

Background: C-terminal SPAK fragments are found in kidney medulla.
Results: We identified Dnpep as the protease responsible for SPAK cleavage, identified the sites of cleavage, and showed unusual preference for α helices.
Conclusion: C-terminal SPAK fragments originate from Dnpep-mediated proteolytic cleavage.
Significance: SPAK and its cleavage are significant for the regulation of renal Na+ reabsorption and control of blood pressure.
Keywords: Metalloprotease, Protein Kinase, Renal Physiology, Signal Transduction, Sodium Transport
Abstract
The Ste20-related kinase SPAK regulates sodium, potassium, and chloride transport in a variety of tissues. Recently, SPAK fragments, which lack the catalytic domain and are inhibitory to Na+ transporters, have been detected in kidney. It has been hypothesized that the fragments originate from alternative translation start sites, but their precise origin is unknown. Here, we demonstrate that kidney lysate possesses proteolytic cleavage activity toward SPAK. Ion exchange and size exclusion chromatography combined with mass spectrometry identified the protease as aspartyl aminopeptidase. The presence of the protease was verified in the active fractions, and recombinant aspartyl aminopeptidase recapitulated the cleavage pattern observed with kidney lysate. Identification of the sites of cleavage by mass spectrometry allowed us to test the function of the smaller fragments and demonstrate their inhibitory action toward the Na+-K+-2Cl− cotransporter, NKCC2.
Introduction
Through reabsorption and secretion mechanisms, the kidney finely regulates body salt balance and thereby extracellular volume and blood pressure. In recent years a kinase cascade involving the Ste20p-like kinases, SPAK2 (STK39) and OSR1 (OxSR1), has emerged as an important regulator of salt transport in the kidney (1, 2). The kinases regulate two transport mechanisms involved in Na+ reabsorption: the Na-K-2Cl cotransporter, NKCC2, expressed in the thick ascending limb of Henle (TAL), and the Na-Cl cotransporter, NCC, expressed in the distal convoluted tubule. SPAK possesses a short N-terminal region rich in the proline/alanine residues (PAPA box) followed by a core kinase domain and a relatively large C-terminal regulatory domain. OSR1 displays the same structure while lacking the PAPA box (Fig. 1A). Examination of fully sequenced genomes revealed that SPAK originated from OSR1 gene duplication during vertebrate evolution (2, 3). In heterologous expression systems, the two kinases behave similarly, anchoring themselves to the N-terminal tail of Na+-dependent cotransporters in order to phosphorylate and stimulate their activities. This regulation has been shown for NKCC1 (4–6), NKCC2 (7), and Na-Cl cotransporter (8, 9). Kinase binding occurs between a conserved protein fold, which is located at the C-terminal end of SPAK and OSR1, and a conserved RFX(V/I) peptide sequence located in the N-terminal tail of the cotransporters (10, 11). Although in sensory neurons the two kinases seem to fulfill a redundant function (12) based on the phenotypes of SPAK and kidney-specific OSR1 knock-out mice, the two kinases in kidney seem to have very distinct roles. Indeed, although the SPAK knock-out mouse reveals a primary role for SPAK in regulating the Na-Cl cotransporter in the distal convoluted tubule (13–16), the kidney-specific OSR1 knock-out mouse displays a phenotype that is consistent with disruption of NKCC2 function in the TAL (17). An intriguing observation from the SPAK knock-out mouse is that NKCC2 in the TAL is hyperphosphorylated (13, 14, 16), which would be inconsistent if the kinase was phosphorylating this cotransporter under normal conditions. In the kidney medulla several smaller SPAK fragments are observed by Western blot analysis, and these fragments have been postulated to act as negative regulators of OSR1 function in the thick ascending limb of Henle (14, 16, 18). Indeed, if these fragments were to inhibit OSR1 function, their absence in the knock-out would lead to increased OSR1 and hyperphosphorylation of NKCC2. Consistent with this hypothesis is the fact that no hyperphosphorylation of NKCC2 was observed in a knock-in mouse model where expression of SPAK and its fragments was left untouched, but the activity of the kinase was eliminated through a single amino acid substitution within the catalytic domain (15). Although a shorter transcript, referred to as kidney-specific or KS-SPAK, has been identified in mouse kidney (14), there is no evidence based on analysis of the human genome that such a transcript exists in non-rodent species. Thus, we considered alternative mechanisms for the production of the fragments and considered the possibility that SPAK is modified by proteolytic cleavage in the kidney.
FIGURE 1.
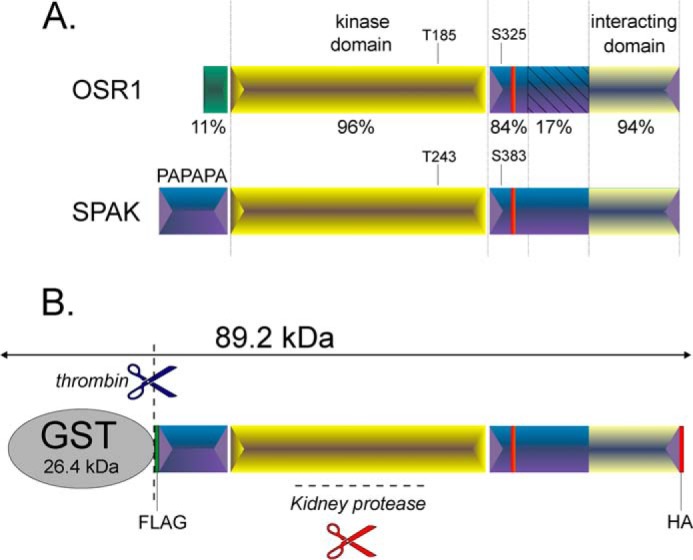
Schematic representation of OSR1 and SPAK kinases. A, the two kinases are characterized by a short and distinct N-terminal region (11% conservations) preceding the highly conserved catalytic domain (96% conservation) and a C-terminal domain divided into 3 regions of various homology. The first region is 84% conserved and contains a key regulatory phosphoserine. The second region lacks homology (17% conservation), and the third region forms a highly conserved domain (94%) that interacts with the kinase substrates. B, engineered SPAK fusion protein. SPAK fusion protein is composed of the full-length SPAK and some epitope tags, such as GST, FLAG (DYKDDDDK), and HA (YPYDVPDYA). The fusion protein is larger than the full-length SPAK and can be cleaved specifically by thrombin. Thrombin cleavage occurs downstream of GST, allowing the release of full-length SPAK as positive control. Note that GST tag is used for fusion protein purification, whereas FLAG and HA epitopes can be used for Western blotting purposes.
EXPERIMENTAL PROCEDURES
Animals
All experiments involving animals were approved by the Vanderbilt Animal Care and Use Committee (IACUC). Organs were collected from in-house-bred C57BL/6J mice. Mice were anesthetized with isoflurane (2–5%) and decapitated under anesthesia. Organs were removed and flash-frozen in liquid nitrogen before use. Two kidneys were obtained from one rabbit that underwent a thoracotomy and heart removal. The rabbit was sedated with ketamine + xylazine (25 + 5 mg/kg), and one ear was cannulated with a 23-gauge butterfly needle to establish intravenous access. The rabbit was given a single dose of heparin (1000 USP units) and deeply anesthetized with pentobarbital (40–60 mg/kg). A midline abdominal incision was made followed by a complete midline thoracotomy to fully expose and remove the heart. After exsanguination, the kidneys were collected, dissected, and flash-frozen in liquid nitrogen. Xenopus laevis frogs were anesthetized with tricaine (1.7 g/liter) buffered with NaHCO3 (3.4 g/liter). Ovarian lobes were surgically externalized and removed.
Human Tissues
With Institutional Review Board approval, renal biopsies from patients were obtained from CHTN (Cooperative Human Tissue Network), Western Division at Vanderbilt University. The samples consisted of liquid nitrogen frozen normal kidney cortex and medulla samples of weights ranging from 0.2–1 g.
Tissue Lysates
Tissue samples were homogenized on ice in 500 μl of lysis buffer (125 mm NaCl, 10% glycerol, 1 mm EGTA, 1 mm EDTA, 1 mm PMSF, 10 μm leupeptin, 4 μm aprotinin, 10 μm pepstatin, 1% Triton X-100, 0.5% SDS, 20 mm HEPES, pH 7.4) per 150 mg of tissue and rotated at 4 °C for 20 min. Soluble lysates were then collected from homogenates by centrifugation at 13,000 × g for 20 min at 4 °C.
