., Richard-Loendt A., Kelly C., Dabaghian R., Boyes L., Linehan J., Simmons M., Webb P., Bellerby P., Andrews N., Hilton D. A., Ironside J. W., Beck J., Poulter M., Mead S., Brandner S. (2013) Prevalent abnormal prion protein in human appendixes after bovine spongiform encephalopathy epizootic: large scale survey. BMJ 347, f5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Knight R. (2010) The risk of transmitting prion disease by blood or plasma products. Transfus. Apher. Sci. 43, 387–391 [DOI] [PubMed] [Google Scholar]
- 5. Peden A., McCardle L., Head M. W., Love S., Ward H. J., Cousens S. N., Keeling D. M., Millar C. M., Hill F. G., Ironside J. W. (2010) Variant CJD infection in the spleen of a neurologically asymptomatic UK adult patient with haemophilia. Haemophilia 16, 296–304 [DOI] [PubMed] [Google Scholar]
- 6. Edgeworth J. A., Farmer M., Sicilia A., Tavares P., Beck J., Campbell T., Lowe J., Mead S., Rudge P., Collinge J., Jackson G. S. (2011) Detection of prion infection in variant Creutzfeldt-Jakob disease: a blood-based assay. Lancet 377, 487–493 [DOI] [PubMed] [Google Scholar]
- 7. Jackson G. S., Burk-Rafel J., Edgeworth J. A., Sicilia A., Abdilahi S., Korteweg J., Mackey J., Thomas C., Wang G., Mead S., Collinge J. (2014) A highly specific blood test for vCJD. Blood 123, 452–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jackson G. S., Burk-Rafel J., Edgeworth J. A., Sicilia A., Abdilahi S., Korteweg J., Mackey J., Thomas C., Wang G., Schott J. M., Mummery C., Chinnery P. F., Mead S., Collinge J. (2014) Population screening for variant Creutzfeldt-Jakob disease using a novel blood test: diagnostic accuracy and feasibility study. JAMA Neurology 71, 421–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wadsworth J. D., Dalmau-Mena I., Joiner S., Linehan J. M., O'Malley C., Powell C., Brandner S., Asante E. A., Ironside J. W., Hilton D. A., Collinge J. (2011) Effect of fixation on brain and lymphoreticular vCJD prions and bioassay of key positive specimens from a retrospective vCJD prevalence study. J. Pathol. 223, 511–518 [DOI] [PubMed] [Google Scholar]
- 10. de Marco M. F., Linehan J., Gill O. N., Clewley J. P., Brandner S. (2010) Large-scale immunohistochemical examination for lymphoreticular prion protein in tonsil specimens collected in Britain. J. Pathol. 222, 380–387 [DOI] [PubMed] [Google Scholar]
- 11. Douet J. Y., Zafar S., Perret-Liaudet A., Lacroux C., Lugan S., Aron N., Cassard H., Ponto C., Corbière F., Torres J. M., Zerr I., Andreoletti O. (2014) Detection of infectivity in blood of persons with variant and sporadic Creutzfeldt-Jakob disease. Emerg. Infect. Dis. 20, 114–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rubenstein R., Chang B. (2013) Re-assessment of PrP(Sc) distribution in sporadic and variant CJD. PLoS One 8, e66352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cervenakova L., Brown P., Soukharev S., Yakovleva O., Diringer H., Saenko E. L., Drohan W. N. (2003) Failure of immunocompetitive capillary electrophoresis assay to detect disease-specific prion protein in buffy coat from humans and chimpanzees with Creutzfeldt-Jakob disease. Electrophoresis 24, 853–859 [DOI] [PubMed] [Google Scholar]
- 14. Dorsey K., Zou S., Schonberger L. B., Sullivan M., Kessler D., Notari E., 4th, Fang C. T., Dodd R. Y. (2009) Lack of evidence of transfusion transmission of Creutzfeldt-Jakob disease in a US surveillance study. Transfusion 49, 977–984 [DOI] [PubMed] [Google Scholar]
- 15. Brown P., Cervenáková L., Diringer H. (2001) Blood infectivity and the prospects for a diagnostic screening test in Creutzfeldt-Jakob disease. J. Lab. Clin. Med. 137, 5–13 [DOI] [PubMed] [Google Scholar]
- 16. Cervenakova L., Yakovleva O., McKenzie C., Kolchinsky S., McShane L., Drohan W. N., Brown P. (2003) Similar levels of infectivity in the blood of mice infected with human-derived vCJD and GSS strains of transmissible spongiform encephalopathy. Transfusion 43, 1687–1694 [DOI] [PubMed] [Google Scholar]
