

Background: Glycogen synthase kinse-3β (GSK-3β) promotes mitochondrial permeability transition (MPT), a mechanism of cell necrosis.
Results: Efficient GSK-3β translocation to mitochondria requires its kinase activity, N-terminal domain, and interaction with VDAC2 protein.
Conclusion: MPT can be suppressed by inhibiting mitochondrial transport of GSK-3β.
Significance: A strategy for cell protection without modifying the physiological function of GSK-3β is proposed.
Keywords: Glycogen Synthase Kinase 3 (GSK-3), Mitochondrial Permeability Transition (MPT), Mitochondrial Transport, Necrosis (Necrotic Death), Protein-Protein Interaction, Voltage-dependent Anion Channel 2
Abstract
Glycogen synthase kinase-3β (GSK-3β) is a major positive regulator of the mitochondrial permeability transition pore (mPTP), a principle trigger of cell death, under the condition of oxidative stress. However, the mechanism by which cytosolic GSK-3β translocates to mitochondria, promoting mPTP opening, remains unclear. Here we addressed this issue by analyses of the effect of site-directed mutations in GSK-3β on mitochondrial translocation and protein/protein interactions upon oxidative stress. H9c2 cardiomyoblasts were transfected with GFP-tagged GSK-3β (WT), a mutant GSK-3β insensitive to inhibitory phosphorylation (S9A), or kinase-deficient GSK-3β (K85R). Time lapse observation revealed that WT and S9A translocated from the cytosol to the mitochondria more promptly than did K85R after exposure to oxidative stress. H2O2 increased the density of nine spots on two-dimensional gel electrophoresis of anti-GSK-3β-immunoprecipitates by more than 3-fold. MALDI-TOF/MS analysis revealed that one of the spots contained voltage-dependent anion channel 2 (VDAC2). Knockdown of VDAC2, but not VDAC1 or VDAC3, by siRNA attenuated both the mitochondrial translocation of GSK-3β and mPTP opening under stress conditions. The mitochondrial translocation of GSK-3β was attenuated also when Lys-15, but not Arg-4 or Arg-6, in the N-terminal domain of GSK-3β was replaced with alanine. The oxidative stress-induced mitochondrial translocation of GSK-3β was associated with an increase in cell death, which was suppressed by lithium chloride (LiCl), a GSK-3β inhibitor. These results demonstrate that GSK-3β translocates from the cytosol to mitochondria in a kinase activity- and VDAC2-dependent manner in which an N-terminal domain of GSK-3β may function as a mitochondrial targeting sequence.
Introduction
Emerging evidence has demonstrated that mitochondria play crucial roles not only in ATP production as a powerhouse but also in mechanisms of cell death. A major mechanism of cell necrosis is opening of the mitochondrial permeability transition pore (mPTP),2 a nonspecific megachannel in the inner mitochondrial membrane (1). Once opened, the mPTP allows entry of any molecule of <1.5 kDa, resulting in matrix swelling and dissipation of mitochondrial membrane potential, which induce outer membrane disruption and ATP depletion, respectively. Opening of the mPTP is triggered by several factors including Ca2+ overload, oxidative stress, elevated phosphate concentration, and adenine nucleotide depletion (1–3). The relationship between mPTP opening factors and cell necrosis has been well characterized in cardiomyocytes undergoing ischemia/reperfusion (4–6). However, involvement of mPTP dysregulation has also been shown in altered response of cancer cells to chemotherapy and etiology of neurodegenerative diseases (7–10). These findings indicate that mPTP is “a master regulator of cell death” in diverse diseases.
Glycogen synthase kinase-3β (GSK-3β) is a multifunctional and constitutively active Ser/Thr kinase that is regulated by multiple mechanisms: phosphorylation (4, 11–13), intracellular translocation (11), binding to other proteins including p53 (14) and STAT3 (15), and formation of a multiprotein complex with axin, casein kinase II, and β-casein (16). Contribution of GSK-3β to mPTP regulation was first demonstrated by Juhaszova et al. (17), who reported that inactivation or knockdown of GSK-3β elevates the threshold for opening of the mPTP. Our previous studies showed that ischemia/reperfusion increased physical interaction of GSK-3β with adenine nucleotide translocase and voltage-dependent anion channel (VDAC), putative regulatory components of the mPTP (18), and that inactivation of mitochondrial GSK-3β was associated with suppression of mPTP opening (19, 20). These findings indicate that GSK-3β in mitochondria is a major regulatory factor of the mPTP.
Inhibition of GSK-3β has been shown to suppress myocardial necrosis after ischemia/reperfusion (4). However, GSK-3β inhibitors potentially have serious adverse effects because of the multifunctional nature and ubiquitous presence of GSK-3β. In fact, knock-out of GSK-3β in mice is embryonic lethal (21). Therefore, for clinical application of GSK-3β inhibition to tissue protection, it is crucial to specifically suppress GSK-3β relevant to promotion of mPTP opening. To this end, we focused on the mechanism of GSK-3β translocation to mitochondria and its impact on cell viability in the present study.
We demonstrated that GSK-3β translocates to the mitochondria in a kinase activity-dependent manner under the condition of oxidative stress. The translocation is facilitated by interaction with VDAC2 in which an N-terminal domain of GSK-3β may also be involved as a mitochondrial targeting sequence. The results indicate that VDAC2 and the N-terminal domain of GSK-3β are novel therapeutic targets for various pathological conditions including myocardial ischemia/reperfusion injury.