Proteolytic Reactions
Typical reactions consisted of the following: 2 μl of kidney lysate (35 μg/μl) or the corresponding volume of other tissue lysates (70 μg) were incubated with 2 μl of SPAK fusion protein (5 μg/μl) in phosphate buffer (140 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.8 mm KH2PO4, pH 7.3) or in HEPES buffer (50 mm Hepes, 140 mm NaCl, pH 7.4) or in lysis buffer in a total volume of 40 μl for 1 h at 37 °C. When required, kidney lysate was first pretreated with different inhibitors or vehicle before reaction with SPAK fusion protein. In the latter case, 2 μl of kidney lysate was first preincubated for 30 min at 37 °C with 4 μl of a 10-fold concentrated inhibitor solution (or corresponding vehicle) in 32 μl of phosphate buffer before the addition of 2 μl of SPAK fusion protein and subsequent reaction for 1 h at 37 °C. To study the effect of temperature on cleavage, two separate protocols were used. For temperatures of 4, 21, and 37 °C, 2 μl of kidney lysate (35 μg/μl) in phosphate buffer was preincubated for 15 min at these temperatures before the addition of SPAK fusion protein (5 μg/μl) and 30 min reactions at same temperatures. For temperatures higher than 37 °C, 2 μl of kidney lysate was treated at 45, 55, 65, 75 °C for 15 min and cooled down to 4 °C before the reaction with SPAK fusion protein (5 μg/μl) at 37 °C for 30 min.
SDS-PAGE, NativePAGE, and Western Blot Analysis
Protein samples were denatured in 2× loading buffer at 75 °C for 15 min and separated on either regular 10% SDS-polyacrylamide gels or 3–12% SDS-polyacrylamide gradient gels or pre-casted Novex NativePAGE 3–12% Bis-Tris gels (Invitrogen). The separated proteins were electroblotted onto 0.45-μm polyvinylidene fluoride membranes (ThermoFisher Scientific) through a semi-dry process, and membranes were incubated for 2 h at room temperature in blocking solution (5% nonfat milk in TBST, i.e. 150 mm NaCl, 10 mm Tris, 0.5% Tween 20). Incubation of primary antibody (1:1000) was performed overnight at 4 °C. Antibodies were rabbit anti-mouse C-terminal SPAK polyclonal antibody (residues 424–556 (10)), anti-mouse OSR1 polyclonal antibody (residues 363–436 (19)), mouse monoclonal anti-FLAG (clone M2, Sigma), HRP-conjugated rat anti-HA monoclonal antibody (clone 3F10, Roche Applied Science), and anti-rabbit aspartyl aminopeptidase (Dnpep) monoclonal antibody (Abcam, Cambridge, MA). Membranes were washed in TBST for 3 h with frequent changes and when appropriate incubated with horseradish peroxidase-conjugated secondary antibody in blocking solution (1:5000) for 1 h at room temperature and washed again for 2 h in TBST. Protein bands were visualized by chemiluminescence (ECL Plus, Amersham Biosciences).
SPAK Fusion Protein Purification
The cDNA encoding mouse SPAK was reported by Piechotta et al. (10). The cDNA was engineered with a FLAG tag and HA tag at 5′ and 3′ ends, respectively (see Fig. 1) and inserted into pGEX4T. The final construct was transformed into BL21 Escherichia coli strain (Invitrogen). Transformants were then cultured in 2XYT medium (16 g/liter BactoTM Tryptone (BD Biosciences), 10 g/liter BactoTM yeast extract, 5 g/liter NaCl) supplemented with 4 mm glucose and 0.1 mg/ml ampicillin in a shaker incubator at 37 °C. When absorbance A600 nm reached 0.5, protein expression was induced by adding 100 μm isopropyl-β-d-thiogalactopyranoside (IPTG, RPI Corp., Chicago, IL), and incubation was continued for 4 h at 30 °C. After incubation, the cell pellet was collected by centrifugation (7000 × g, 15 min, 4 °C) and resuspended in cold phosphate buffer supplemented with protease inhibitors (1 mm PMSF, 10 μm leupeptin, 4 μm aprotinin). Lysis was performed by two passes through a French pressure cell, and lysate was centrifuged (12,000 × g, 20 min, 4 °C). Supernatant was loaded onto a glutathione-Sepharose column (GE Healthcare). Finally, GST-SPAK fusion protein was eluted by a 10 mm glutathione, pH 8.0, solution.
Cloning of Mouse Dnpep
Total RNA was isolated from mouse kidney using RNeasy kit (Qiagen, Valencia, CA) following the manufacturer's instructions, and 5 μg of was reverse-transcribed into cDNA using SuperScript III reverse transcriptase (Invitrogen). The cDNA encoding the mouse Dnpep was amplified in three pieces using specific oligonucleotides. The first sense oligonucleotide contained a 5′ EcoRI restriction site (underlined sequence) for cloning in different vectors, including an in-frame insertion into the GST fusion protein vector, pGEX4T, and the His6 fusion protein vector, pET14b (5′-GAATTCATGGCTATGAACGGCAGGGC-3′). The first antisense oligonucleotide introduced two silent mutations at its 5′ end, one eliminating a MluI site and the other introducing a new NheI site (5′ GCTAGCGTCAGGTCCCGGTCGAACC-3′). The sense oligonucleotide to amplify the middle piece started with the NheI site (5′-GCTAGCTGGTCGCGTCATTATCAAGTGCCCTAC-3′), whereas the antisense oligonucleotide contained a BamHI site, native to the Dnpep sequence (5′-GGATCCCTGGCCAGAGAGGCAGG 3′). The third fragment was amplified from the BamHI site (5′-GGATCCACATGTGCGCATGGTC 3′) to the end of the open reading frame was followed by a stop codon (written in bold letters) and a XhoI site (5′-CTCGAGTCAGTCCACTAAGAGGTTCCGGC-3′). The three PCR fragments were then gel-extracted, ligated into the cloning vector pGEM, and sequenced individually. The full-length clone was then constructed into a modified pBluescript that contains EcoRI-NheI-BamHI-XhoI sites and then moved to pGEX4T1, pBF, and pCDNA3 using EcoRI and XhoI restriction sites.
Dnpep Fusion Protein Purification
Dnpep cDNA inserted in pGEX4T or pET28b was transformed into BL21 E. coli strain, and transformants were cultured as described above. Incubation after protein expression induction (100 μm isopropyl-β-d-thiogalactopyranoside) was performed at 18 °C in a shaker incubator. After centrifugation, the pellet was resuspended in phosphate buffer (GST-Dnpep fusion protein) or in HEPES buffer (His-Dnpep fusion protein) supplemented with protease inhibitors. After lysis via a French press and centrifugation, the supernatant was loaded onto a glutathione-Sepharose column (GST-Dnpep fusion protein) or a nickel-nitrilotriacetic acid-agarose column (His-Dnpep fusion protein). Elution was then performed using either 10 mm glutathione, phosphate buffer, pH 8.0; GST-Dnpep fusion protein), or 400 mm imidazole, HEPES buffer, pH 8.0; His-Dnpep fusion protein).
Ion Exchange and Size Exclusion Chromatography
For ion exchange chromatography, diluted samples were loaded on a 5-ml HiTrap High Performance Q column (GE Healthcare) equilibrated in 25 mm Tris, 50 mm NaCl, pH 8.5. Proteins were eluted with a 50–500 mm NaCl gradient run at 1 ml/min and maintained at 4 °C. Gel filtration (size exclusion chromatography) was performed by loading 1 ml of clarified (i.e. dialyzed through a 14-kDa dialysis membrane (Spectrum Laboratories) for 2 days) kidney extract onto a 24-ml Superdex 200 10/300 GL column (GE Healthcare) equilibrated in 10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine run at 0.5 ml/min and maintained at 4 °C.