- 17. Andréoletti O., Litaise C., Simmons H., Corbière F., Lugan S., Costes P., Schelcher F., Vilette D., Grassi J., Lacroux C. (2012) Highly efficient prion transmission by blood transfusion. PLoS Pathog. 8, e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCutcheon S., Alejo Blanco A. R., Houston E. F., de Wolf C., Tan B. C., Smith A., Groschup M. H., Hunter N., Hornsey V. S., MacGregor I. R., Prowse C. V., Turner M., Manson J. C. (2011) All clinically-relevant blood components transmit prion disease following a single blood transfusion: a sheep model of vCJD. PLoS One 6, e23169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mathiason C. K., Powers J. G., Dahmes S. J., Osborn D. A., Miller K. V., Warren R. J., Mason G. L., Hays S. A., Hayes-Klug J., Seelig D. M., Wild M. A., Wolfe L. L., Spraker T. R., Miller M. W., Sigurdson C. J., Telling G. C., Hoover E. A. (2006) Infectious prions in the saliva and blood of deer with chronic wasting disease. Science 314, 133–136 [DOI] [PubMed] [Google Scholar]
- 20. Hunter N., Foster J., Chong A., McCutcheon S., Parnham D., Eaton S., MacKenzie C., Houston F. (2002) Transmission of prion diseases by blood transfusion. J. Gen. Virol. 83, 2897–2905 [DOI] [PubMed] [Google Scholar]
- 21. Tattum M. H., Jones S., Pal S., Collinge J., Jackson G. S. (2010) Discrimination between prion-infected and normal blood samples by protein misfolding cyclic amplification. Transfusion 50, 996–1002 [DOI] [PubMed] [Google Scholar]
- 22. Terry L. A., Howells L., Hawthorn J., Edwards J. C., Moore S. J., Bellworthy S. J., Simmons H., Lizano S., Estey L., Leathers V., Everest S. J. (2009) Detection of PrPsc in blood from sheep infected with the scrapie and bovine spongiform encephalopathy agents. J. Virol. 83, 12552–12558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Elder A. M., Henderson D. M., Nalls A. V., Wilham J. M., Caughey B. W., Hoover E. A., Kincaid A. E., Bartz J. C., Mathiason C. K. (2013) In vitro detection of prionemia in TSE-infected cervids and hamsters. PLoS One 8, e80203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Castilla J., Saá P., Soto C. (2005) Detection of prions in blood. Nat. Med. 11, 982–985 [DOI] [PubMed] [Google Scholar]
- 25. Saá P., Castilla J., Soto C. (2006) Presymptomatic detection of prions in blood. Science 313, 92–94 [DOI] [PubMed] [Google Scholar]
- 26. Thorne L., Terry L. A. (2008) In vitro amplification of PrPSc derived from the brain and blood of sheep infected with scrapie. J. Gen. Virol. 89, 3177–3184 [DOI] [PubMed] [Google Scholar]
- 27. Vascellari S., Orrù C. D., Hughson A. G., King D., Barron R., Wilham J. M., Baron G. S., Race B., Pani A., Caughey B. (2012) Prion seeding activities of mouse scrapie strains with divergent PrPSc protease sensitivities and amyloid plaque content using RT-QuIC and eQuIC. PLoS One 7, e48969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rubenstein R., Chang B., Gray P., Piltch M., Bulgin M. S., Sorensen-Melson S., Miller M. W. (2010) A novel method for preclinical detection of PrPSc in blood. J. Gen. Virol. 91, 1883–1892 [DOI] [PubMed] [Google Scholar]
- 29. Orrú C. D., Wilham J. M., Raymond L. D., Kuhn F., Schroeder B., Raeber A. J., Caughey B. (2011) Prion disease blood test using immunoprecipitation and improved quaking-induced conversion. mBio 2, e00078–e00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bannach O., Birkmann E., Reinartz E., Jaeger K. E., Langeveld J. P., Rohwer R. G., Gregori L., Terry L. A., Willbold D., Riesner D. (2012) Detection of prion protein particles in blood plasma of scrapie infected sheep. PLoS One 7, e36620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pan B.-T., Johnstone R. M. (1983) Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: Selective externalization of the receptor. Cell 33, 967–978 [DOI] [PubMed] [Google Scholar]