Mass Spectrometry
Shotgun proteomic analysis was performed on 100 μl of fraction #15 first by TCA precipitating the proteins by the addition of 33 μl of 100% w/v solution of TCA. After 2 ice-cold acetone washes, the precipitated proteins were resolubilized in a solution of 8 m urea and 100 mm Tris, pH 8.5, reduced with Tris(2-carboxyethyl)phosphine (10 mm), and alkylated with iodoacetamide (20 mm). After diluting the sample to 2 m urea, trypsin was added and allowed to proceed overnight at 37 °C. Resulting peptides were analyzed via MudPIT (Multidimensional Protein Identification Technology) essentially as described in Refs. 20 and 21). Briefly, digested peptides were loaded onto a biphasic precolumn consisting of 4 cm of reversed phase material followed by 4 cm of strong cation exchange material. Once loaded, this column was placed in line with a 20-cm reversed phase analytical column packed into a nanospray emitter tip directly coupled to a linear ion trap mass spectrometer (LTQ). A subset of peptides were eluted from the strong cation exchange material onto the reversed phase analytical via a pulse of volatile salt those peptides separated by an reversed phase gradient and then ionized directly into the mass spectrometry where both the intact masses (MS) and fragmentation patters (MS/MS) of the peptides were collected.
For the analysis of proteolytic fragments, NuPAGE 4–12% Bis-Tris resolved bands were excised and subjected to in-gel trypsin digestion. Data for these peptides were collected using a similar low flow HPLC separation but with the acquisition instrument being an LTQ-Velos Orbitrap. This allowed for the acquisition of both ion trap resolution MS/MS spectra and high resolution orbitrap spectra of the intact peptide masses. Peptide MS/MS spectral data were searched against a protein database using Sequest (22), and the resulting identifications were collated and filtered using IDPicker (23) and Scaffold. Extracted ion chromatograms of high resolution spectral data and subsequent quantitation were performed using the Skyline software package.
cRNA Transcription
Complementary DNA clones (20 μg) inserted into the X. laevis oocyte expression vector pBF and encoding for mouse NKCC2, full-length SPAK, and SPAK C-terminal fragments and subcloned were linearized by incubation at 37 °C overnight with MluI (New England Biolabs, Beverly, MA), purified using a QIAquick PCR Purification Kit (Qiagen), and transcribed into cRNA using a mMESSAGE mMACHINE SP6 transcription kit (Invitrogen). cRNA was then purified by using the RNeasy Mini kit (Qiagen) and eluted in diethylpyrocarbonate-treated water. RNA preparation was assessed for quality and quantity through denaturing agarose gel electrophoresis and spectrometric methods.
Injection of X. laevis Oocytes
Individual oocytes were dissociated through the use of collagenase (5 mg/ml, Sigma) and incubated overnight in modified L15 solution (250 ml of Leibovitz L15 Ringer (Invitrogen), 200 ml of deionized water, 952 mg of HEPES (acid form), and 44 mg/liter gentamycin (Invitrogen), pH 7.0, 195–200 mosm) at 16 °C. The following day, groups of 25 oocytes were injected with 15 ng of NKCC2 cRNA and returned to the incubator. On day 3, they were injected with 10 ng of kinase cRNA. Western blot analysis and 86Rb tracer flux studies to measure NKCC2 cotransport expression and function, respectively, were assessed on day 5.
NKCC2 Cotransporter Function
Groups of 20–25 oocytes in 35-mm dishes were washed once with 3 ml of isosmotic saline (96 mm NaCl, 4 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 5 mm HEPES, pH 7.4, 200 mosm) and preincubated for 15 min in 1 ml of the same solution containing 1 mm ouabain. Then the solution was aspirated and replaced with 1 ml of isosmotic flux solution containing 5 μCi of 86Rb. Two aliquots (5 μl each) of flux solution were sampled at the beginning of each uptake period and used as standards. After a 1-h uptake, the radioactive solution was aspirated, and the oocytes were washed 4 times with 3 ml of ice-cold isosmotic solution. Single oocytes were transferred into glass vials, lysed for 1 h with 200 μl of 0.25 n NaOH, and neutralized with 100 μl of glacial acetic acid, and tracer activity was measured by β-scintillation counting. NKCC2-mediated K+ flux is expressed in nmol of K+ per oocyte/h.
RESULTS
To assess proteolytic activity, a GST fusion protein comprising of the entire open reading frame of SPAK flanked by 5′ FLAG and 3′ HA epitopes (Fig. 1B) was incubated with a kidney lysate, and the SPAK reactants were subjected to Western blot analysis with either FLAG or HA antibodies or an antibody that recognizes the C-terminal domain of SPAK (see “Experimental Procedures”). As a control, the fusion protein was also treated with thrombin, a protease that recognizes a site engineered between the GST moiety and SPAK. As seen in Fig. 2A, incubation with kidney lysate produces several C-terminal fragments of sizes smaller than the proteolytic product of thrombin, indicating cleavages of the N terminus of SPAK by a different protease. As controls, we found that the observed signal does not originate from the kidney lysate itself and is not produced when using a GST-OSR1 fusion protein (Fig. 2B). The proteolytic activity is observed in kidney but not in brain, liver, or spleen (Fig. 2C) and is not species-specific as it was also observed with rabbit and human kidney lysates (Fig. 2D). The activity is maximal in the range of 4–45 °C, significantly reduced at 55 °C, and absent at 65 °C and 75 °C (Fig. 2E), consistent with protein temperature optima.
FIGURE 2.

Kidney lysate cleaves SPAK fusion protein. A, the addition of kidney lysate (70 μg) to SPAK fusion protein (SPAK-FP, 10 μg) and subsequent reaction for 1 h at 37 °C resulted in the appearance of several bands above the 50-kDa mark. The pattern is different from thrombin proteolytic cleavage. Open arrowheads indicate sizes of the full-length fusion protein and C-terminal fragment after thrombin. The thin lines under arrowheads highlight the presence of at least five fragments of B. IB, immunoblot. The pattern was not observed without SPAK fusion protein and was also not observed for reaction between kidney lysate and GST-OSR1 (10 μg) fusion protein. C, reaction between SPAK fusion protein and different tissue lysates. All reactions were performed with identical protein amounts of tissue lysate (100 μg). IB, immunoblot. D, proteolytic cleavage from mouse, rabbit, and human kidney lysates (70 μg). E, effect of temperature on the reaction. Two different protocols were used. For temperatures of 4, 20, and 37 °C, kidney lysates were added to GST-SPAK fusion protein, and a subsequent reaction was carried out at these temperatures. For temperatures higher than 37 °C, kidney lysate was first preincubated at these temperatures before reacting at 37 °C with GST-SPAK fusion protein. Western blot analyses were done using 1:1000 anti-SPAK (C-terminal) antibody followed by 1:5000 HRP-conjugated anti-rabbit antibody.
To ensure that the activity is not related to the GST moiety or the FLAG epitope, we repeated the experiment using N-terminal HA tagged full-length SPAK heterologously expressed in HEK293 cells. As seen in Fig. 3A, we observe similar kidney lysate-induced cleavage patterns with GST-SPAK and HA-SPAK isolated from two different sources. Note the presence of not only the major bands at molecular sizes higher than 50 kDa but also fragments at lower sizes. Because the activity is also time (Fig. 3B)- and concentration-dependent (Fig. 3C), it has the hallmark of an enzymatic reaction. Cleavage occurs within a wide range of pH (5.5–9.5) with intensities of the small bands increasing at more acidic pH (Fig. 3D). As detergent was present in the tissue lysis buffer, we tested the resistance of the enzyme to different concentrations of SDS and Triton X-100. As seen in Fig. 4A, the reaction was not affected by low concentrations of SDS or Triton. Only high 1% SDS and 10% Triton significantly decreased cleavage activity. To assess whether the activity is associated with plasma membranes, we prepared some lysates in 0.32 m sucrose and centrifuged up to 100,000 × g to isolate microsomes. Although cleavage was observed with all fractions, the strongest cleavage was observed in the microsomal fraction (Fig. 4B). Finally, we demonstrated that the activity was resistant to widely used protease inhibitors such as PMSF, leupeptin, aprotinin, pepstatin, EGTA, and EDTA at concentrations, which typically inhibit Ca2+-sensitive proteases (Fig. 4C).
FIGURE 3.

Properties of kidney lysate cleavage of SPAK fusion protein. A, incubation of GST-SPAK isolated from bacteria and HA-SPAK isolated from HEK293 cells with kidney lysate (2 μl) cells results in proteolytic cleavage independent of the nature of the added 5′ sequence. B, incubation of GST-SPAK with kidney lysate (2 μl) for short incubation times shows time dependence. C, incubation of GST-SPAK with increasing amounts of kidney lysate shows dose dependence. D, reaction between GST-SPAK and kidney lysate (2 μl) at three different pHs. The pH was adjusted to 7.5 at the end of the reaction before protein denaturation and gel loading.
FIGURE 4.

Additional properties of kidney lysate cleavage of SPAK fusion protein. A, incubation of GST-SPAK with kidney lysate (2 μl) in the presence of various concentrations of SDS or Triton. Note that the lysate contains 0.5% SDS, and the volume of the reaction is 40 μl, and therefore, all reactions contain 0.025% SDS. B, incubation of GST-SPAK with 50 μg of protein from 3,000 × g pellet, 20,000 × g pellet, 100,000 × g pellet, and 100,000 × g supernatant obtained from a 0.32 m sucrose kidney lysate. C, cleavage was resistant to traditional protease inhibitors. Samples containing 70 μg of kidney lysate were preincubated for 30 min at 37 °C in phosphate buffer with different inhibitors (final concentration: 1 mm PMSF, 10 μm leupeptin, 1 μm pepstatin, 0.1 mm EDTA, 1 mm EDTA, 1 mm EGTA) or with vehicle before reactions for 1 h at 37 °C with 2 μl of SPAK fusion protein (5 μg/μl). Western blot (IB) analysis was done using 1:1000 anti-SPAK (C-terminal) antibody followed by 1:5000 HRP-conjugated anti-rabbit antibody.
To identify the kidney protein responsible for this cleavage, we subjected 1 ml of kidney lysate (the equivalent of 2 mouse kidneys) on a cationic exchange column in a Tris buffer containing 50 mm NaCl, pH 8.5. Once loaded, the column was washed with the same buffer then exposed to a gradient of 50 mm to 500 mm NaCl, and fractions (Fig. 5A) were collected and tested for activity. As seen in Fig. 5B, peak activity was detected in fraction 15, which corresponds to a salt concentration of 240 mm. This fraction was subjected to multidimensional liquid chromatography tandem mass spectrometry (MudPIT) analysis, and 1119 proteins were identified. Using an extensive list of rat proteases (24), we recognized among them 44 proteases. The top 12 (ranking based on total spectrum count) are listed in Table 1, whereas the others are inventoried in Supplemental Table 1. To narrow down the list, we performed size exclusion chromatography to ascertain the size of the protease or protease complex. One milliliter of kidney lysate dialyzed using a 14-kDa size exclusion Spectra/Por membrane, was separated by gel filtration over a Sephadex 200 column, and 0.5-ml fractions were collected. Fig. 5C shows the optical density trace of the sample and of molecular standards. When equal amounts of protein (9 μg) from fractions were tested for activity, signal was observed in many early fractions, indicating large molecular weight similar to thyroglobulin, i.e. much larger than most of the proteases identified by MS, this being consistent with a multimeric protein complex (Fig. 5D).
FIGURE 5.

Ion exchange and size exclusion chromatography. A, 1 ml of kidney lysate (25 μg/μl) was diluted on ice into 10 mm Tris/HCl, pH 8.5, and loaded onto a 5-ml Q-column. After washing the column with this buffer, a salt gradient (50 mm to 500 mm NaCl) was applied (see the thin black line). Absorbance at 280 nm and conductivity were followed over time (thick black line and red line, respectively). B, equal fraction volumes (20 μl) were diluted in phosphate buffer and incubated for 1 h at 37 °C with 2 μl of SPAK fusion protein (5 μg/μl). ft is the flow-through, proteins that are not bound to the cation exchange column. IB, immunoblot. C, kidney lysate was dialyzed through a 14-kDa dialysis membrane for 2 days. After recovery, 1 ml of dialyzed sample was subjected to Superdex 200 gel filtration, and 0.5-ml fractions were collected. After the run of the sample, an identical run was performed with Bio-Rad standards (red curve). Note that for clarity of the figure, the standard curve was reduced vertically to 40% of its original size. Proteins in standard were thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobulin (17 kDa), and vitamin B (1.35 kDa). D, equal amounts of protein (9 μg) from each fraction was diluted in phosphate buffer and incubated for 1 h at 37 °C with 2 μl of SPAK fusion protein (40 μl total reaction volume). Samples were subjected to 10% SDS-PAGE and Western blot analysis using 1:1000 anti-SPAK C-terminal antibody followed by 1:5000 HRP-conjugated anti-rabbit antibody.
TABLE 1.
Top 12 peptidases/proteases identified by mass spectrometry analysis
Data are from 1119 proteins identified by MudPIT. Rank, position of the protein in the list sorted by total spectrum count. A, exclusive unique peptides; B, exclusive unique spectra; C, total spectra; D, % coverage.
| Peptidase/protease | Rank | Accession no. | Name | Size | A | B | C | D |
|---|---|---|---|---|---|---|---|---|
| kDa | % | |||||||
| Cytosol aminopeptidase | 5 | Q9CPY7 | Lap3 | 56 | 35 | 79 | 508 | 84 |
| Secernin-2 | 13 | Q8VCA8 | Scrn2 | 47 | 23 | 46 | 281 | 76 |
| Aminoacylase-1 | 20 | O99JW2 | Acy1 | 46 | 22 | 45 | 205 | 65 |
| Cathepsin B | 40 | P10605 | Ctsb | 37 | 16 | 30 | 111 | 51 |
| Meprin A | 48 | P28825 | Mep1a | 84 | 20 | 32 | 88 | 37 |
| Presequence protease | 58 | Q8K411 | Pitrm1 | 117 | 40 | 59 | 85 | 59 |
| Ubiquitin conjugated enzyme | 74 | P61087 | Ube2k | 22 | 14 | 26 | 63 | 80 |
| Parkinson disease 7 domain-containing protein | 116 | Q8BFQ8 | Pddc1 | 23 | 7 | 12 | 45 | 61 |
| Plasma kallikrein | 161 | P26262 | Klkb1 | 71 | 19 | 23 | 31 | 41 |
| Aspartyl aminopeptidase | 176 | Q9Z2W0 | Dnpep | 52 | 10 | 14 | 23 | 35 |
| Mitochondrial processing peptidase | 245 | Q9CXT8 | Pmpcb | 55 | 10 | 15 | 17 | 30 |
| Renin-1 | 282 | P06281 | Ren1 | 44 | 9 | 12 | 13 | 22 |
To explore whether the proteasome complex is involved in cleavage, we tested the proteasome inhibitor MG132 and found no effect (Fig. 6A). Other inhibitors were also tested to match a pharmacological profile to a MS-identified protease. N-Ethylmaleimide, which inhibits cysteine proteases by alkylation of thiol group, also does not prevent SPAK cleavage (Fig. 6A). As cytosolic aminopeptidase Lap3 was the top candidate of the MS list and this aminopeptidase is highly sensitive to bestatin, we tested this inhibitor at different concentrations and observed no effect (Fig. 6, A and B). In contrast, the proteolytic activity was significantly diminished by EDTA at 5 mm, dithiothreitol (DTT) at 1–10 mm, and 1,10-phenanthroline at 5 mm, all pointing to the involvement of a Zn2+ metalloprotease. Indeed, EDTA at high concentrations chelates not only Ca2+ and Mg2+ but also other divalent cations. Although DTT is commonly used to reduce disulfide bonds (25), it is also able to chelate metals such as Zn2+ but not Ca2+ (26). Additionally 1,10-phenanthroline is a specific inhibitor of Zn2+ metalloprotease (27). To confirm that Zn2+ is required for function, we incubated a kidney lysate with 20 mm EDTA. After dialyzing the sample to eliminate the chelating agent, we tested the activity of the lysate. As seen in Fig. 6C, preincubation with EDTA abolished the activity of the lysate. Activity could be restored by supplying before the reaction 1 mm ZnCl2 to the EDTA-treated/dialyzed lysate, whereas preincubation with other divalent cations (Mn2+, Cu2+, Mg2+, Ca2+) failed to restore activity (Fig. 6C).
FIGURE 6.