- 32. Pan B. T., Johnstone R. (1984) Selective externalization of the transferrin receptor by sheep reticulocytes in vitro. Response to ligands and inhibitors of endocytosis. J. Biol. Chem. 259, 9776–9782 [PubMed] [Google Scholar]
- 33. Johnstone R. M., Adam M., Hammond J. R., Orr L., Turbide C. (1987) Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 262, 9412–9420 [PubMed] [Google Scholar]
- 34. Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J. J., Lötvall J. O. (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 9, 654–659 [DOI] [PubMed] [Google Scholar]
- 35. Zitvogel L., Regnault A., Lozier A., Wolfers J., Flament C., Tenza D., Ricciardi-Castagnoli P., Raposo G., Amigorena S. (1998) Eradication of established murine tumors using a novel cell-free vaccine: dendritic cell-derived exosomes. Nat. Med. 4, 594–600 [DOI] [PubMed] [Google Scholar]
- 36. Raposo G., Nijman H. W., Stoorvogel W., Liejendekker R., Harding C. V., Melief C. J., Geuze H. J. (1996) B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 183, 1161–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vlassov A. V., Magdaleno S., Setterquist R., Conrad R. (2012) Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948 [DOI] [PubMed] [Google Scholar]
- 38. Nguyen D. G., Booth A., Gould S. J., Hildreth J. E. (2003) Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J. Biol. Chem. 278, 52347–52354 [DOI] [PubMed] [Google Scholar]
- 39. Gould S. J., Booth A. M., Hildreth J. E. (2003) The Trojan exosome hypothesis. Proc. Natl. Acad. Sci. U.S.A. 100, 10592–10597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider A., Simons M. (2013) Exosomes: vesicular carriers for intercellular communication in neurodegenerative disorders. Cell Tissue Res. 352, 33–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tasaki M., Ueda M., Ochiai S., Tanabe Y., Murata S., Misumi Y., Su Y., Sun X., Shinriki S., Jono H., Shono M., Obayashi K., Ando Y. (2010) Transmission of circulating cell-free AA amyloid oligomers in exosomes vectors via a prion-like mechanism. Biochem. Biophys. Res. Commun. 400, 559–562 [DOI] [PubMed] [Google Scholar]
- 42. Vingtdeux V., Sergeant N., Buee L. (2012) Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer's disease. Front. Physiol. 3, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rajendran L., Honsho M., Zahn T. R., Keller P., Geiger K. D., Verkade P., Simons K. (2006) Alzheimer's disease β-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 103, 11172–11177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Surgucheva I., Sharov V. S., Surguchov A. (2012) γ-Synuclein: seeding of α-synuclein aggregation and transmission between cells. Biochemistry 51, 4743–4754 [DOI] [PubMed] [Google Scholar]
- 45. Saman S., Kim W., Raya M., Visnick Y., Miro S., Saman S., Jackson B., McKee A. C., Alvarez V. E., Lee N. C., Hall G. F. (2012) Exosome-associated Tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J. Biol. Chem. 287, 3842–3849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ecroyd H., Sarradin P., Dacheux J.-L., Gatti J.-L. (2004) Compartmentalization of prion isoforms within the reproductive tract of the ram. Biol. Reprod. 71, 993–1001 [DOI] [PubMed] [Google Scholar]
- 47. Fevrier B., Vilette D., Archer F., Loew D., Faigle W., Vidal M., Laude H., Raposo G. (2004) Cells release prions in association with exosomes. Proc. Natl. Acad. Sci. U.S.A. 101, 9683–9688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Robertson C., Booth S. A., Beniac D. R., Coulthart M. B., Booth T. F., McNicol A. (2006) Cellular prion protein is released on exosomes from activated platelets. Blood 107, 3907–3911 [DOI] [PubMed] [Google Scholar]
- 49. Vella L. J., Sharples R. A., Lawson V. A., Masters C. L., Cappai R., Hill A. F. (2007) Packaging of prions into exosomes is associated with a novel pathway of PrP processing. J. Pathol. 211, 582–590 [DOI] [PubMed] [Google Scholar]