Cleavage is prevented by excess EDTA and by 1,10-phenanthroline and DTT but not by bestatin. A, samples containing 70 μg of kidney lysate were preincubated for 30 min at 37 °C in phosphate buffer with different inhibitors (final concentration: 100 μm bestatin, 10 μm MG132, 10 mm N-ethylmaleimide (NEM), 5 mm EDTA, 5 mm 1, 10-phenanthroline, 1 mm DTT, 5 mm DTT, 10 mm DTT) or with vehicle before reactions for 1 h at 37 °C with 2 μl of SPAK fusion protein (5 μg/μl). IB, immunoblot. B, absence of bestatin effect at concentrations ranging from 1 nm to 100 μm. Western blot analysis was done using 1:1000 anti-SPAK (C-terminal) antibody followed by 1:5000 HRP-conjugated anti-rabbit antibody. FP, fusion protein. C, the protease that cleaves SPAK is a Zn2+ metalloprotease. Kidney lysate was incubated with 20 mm EDTA for 1 h at 37 °C followed by 2 days of dialysis at 4 °C to wash away the EDTA. Recovered lysate was then concentrated using a 30-kDa centrifugal filter (Millipore) and preincubated (4 μg) with or without 1 mm concentrations of different divalent cations for 30 min before reaction with GST-SPAK fusion protein for 1 h at 37 °C. Reaction between non-EDTA-treated kidney lysate (4 μg, diluted) and SPAK fusion protein was used as positive control. Typical reactions between 1 μl of kidney lysate (22 μg/μl, original) and 2 μl of SPAK fusion protein was also used as a positive control. Samples were subjected to 10% SDS-PAGE followed by Western blot analysis as described before.
Seven of MS-identified proteases are Zn2+ metalloproteases. One cytosolic metalloprotease in particular, aspartyl aminopeptidase or Dnpep (EC 3.4.11.21), which is the sole mammalian member of the poorly understood M18 peptidase family, caught our attention as it is inhibited by 1,10-phenanthroline, DTT, and EDTA but not bestatin (28–30). Furthermore, although the molecular size of its monomer is relatively small (∼52 kDa), the crystal structure of Dnpep revealed that it forms a homododecamer tetrahedron complex, bringing the molecular mass of the native protein to 624 kDa (31). If Dnpep is the enzyme that degrades SPAK in the kidney, expression of aspartyl aminopeptidase should closely trace the cleavage activity that we observed with the chromatography fractions. As seen in Fig. 7, A and B, Dnpep expression traces well with the proteolytic activity in the ion exchange chromatography fractions. In addition, the peptidase is detected from gel filtration fractions 5–19, consistent with Dnpep being a multimeric protein (Fig. 5, B and D). In fact, proteolytic activity of the fractions tracked Dnpep in the higher molecular complexes rather than the more abundant smaller complexes. To confirm the large size of the Dnpep complex, we loaded an aliquot of fraction 9 from gel filtration on a NativePage 3–12% Bis-Tris gel and observed a protein complex that migrated at ∼620 kDa (Fig. 7, C and D).
FIGURE 7.

Dnpep is involved in the cleavage of SPAK. Western blot analysis detects Dnpep in ion exchange (A) and gel filtration (B) fractions. Equal fraction volumes (10 μl) were separated on 10% SDS-PAGE and subjected to Western blot (IB) analysis using 1:1000 anti-Dnpep antibody followed by 1:5000 HRP-conjugated anti-rabbit antibody. M, molecular mass markers. C, 9 μg of fraction #9 from gel permeation (see panel C) was loaded into Novex NativePAGE 3–12% Bis-Tris gels. Immunoblotting with anti-Dnpep antibody shows one single broad band. Protein markers (NativeMarkTM, Invitrogen) ranged from 1236 kDa to 20 kDa. Western blot analysis used 1:1000 anti-Dnpep antibody followed by 1:5000 HRP-conjugated anti-rabbit antibody. D, size determination of bands based on migration of molecular markers. The molecular mass (kDa, y axis) is plotted against the distance from the top of the gel (mm, x axis). The data were fitted using non-linear (exponential) regression analysis (GraphPad Prism 3.0).
Next, Dnpep was PCR-amplified from mouse kidney cDNA and inserted into a GST fusion protein expression vector. GST-Dnpep was produced in bacteria and purified on a glutathione-Sepharose column. As seen in Fig. 8A, proteolytic activity toward SPAK was confirmed with the recombinant fusion protein. As the activity was labile, possibly due to the large GST moiety, we also produced a His6-Dnpep fusion protein. Despite the fact that the fusion protein was confined to the bacterial pellet, we were able to confirm Dnpep-specific proteolytic activity toward SPAK (Fig. 8, B–D).
FIGURE 8.

Dnpep fusion protein cleaves SPAK fusion protein. A, reaction between GST-SPAK and GST-Dnpep. GST-Dnpep fusion protein (FP) was detected in the supernatant of lysed Dnpep-transformed bacteria and purified by a glutathione-Sepharose column. SPAK fusion protein (2 μl) was incubated (or not) in HEPES buffer with 10 μl of GST-Dnpep fusion protein for 1 h at 37 °C. IB, immunoblot. B, reaction between GST-SPAK and His-Dnpep. His-Dnpep fusion protein was detected in inclusion bodies in the pellet of lysed Dnpep-transformed bacteria. Different dilutions (4–512×) of pellet resuspended in lysis buffer (38 μl) were incubated with 2 μl of SPAK fusion protein for 1 h at 37 °C. The reaction between 2 μl of kidney lysate and 2 μl of SPAK fusion protein was used as positive control. The reaction between the pellet of GST-unspecific fusion protein and SPAK fusion protein was used as negative control. C, detection of Dnpep in SDS-PAGE Gel. Dnpep was detected in the pellet of Dnpep-transformed bacteria whereas it was absent in the pellet of unspecific protein-transformed bacteria. D, detection of Dnpep in NativePAGE 3–12% Bis-Tris gel. Dnpep was detected in the pellet of Dnpep-transformed bacteria.
To determine the sites of cleavage, we modified the fusion protein to allow for easier mass spectrometry of the PAPA box. This region is in fact typically not detected by trypsin using digestion due to the large size of the PAPA-containing tryptic peptide. We mutated residues Thr-21 and Ser-46 to Lys and demonstrated that these mutations did not affect the cleavage pattern (data not shown). The new GST-SPAK fusion protein was further purified by gel filtration and incubated for 2 h with a dilute kidney lysate. After proteolysis, the C-terminal fragments were purified by HA immunoprecipitation, separated by SDS-PAGE, extracted from the gel, and subjected to MS analysis. Tandem mass spectrometry data were analyzed using Scaffold 4 (Proteome Software Inc.), and Skyline 2.5 (MacCoss laboratory, University of Washington) was used to extract and integrate ion chromatograms for the peptide masses. Sites of cleavage are visualized in Fig. 9 and detailed in Table 2. It can be seen that the most N-terminal sites of cleavage occur in the PAPA box at two very specific locations. Analysis of secondary structure of the PAPA region using Phyre2 (32) revealed that the protease preferentially cleaves at the C-terminal tail of α-helices (Fig. 9, A and B). Downstream digests within the catalytic and the C-terminal regulatory domain also seem to occur preferentially at the C-terminal end of α-helices.
FIGURE 9.

Schematic representation of SPAK cleavage sites. A, the sequence of mouse SPAK is given with β-sheets and α-helices highlighted in blue bars and green helices, respectively. Sites of non-trypsin cleavage identified by mass spectrometry are indicated by vertical colored bars. Integrated peptide intensity levels are indicated by yellow bars (>E8), orange bars (>E7), red bars (>E6), and blue bars (>E5). Calculated sizes of the C-terminal fragments are indicated by a flag. B, schematic of the catalytic domain of SPAK based on OSR1 crystal structure. Secondary structure shows β-sheets and α-helices in orange. The two major sites of cleavage are indicated by bulky blue leucine residues.
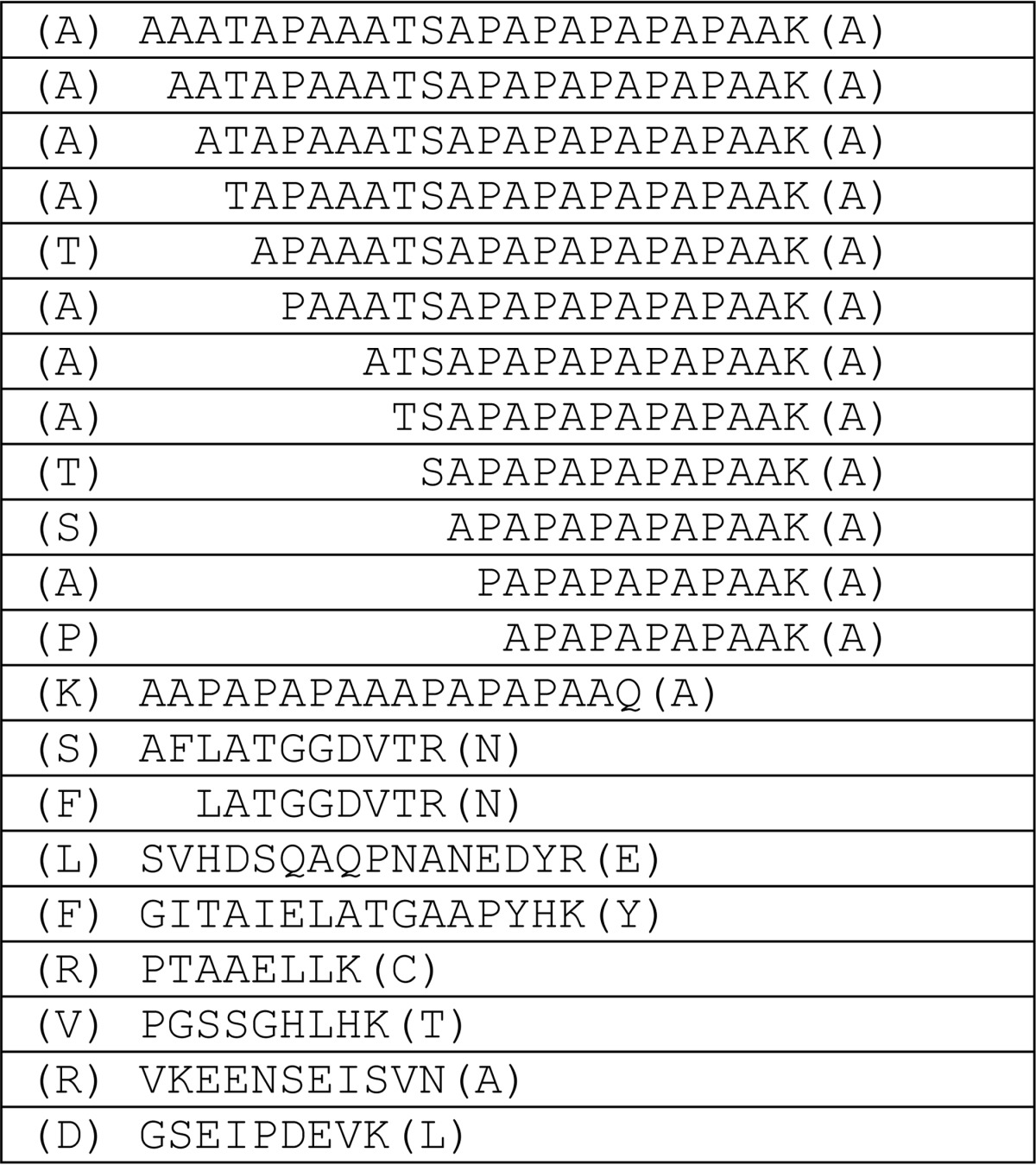
TABLE 2.
List of peptides identified by mass Spectrometry analysis
Sequences of the peptides identified by mass spectrometry with flanking residues are indicated in parenthesis. The peptides were from four bands excised from the gel as described under “Experimental Procedures.”
Cleavage of SPAK within the PAPA box was confirmed by experiments in which we removed the proline/alanine-rich sequence from SPAK and lost the cleavage pattern or in which we inserted this amino acids sequence to OSR1 and gained it (Fig. 10, A and B). Our study also demonstrated cleavage within the catalytic domain. In fact two sites yield C-terminal fragments with sizes close to the native fragments that are observed by Western blot analyses of kidney samples. The predicted molecular weights of 40.8 (V↓LEG) and 36.7 (F↓LAT) are close to the size of the inhibitory KS fragments measured previously (14, 16). We created the cDNA encoding these fragments (Fig. 10C) and tested their function on NKCC2 in X. laevis oocytes. We also created a protein devoid of the PAPA box. As seen in Fig. 10D, co-injection of full-length SPAK or of the SPAK fragment devoid of the PAPA box had no inhibitory effect on NKCC2 function. In contrast, co-injection of the two smaller fragments led to a significant decrease in NKCC2 function consistent with dominant negative effect of SPAK fragments on NKCC2 function. This observation is in agreement with the dominant negative effect of other fragments previously studied (16, 18).
FIGURE 10.

SPAK and OSR1 chimera and inhibitory action of SPAK fragments on NKCC2 function. A, different SPAK and OSR1 chimera were built and purified using a glutathione-Sepharose column. All chimeras have the PF2 domain, required for detection by the C-terminal SPAK antibody. OSR1A represents the wild-type OSR1 kinase with PF2 domain, OSR1B is the same kinase but with the PAPA box of SPAK attached at its 5′ end, and OSR1C is also OSR1A but with a portion of the SPAK kinase domain but not the PAPA box. B, Western blot analysis of reactions between SPAK or OSR1 chimera (10 μg) with kidney lysate (70 μg) for 1 h at 37 °C. FP, fusion protein. C, SPAK fusion proteins that lacked the entire PAPA box (SPAKΔ02–64) and also different portions of the kinase domain (SPAKΔ02–188, SPAKΔ02–228) were built and purified using a glutathione-Sepharose column. Note that three potential sites of cleavage were identified in the PAPA box, whereas sequences LEGLD and LATGGD corresponded to sites of cleavage within the kinase domain. D, NKCC2 and different SPAK fragments cRNAs were injected into oocytes and incubated with 86Rb for NKCC2-mediated flux measurements (expressed as nmol K+/oocyte/h). *** is highly significant (p < 0.001, analysis of variance).
DISCUSSION
In this study we showed that SPAK is cleaved to give rise to a reproducible pattern of degradation when it is incubated with kidney but not other tissue lysates. Of interest, this cleavage was not observed with OSR1. Because pretreatment of kidney lysate at a temperature of 55 °C or larger prevented this degradation, the activity was likely mediated by a protein, i.e. a protease. Effort was then made in finding experimental conditions that prevent the degradation pattern, finding inhibitors, and identifying the protease as well as its sites of cleavage. Although pretreatment of kidney lysate with regular protease inhibitors or cocktails did not prevent SPAK cleavage, 5 mm phenanthroline and 1 mm dithiothreitol effectively inhibited SPAK degradation. Phenanthroline is a specific inhibitor of Zn2+ metalloprotease (27). Dithiothreitol, which is commonly used to reduce disulfide bonds, is also able to chelate heavy metals such as Zn2+ but not Ca2+ (26). The observation that preincubation and a high concentration of EDTA (≥5 mm) are both required to inhibit the action of the protease on SPAK indicates that a divalent cation other than Ca2+ is involved. When kidney lysate was treated with 20 mm EDTA and then dialyzed to eliminate the chelating agent, we were able to recover the proteolytic activity only when Zn2+ was added to the reaction. There was no recovery with other divalent cations. Altogether those data strongly support that it is a Zn2+ metalloprotease that cleaves SPAK. Using traditional chromatography techniques combined to MS, we were able to identify Dnpep as the protease involved in the degradation of the kinase. Note that the identification of Dnpep does not exclude the possibility that another protease is also involved, particularly as we identified multiple sites of cleavage. Through the use of inhibitors, however, we were able to exclude the participation of other metalloproteases that were present in the MS list. For instance, we were able to eliminate lap3 based on insensitivity of our reaction to bestatin. Similarly, meprin A, which forms a high molecular weight complex at the membrane, is sensitive to micromolar concentrations of EDTA (33), whereas our reaction required several order of magnitude more EDTA to inhibit its activity. Finally, we were also able to eliminate aminoacylase-1, a cytosolic homodimer that is inhibited by lactate (34), and presequence protease, which is located in mitochondria and inhibited by Ni2+ and Zn2+ (35).