- 50. Alais S., Simoes S., Baas D., Lehmann S., Raposo G., Darlix J. L., Leblanc P. (2008) Mouse neuroblastoma cells release prion infectivity associated with exosomal vesicles. Biol. Cell 100, 603–615 [DOI] [PubMed] [Google Scholar]
- 51. Castro-Seoane R., Hummerich H., Sweeting T., Tattum M. H., Linehan J. M., Fernandez de Marco M., Brandner S., Collinge J., Klöhn P.-C. (2012) Plasmacytoid dendritic cells sequester high prion titres at early stages of prion infection. PLoS Pathog. 8, e1002538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coleman B. M., Hanssen E., Lawson V. A., Hill A. F. (2012) Prion-infected cells regulate the release of exosomes with distinct ultrastructural features. FASEB J. 26, 4160–4173 [DOI] [PubMed] [Google Scholar]
- 53. Wik L., Klingeborn M., Willander H., Linne T. (2012) Separate mechanisms act concurrently to shed and release the prion protein from the cell. Prion 6, 498–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Veith N. M., Plattner H., Stuermer C. A., Schulz-Schaeffer W. J., Bürkle A. (2009) Immunolocalisation of PrPSc in scrapie-infected N2a mouse neuroblastoma cells by light and electron microscopy. Eur. J. Cell Biol. 88, 45–63 [DOI] [PubMed] [Google Scholar]
- 55. Mattei V., Barenco M. G., Tasciotti V., Garofalo T., Longo A., Boller K., Löwer J., Misasi R., Montrasio F., Sorice M. (2009) Paracrine diffusion of PrPC and propagation of prion infectivity by plasma membrane-derived microvesicles. PLoS One 4, e5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Simák J., Holada K., D'Agnillo F., Janota J., Vostal J. G. (2002) Cellular prion protein is expressed on endothelial cells and is released during apoptosis on membrane microparticles found in human plasma. Transfusion 42, 334–342 [DOI] [PubMed] [Google Scholar]
- 57. Brouckova A., Holada K. (2009) Cellular prion protein in blood platelets associates with both lipid rafts and the cytoskeleton. Thromb. Haemost. 102, 966–974 [DOI] [PubMed] [Google Scholar]
- 58. Février B., Vilette D., Laude H., Raposo G. (2005) Exosomes: a bubble ride for prions? Traffic 6, 10–17 [DOI] [PubMed] [Google Scholar]
- 59. Skog J., Würdinger T., van Rijn S., Meijer D. H., Gainche L., Sena-Esteves M., Curry W. T., Jr., Carter B. S., Krichevsky A. M., Breakefield X. O. (2008) Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 10, 1470–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kalra H., Adda C. G., Liem M., Ang C.-S., Mechler A., Simpson R. J., Hulett M. D., Mathivanan S. (2013) Comparative proteomics evaluation of plasma exosome isolation techniques and assessment of the stability of exosomes in normal human blood plasma. Proteomics 13, 3354–3364 [DOI] [PubMed] [Google Scholar]
- 61. Cheng L., Sharples R. A., Scicluna B. J., Hill A. F. (2014) Exosomes provide a protective and enriched source of miRNA for biomarker profiling compared to intracellular and cell-free blood. J. Extracell. Vesicles 3, 23743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brown P., Cervenáková L., McShane L. M., Barber P., Rubenstein R., Drohan W. N. (1999) Further studies of blood infectivity in an experimental model of transmissible spongiform encephalopathy, with an explanation of why blood components do not transmit Creutzfeldt-Jakob disease in humans. Transfusion 39, 1169–1178 [DOI] [PubMed] [Google Scholar]
- 63. Brown P., Rohwer R. G., Dunstan B. C., MacAuley C., Gajdusek D. C., Drohan W. N. (1998) The distribution of infectivity in blood components and plasma derivatives in experimental models of transmissible spongiform encephalopathy. Transfusion 38, 810–816 [DOI] [PubMed] [Google Scholar]
- 64. Reed L. J., Muench H. (1938) A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27, 493–497 [Google Scholar]