Although Dnpep has been reported to cleave acidic amino acid-rich sequences in vitro with a preference for aspartyl over glutamyl at the N-terminal tail of the short peptide (28), our data reveal that the protease also has an unusual structural preference. Indeed, SPAK is cleaved at the N-terminal tail of the kinase within the proline/alanine-rich region and within the kinase domain, regions that share no obvious sequence homology. Secondary structure prediction of SPAK revealed these cleavage sensitive regions as putative α-helices. A look at the catalytic domain structure (PDB codes 3DAK and 2VWI) confirmed this idea, localizing the cleavage sites to the C-terminal end of α-helices (Fig. 9B). Although proteases do not typically target α-helices, such behavior has been described for caspase-3 and the bacterial protease glutamyl endopeptidase, GluC (36). Perhaps the homododecameric structure of Dnpep, constituted by six dimers, each of which diagonally sits on a face of a tetrahedron (31), provides a means to recognize C-terminal ends of specific α-helices. Although unusual, this multimeric configuration is also found in an unrelated holobacterium aminopeptidase (37).
The proline/alanine-rich region of SPAK is of interest as it constitutes the differentiating feature from its ancestor kinase OSR1 (2). As demonstrated by our study, this unique region is primarily targeted by the protease, suggesting it may be involved in regulating the kinase. As the proline-rich region is located at the N terminus, a possibility could be that its presence is a prerequisite for the recognition and cleavage at downstream sites. The fact that the PAPA box is an unfolded region might help targeting the protease to the kinase. Proline repeat sequences have been described as “sticky,” and because the interaction of proline stretches with other proteins is weak, it is easily modulated (38). Indeed, proline-rich regions are believed to bring proteins together to increase the likelihood of subsequent interactions (39). The PAPA box, (AP)6, of the light chain myosin kinase was shown to target the kinase to actin, thereby allowing their direct interaction (40). Hence we can speculate that the presence of the PAPA box could facilitate the transport of SPAK to the apical membrane, where it interacts with the sodium chloride cotransporter NKCC2. For instance, the PAPA box could direct SPAK to apical actin bundles, and the action of Dnpep could release this interaction and allow SPAK to move away from the membrane. The PAPA box could account for the significant differences in cellular distribution of SPAK and OSR1 that have been observed in renal tubule cells (14, 16, 41).
Our study demonstrates that Dnpep provides another level of regulation to the signaling cascade that leads to activation of Na+ transporters. The aminopeptidase targets the proline/alanine-rich region of SPAK as well as the catalytic domain, resulting in shorter C-terminal fragments. Whether expression and/or activity of this aminopeptidase in the distal portion of the nephron are physiologically regulated still needs to be tested. Furthermore, as Dnpep is also expressed outside the kidney, whereas our results indicate “kidney-specific” proteolytic activity, suggests additional factors must be at play. These could be adaptor proteins that facilitate the localization and interaction between the protease and the kinase or a co-factor that is required for the protease activity. In this regard the level of Zn2+ in cells might be critical, as the divalent cation is required but also inhibitory at high concentrations. Zn2+ is transported by many mechanisms along the nephron and likely to be tightly regulated (42, 43).
Further studies will be required to determine the physiological relevance of the Dnpep fragments. Nevertheless, it should be pointed out that the proteolytic fragments are roughly equivalent to the sizes of the endogenous inhibitory SPAK fragments in the kidney, which are more prevalent in the medulla than the cortex (14) and have been suggested to dampen OSR1 (full-length) SPAK-mediated phosphorylation of NCCK2. Although Dnpep is found in both the cortex and the medulla, activity of the protease may be controlled in a nephron segment-specific manner by dietary conditions and/or the cofactor, Zn2+. There is currently no knock-out mouse model of Dnpep available to test these ideas. Whether deletion of the protease will lead to a viable mouse is, therefore, unknown, and future studies might require the development of a kidney-specific Dnpep knock-out mouse. Our data, which suggest a novel role for Dnpep in regulating Na+ transport in the nephron through specific targeting and cleavage of SPAK, are intriguing. Action of aminopeptidase or protease in renal physiology would, however, not be a precedent as aminopeptidase A (which in this case acts extracellularly) plays among other functions a key role in regulating blood pressure through the cleavage and degradation of angiotensin II (44, 45). There are also many examples of much larger proteins undergoing changes in activity upon proteolytic cleavage. This is the case for the epithelial Na+ channel, ENaC, which is cleaved by different proteases once it has been inserted in the plasma membrane of epithelial cells (46). ENaC activity increases severalfold upon proteolytic cleavage at the membrane, mediating reabsorption of Na+ in the collecting duct (47, 48).
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants DK093501 (to E. D. and P. A. W.) and GM081778 (to B. W. S.).

This article contains supplemental Table 1.
- SPAK
- Ste20-related proline/alanine-rich kinase
- TAL
- thick ascending limb of Henle
- Dnpep
- aspartyl aminopeptidase
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. McCormick J. A., Ellison D. H. (2011) The WNKs: atypical protein kinases with pleiotropic actions. Physiol. Rev. 91, 177–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gagnon K. B., Delpire E. (2012) Molecular physiology of SPAK and OSR1: two Ste20-related protein kinases regulating ion transport. Physiol. Rev. 92, 1577–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Delpire E., Gagnon K. B. (2008) SPAK and OSR1: STE20 kinases involved in the regulation of ion homoeostasis and volume control in mammalian cells. Biochem. J. 409, 321–331 [DOI] [PubMed] [Google Scholar]
- 4. Gagnon K. B., England R., Delpire E. (2006) Volume sensitivity of cation chloride cotransporters is modulated by the interaction of two kinases: SPAK and WNK4. Am. J. Physiol. Cell Physiol. 290, C134–C142 [DOI] [PubMed] [Google Scholar]
- 5. Gagnon K. B., England R., Delpire E. (2006) Characterization of SPAK and OSR1, regulatory kinases of the Na-K-2Cl cotransporter. Mol. Cell. Biol. 26, 689–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gagnon K. B., Rios K., Delpire E. (2011) Functional insights into the activation mechanism of Ste20-related kinases. Cell. Physiol. Biochem. 28, 1219–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ponce-Coria J., San-Cristobal P., Kahle K. T., Vazquez N., Pacheco-Alvarez D., de Los Heros P., Juárez P., Muñoz E., Michel G., Bobadilla N. A., Gimenez I., Lifton R. P., Hebert S. C., Gamba G. (2008) Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. U.S.A. 105, 8458–8463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Richardson C., Rafiqi F. H., Karlsson H. K., Moleleki N., Vandewalle A., Campbell D. G., Morrice N. A., Alessi D. R. (2008) Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J. Cell Sci. 121, 675–684 [DOI] [PubMed] [Google Scholar]
- 9. San-Cristobal P., Pacheco-Alvarez D., Richardson C., Ring A. M., Vazquez N., Rafiqi F. H., Chari D., Kahle K. T., Leng Q., Bobadilla N. A., Hebert S. C., Alessi D. R., Lifton R. P., Gamba G. (2009) Angiotensin II signaling increases activity of the renal Na-Cl cotransporter through a WNK4-SPAK-dependent pathway. Proc. Natl. Acad. Sci. U.S.A. 106, 4384–4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Piechotta K., Lu J., Delpire E. (2002) Cation-chloride cotransporters interact with the stress-related kinases SPAK and OSR1. J. Biol. Chem. 277, 50812–50819 [DOI] [PubMed] [Google Scholar]