- 65. Cervenakova L., Akimov S., Vasilyeva I., Yakovleva O., McKenzie C., Cervenak J., Piccardo P., Asher D. M. (2011) Fukuoka-1 strain of transmissible spongiform encephalopathy agent infects murine bone marrow-derived cells with features of mesenchymal stem cells. Transfusion 51, 1755–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Akimov S., Yakovleva O., Vasilyeva I., McKenzie C., Cervenakova L. (2008) Persistent propagation of variant Creutzfeldt-Jakob disease agent in murine spleen stromal cell culture with features of mesenchymal stem cells. J. Virol. 82, 10959–10962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Akimov S., Vasilyeva I., Yakovleva O., McKenzie C., Cervenakova L. (2009) Murine bone marrow stromal cell culture with features of mesenchymal stem cells susceptible to mouse-adapted human TSE agent, Fukuoka-1. Folia Neuropathol. 47, 205–214 [PubMed] [Google Scholar]
- 68. Thery C., Clayton A., Amogorena S., Raposo G. (2006) Current Protocols in Cell Biology, pp. 3.22.1–3.22.29, John Wiley & Sons, Inc., New York: [DOI] [PubMed] [Google Scholar]
- 69. Castilla J., Saá P., Hetz C., Soto C. (2005) In vitro generation of infectious scrapie prions. Cell 121, 195–206 [DOI] [PubMed] [Google Scholar]
- 70. Saá P., Castilla J., Soto C. (2006) Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J. Biol. Chem. 281, 35245–35252 [DOI] [PubMed] [Google Scholar]
- 71. Fujihara A., Atarashi R., Fuse T., Ubagai K., Nakagaki T., Yamaguchi N., Ishibashi D., Katamine S., Nishida N. (2009) Hyperefficient PrP Sc amplification of mouse-adapted BSE and scrapie strain by protein misfolding cyclic amplification technique. FEBS J. 276, 2841–2848 [DOI] [PubMed] [Google Scholar]
- 72. Safar J. G., Wille H., Geschwind M. D., Deering C., Latawiec D., Serban A., King D. J., Legname G., Weisgraber K. H., Mahley R. W., Miller B. L., Dearmond S. J., Prusiner S. B. (2006) Human prions and plasma lipoproteins. Proc. Natl. Acad. Sci. U.S.A. 103, 11312–11317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Dragovic R. A., Gardiner C., Brooks A. S., Tannetta D. S., Ferguson D. J., Hole P., Carr B., Redman C. W., Harris A. L., Dobson P. J., Harrison P., Sargent I. L. (2011) Sizing and phenotyping of cellular vesicles using nanoparticle tracking analysis. Nanomedicine 7, 780–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rubenstein R., Chang B., Gray P., Piltch M., Bulgin M. S., Sorensen-Melson S., Miller M. W. (2011) Prion disease detection, PMCA kinetics, and IgG in urine from sheep naturally/experimentally infected with scrapie and deer with preclinical/clinical chronic wasting disease. J. Virol. 85, 9031–9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Atarashi R., Satoh K., Sano K., Fuse T., Yamaguchi N., Ishibashi D., Matsubara T., Nakagaki T., Yamanaka H., Shirabe S., Yamada M., Mizusawa H., Kitamoto T., Klug G., McGlade A., Collins S. J., Nishida N. (2011) Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat. Med. 17, 175–178 [DOI] [PubMed] [Google Scholar]
- 76. Dabaghian R., Zerr I., Heinemann U., Zanusso G. (2008) Detection of proteinase K-resistant proteins in the urine of patients with Creutzfeldt-Jakob and other neurodegenerative diseases. Prion 2, 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Gonzalez-Romero D., Barria M. A., Leon P., Morales R., Soto C. (2008) Detection of infectious prions in urine. FEBS Lett. 582, 3161–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tadokoro H., Umezu T., Ohyashiki K., Hirano T., Ohyashiki J. H. (2013) Exosomes derived from hypoxic leukemia cells enhance tube formation in endothelial cells. J Biol. Chem. 288, 34343–34351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Huang X., Yuan T., Tschannen M., Sun Z., Jacob H., Du M., Liang M., Dittmar R. L., Liu Y., Liang M., Kohli M., Thibodeau S. N., Boardman L., Wang L. (2013) Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genomics 14, 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Taylor D., Zacharias W., Gercel-Taylor C. (2011) in Serum/Plasma Proteomics (Simpson R. J., Greening D. W., eds) pp. 235–246, Humana Press Inc., Totowa, NJ [Google Scholar]