- 11. Villa F., Goebel J., Rafiqi F. H., Deak M., Thastrup J., Alessi D. R., van Aalten D. M. (2007) Structural insights into the recognition of substrates and activators by the OSR1 kinase. EMBO Rep. 8, 839–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geng Y., Hoke A., Delpire E. (2009) The Ste20 kinases SPAK and OSR1 regulate NKCC1 function in sensory neurons. J. Biol. Chem. 284, 14020–14028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang S. S., Lo Y. F., Wu C. C., Lin S. W., Yeh C. J., Chu P., Sytwu H. K., Uchida S., Sasaki S., Lin S. H. (2010) SPAK-knockout mice manifest Gitelman syndrome and impaired vasoconstriction. J. Am. Soc. Nephrol. 21, 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McCormick J. A., Mutig K., Nelson J. H., Saritas T., Hoorn E. J., Yang C.-L., Rogers S., Curry J., Delpire E., Bachmann S., Ellison D. H. (2011) A SPAK isoform switch modulates renal salt transport and blood pressure. Cell Metab. 14, 352–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rafiqi F. H., Zuber A. M., Glover M., Richardson C., Fleming S., Jovanović S., Jovanović A., O'Shaughnessy K. M., Alessi D. R. (2010) Role of the WNK-activated SPAK kinase in regulating blood pressure. EMBO Mol. Med. 2, 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grimm P. R., Taneja T. K., Liu J., Coleman R., Chen Y. Y., Delpire E., Wade J. B., Welling P. A. (2012) SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J. Biol. Chem. 287, 37673–37690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lin S. H., Yu I. S., Jiang S. T., Lin S. W., Chu P., Chen A., Sytwu H. K., Sohara E., Uchida S., Sasaki S., Yang S. S. (2011) Impaired phosphorylation of Na+-K+-2Cl− cotransporter by oxidative stress-responsive kinase-1 deficiency manifests hypotension and Bartter-like syndrome. Proc. Natl. Acad. Sci. U.S.A. 108, 17538–17543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park H. J., Curry J. N., McCormick J. A. (2013) Regulation of NKCC2 activity by inhibitory SPAK isoforms: KS-SPAK is a more potent inhibitor than SPAK2. Am. J. Physiol. Renal Physiol. 305, F1687–F1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Piechotta K., Garbarini N., England R., Delpire E. (2003) Characterization of the interaction of the stress kinase SPAK with the Na+-K+-2Cl− cotransporter in the nervous system: evidence for a scaffolding role of the kinase. J. Biol. Chem. 278, 52848–52856 [DOI] [PubMed] [Google Scholar]
- 20. MacCoss M. J., McDonald W. H., Saraf A., Sadygov R., Clark J. M., Tasto J. J., Gould K. L., Wolters D., Washburn M., Weiss A., Clark J. I., Yates J. R., 3rd (2002) Shotgun identification of protein modifications from protein complexes and lens tissue. Proc. Natl. Acad. Sci. U.S.A. 99, 7900–7905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Martinez M. N., Emfinger C. H., Overton M., Hill S., Ramaswamy T. S., Cappel D. A., Wu K., Fazio S., McDonald W. H., Hachey D. L., Tabb D. L., Stafford J. M. (2012) Obesity and altered glucose metabolism impact HDL composition in CETP transgenic mice: a role for ovarian hormones. J. Lipid Res. 53, 379–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yates J. R., Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 23. Ma Z. Q., Dasari S., Chambers M. C., Litton M. D., Sobecki S. M., Zimmerman L. J., Halvey P. J., Schilling B., Drake P. M., Gibson B. W., Tabb D. L. (2009) IDPicker 2.0: improved protein assembly with high discrimination peptide identification filtering. J. Proteome Res. 8, 3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Puente X. S., López-Otín C. (2004) A genomic analysis of rat proteases and protease inhibitors. Genome Res. 14, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cleland W. W. (1964) Dithiothreitol. A new protective reagent for SH groups. Biochemistry 3, 480–482 [DOI] [PubMed] [Google Scholar]
- 26. Kr zel A., Lesniak W., Jezowska-Bojczuk M., Mlynarz P., Brasuñ J., Kozlowski H., Bal W. (2001) Coordination of heavy metals by dithiothreitol, a commonly used thiol group protectant. J. Inorg. Biochem. 84, 77–88 [DOI] [PubMed] [Google Scholar]
- 27. Correa L. M., Cho C., Myles D. G., Primakoff P. (2000) A role for a TIMP-3-sensitive, Zn2+-dependent metalloprotease in mammalian gamete membrane fusion. Dev. Biol. 225, 124–134 [DOI] [PubMed] [Google Scholar]
- 28. Wilk S., Wilk E., Magnusson R. P. (1998) Purification, characterization, and cloning of a cytosolic aspartyl aminopeptidase. J. Biol. Chem. 273, 15961–15970 [DOI] [PubMed] [Google Scholar]
- 29. Yokoyama R., Kawasaki H., Hirano H. (2006) Identification of yeast aspartyl aminopeptidase gene by purifying and characterizing its product from yeast cells. FEBS J. 273, 192–198 [DOI] [PubMed] [Google Scholar]
- 30. Chen Y., Farquhar E. R., Chance M. R., Palczewski K., Kiser P. D. (2012) Insights into substrate specificity and metal activation of mammalian tetrahedral aspartyl aminopeptidase. J. Biol. Chem. 287, 13356–13370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaikuad A., Pilka E. S., De Riso A., von Delft F., Kavanagh K. L., Vénien-Bryan C., Oppermann U., Yue W. W. (2012) Structure of human aspartyl aminopeptidase complexed with substrate analogue: insight into catalytic mechanism, substrate specificity and M18 peptidase family. BMC Struct. Biol. 12, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 33. Bylander J. E., Bertenshaw G. P., Matters G. L., Hubbard S. J., Bond J. S. (2007) Human and mouse homo-oligomeric meprin A metalloendopeptidase: substrate and inhibitor specificities. Biol. Chem. 388, 1163–1172 [DOI] [PubMed] [Google Scholar]
- 34. Tamura T., Oki Y., Yoshida A., Kuriyama T., Kawakami H., Inoue H., Inagaki K., Tanaka H. (2000) Noncompetitive, reversible inhibition of aminoacylase-1 by a series of l-α-hydroxyl and l-α-fluoro fatty acids: ligand specificity of aspergillus oryzae and porcine kidney enzymes. Arch. Biochem. Biophys. 379, 261–266 [DOI] [PubMed] [Google Scholar]
- 35. Chow K. M., Gakh O., Payne I. C., Juliano M. A., Juliano L., Isaya G., Hersh L. B. (2009) Mammalian pitrilysin: substrate specificity and mitochondrial targeting. Biochemistry 48, 2868–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Timmer J. C., Zhu W., Pop C., Regan T., Snipas S. J., Eroshkin A. M., Riedl S. J., Salvesen G. S. (2009) Structural and kinetic determinants of protease substrates. Nat. Struct. Mol. Biol. 16, 1101–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Franzetti B., Schoehn G., Hernandez J. F., Jaquinod M., Ruigrok R. W., Zaccai G. (2002) Tetrahedral aminopeptidase: a novel large protease complex from archaea. EMBO J. 21, 2132–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Williamson M. P. (1994) The structure and function of proline-rich regions in proteins. Biochem. J. 297, 249–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kay B. K., Williamson M. P., Sudol M. (2000) The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 14, 231–241 [PubMed] [Google Scholar]
- 40. Frank G., Weeds A. G. (1974) The amino-acid sequence of the alkali light chains of rabbit skeletal-muscle myosin. Eur. J. Biochem. 44, 317–334 [DOI] [PubMed] [Google Scholar]
- 41. Terker A. S., Yang C.-L., McCormick J. A., Meermeier N. P., Rogers S. L., Grossmann S., Trompf K., Delpire E., Loffing J., Ellison D. H. (2014) Sympathetic stimulation of thiazide-sensitive sodium-chloride cotransport in the generation of salt-sensitive hypertension. Hypertension 64, 178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cousins R. J., McMahon R. J. (2000) Integrative aspects of zinc transporters. J. Nutr. 130, 1384S–1387S [DOI] [PubMed] [Google Scholar]
- 43. Ranaldi G., Perozzi G., Truong-Tran A., Zalewski P., Murgia C. (2002) Intracellular distribution of labile Zn(II) and zinc transporter expression in kidney and MDCK cells. Am. J. Physiol. Renal Physiol. 283, F1365–F1375 [DOI] [PubMed] [Google Scholar]
- 44. Wolf G., Mentzel S., Assmann K. J. (1997) Aminopeptidase A: a key enzyme in the intrarenal degradation of angiotensin II. Exp. Nephrol. 5, 364–369 [PubMed] [Google Scholar]
- 45. Schwacke J. H., Spainhour J. C., Ierardi J. L., Chaves J. M., Arthur J. M., Janech M. G., Velez J. C. (2013) Network modeling reveals steps in angiotensin peptide processing. Hypertension 61, 690–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gondzik V., Weber W. M., Awayda M. S. (2012) Coupling of epithelial Na+ and Cl− channels by direct and indirect activation by serine proteases. Am. J. Physiol. Cell Physiol. 303, C936–C946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kleyman T. R., Carattino M. D., Hughey R. P. (2009) ENaC at the cutting edge: regulation of epithelial sodium channels by proteases. J. Biol. Chem. 284, 20447–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rossier B. C., Stutts M. J. (2009) Activation of the epithelial sodiumchannel (ENaC) by serine proteases. Annu. Rev. Physiol. 71, 361–379 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.