- 81. Epple L. M., Griffiths S. G., Dechkovskaia A. M., Dusto N. L., White J., Ouellette R. J., Anchordoquy T. J., Bemis L. T., Graner M. W. (2012) Medulloblastoma exosome proteomics yield functional roles for extracellular vesicles. PLoS ONE 7, e42064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Haney M. J., Suresh P., Zhao Y., Kanmogne G. D., Kadiu I., Sokolsky-Papkov M., Klyachko N. L., Mosley R. L., Kabanov A. V., Gendelman H. E., Batrakova E. V. (2012) Blood-borne macrophage–neural cell interactions hitchhike on endosome networks for cell-based nanozyme brain delivery. Nanomedicine 7, 815–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Cocucci E., Racchetti G., Meldolesi J. (2009) Shedding microvesicles: artefacts no more. Trends Cell Biol. 19, 43–51 [DOI] [PubMed] [Google Scholar]
- 84. Porto-Carreiro I., Février B., Paquet S., Vilette D., Raposo G. (2005) Prions and exosomes: from PrPc trafficking to PrPsc propagation. Blood Cells Mol. Dis. 35, 143–148 [DOI] [PubMed] [Google Scholar]
- 85. Vella L. J., Sharples R. A., Nisbet R. M., Cappai R., Hill A. F. (2008) The role of exosomes in the processing of proteins associated with neurodegenerative diseases. Eur. Biophys. J. 37, 323–332 [DOI] [PubMed] [Google Scholar]
- 86. Vella L. J., Greenwood D. L., Cappai R., Scheerlinck J. P., Hill A. F. (2008) Enrichment of prion protein in exosomes derived from ovine cerebral spinal fluid. Vet. Immunol. Immunopathol. 124, 385–393 [DOI] [PubMed] [Google Scholar]
- 87. Ritchie A. J., Crawford D. M., Ferguson D. J., Burthem J., Roberts D. J. (2013) Normal prion protein is expressed on exosomes isolated from human plasma. Br. J. Haematol. 163, 678–680 [DOI] [PubMed] [Google Scholar]
- 88. Silva C. J., Dynin I., Erickson M. L., Requena J. R., Balachandran A., Hui C., Onisko B. C., Carter J. M. (2013) Oxidation of methionine 216 in sheep and elk prion protein is highly dependent upon the amino acid at position 218 but is not important for prion propagation. Biochemistry 52, 2139–2147 [DOI] [PubMed] [Google Scholar]
- 89. Gonzalez-Begne M., Lu B., Han X., Hagen F. K., Hand A. R., Melvin J. E., Yates J. R. (2009) Proteomic analysis of human parotid gland exosomes by multidimensional protein identification technology (MudPIT). J. Proteome Res. 8, 1304–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Admyre C., Johansson S. M., Qazi K. R., Filén J.-J., Lahesmaa R., Norman M., Neve E. P., Scheynius A., Gabrielsson S. (2007) Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 179, 1969–1978 [DOI] [PubMed] [Google Scholar]
- 91. Mathivanan S., Lim J. W., Tauro B. J., Ji H., Moritz R. L., Simpson R. J. (2010) Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell. Proteomics 9, 197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bard M. P., Hegmans J. P., Hemmes A., Luider T. M., Willemsen R., Severijnen L.-A., van Meerbeeck J. P., Burgers S. A., Hoogsteden H. C., Lambrecht B. N. (2004) Proteomic analysis of exosomes isolated from human malignant pleural effusions. Am. J. Respir. Cell Mol. Biol. 31, 114–121 [DOI] [PubMed] [Google Scholar]
- 93. Stoorvogel W., Kleijmeer M. J., Geuze H. J., Raposo G. (2002) The biogenesis and functions of exosomes. Traffic 3, 321–330 [DOI] [PubMed] [Google Scholar]
- 94. Kalani A., Tyagi A., Tyagi N. (2014) Exosomes: mediators of neurodegeneration, neuroprotection and therapeutics. Mol. Neurobiol. 49, 590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sun D., Zhuang X., Xiang X., Liu Y., Zhang S., Liu C., Barnes S., Grizzle W., Miller D., Zhang H.-G. (2010) A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol. Ther. 18, 1606–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]