EXPERIMENTAL PROCEDURES
Cell Culture
H9c2 or HEK293 cells (American Type Culture Collection) were cultured in DMEM (Sigma-Aldrich) supplemented with 10% FBS at 37 °C with 5% CO2. The cells were used for experiments when they were 70–90% confluent.
Cell Fractionation and Western Blotting
Mitochondrial and cytosolic fractions were prepared by using a mitochondrial isolation kit (Pierce) according to the manufacturer's protocol. Western blotting and immunoprecipitation were performed as reported previously (22). Antibodies used were rabbit monoclonal anti-GSK-3β (9315, Cell Signaling Technology), rabbit polyclonal anti-phospho-(Ser-9) GSK-3β (9336, Cell Signaling Technology), rabbit polyclonal anti-glycogen synthase (3893, Cell Signaling Technology), rabbit polyclonal anti-phospho-(Ser-641/645) glycogen synthase (44-1092G, Invitrogen), mouse monoclonal anti-Rieske (ab14746, Abcam), mouse monoclonal anti-VDAC1 (ab14734, Abcam), anti-VDAC2 (ab118872, Abcam), anti-VDAC3 (NBP1-80069, Novus Biologicals), mouse monoclonal anti-cyclophilin D (AP1035, Calbiochem), and mouse monoclonal anti-prohibitin (sc-56346, Santa Cruz Biotechnology).
Plasmid Construction and Transfection
To construct a GSK-3β-enhanced green fluorescent protein (GFP) fusion protein, the coding region of rat GSK-3β cDNA lacking its stop codon was cloned into the vector pEGFP-N3 (Clontech) at the BglII and SalI sites and in-frame with the enhanced GFP coding region. To construct a constitutively active (S9A), kinase-deficient (K85R), and mitochondrial targeting sequence (MTS) mutant (R4A, R6A, and K15A) GSK-3β, site-directed mutagenesis was carried out using the QuikChange XL mutagenesis kit (Agilent Technologies). Successful mutagenesis was confirmed by sequencing. H9c2 cells were transfected with each plasmid using Cell Line Nucleofector kit L (Lonza) and used for further analyses. Transfection efficiencies were ∼10% for wild type, S9A, K85R, R4A, R6A, and K15A and ∼25% for GFP control.
Generation of FLAG-tagged VDAC2
To generate an expression vector for FLAG-tagged VDAC2, the coding region of VDAC2 was amplified from rat cDNA by PCR using LA Taq with GC buffer (Takara Bio Inc., Shiga, Japan) and the following primers: 5′-AAAAGCGGCCGCGATGGCTGAATGTTGTGTACC-3′ (forward) and 5′-AAAAGGATCCTTAAGCCTCCAATTCCAAGG-3′ (reverse). The PCR fragment was cloned into the NotI and BamHI sites of p3xFLAG-CMVTM-7.1 expression vector (Sigma-Aldrich). The cDNA sequence was verified by nucleotide sequencing.
siRNA
Three different siRNAs for VDAC isoforms were synthesized by Sigma-Genosys siRNA Service, and the one that most efficiently suppressed the expression of VDAC mRNA was used in the present study. The sequences of sense and antisense siRNA were: VDAC1, GAUACACUCAGACUCUAAATT (sense) and UUUAGAGUCUGAGUGUAUCTT (antisense); VDAC2, CAGAAAGUAUGUGAAGAUUTT (sense) and AAUCUUCACAUACUUUCUGTT (antisense); and VDAC3, GACUCUUGAUACCAUAUUUTT (sense) and AAAUAUGGUAUCAAGAGUCTT (antisense). Transfection of siRNA (100 nmol/liter) into cells was performed using a Nucleofector kit (Lonza).
Fluorescence Microscopy Analysis of Living Cells
Two days after transfection, cells were stained with MitoTracker Red (Invitrogen) for 7 min and put in an incubator on an inverted microscope (TE2000-E, Nikon) in the presence or absence of H2O2 (10 μmol/liter). Images were obtained by time lapse laser scanning for 40 ms every 6 s using a charge-coupled device camera (ORCA-AG, Hamamatsu Photonics) for 15 min and then analyzed with AQUACOSMOS software (Hamamatsu Photonics). Still images of MitoTracker Red staining and GFP-GSK-3β signal before and 3 min after exposure to H2O2 at a magnification of 400× were further analyzed by image analysis software (Photoshop CS6, Adobe). The background fluorescence was cut off using a constant threshold value before the images were merged. Pixels positive for MitoTracker Red (total mitochondria) and pixels positive for both MitoTracker Red and GFP-GSK-3β (GFP-positive mitochondria) were counted, and the ratio of the latter to the former (GFP-positive mitochondria/total mitochondria) was used as an index for the mitochondrial localization of GFP-GSK-3β constructs.
Observation of Mitochondrial Morphology
H9c2 cells were transfected with VDAC2 siRNA or control siRNA and stained with Mito Tracker Red (0.2 μmol/liter) for 15 min, and time lapse observation of the mitochondrial morphology was achieved by N-SIM super-resolution microscopy (Nikon). To construct a super-resolution image, nine images (three different directions for three different phases) were captured in 2D-SIM mode with a temporal resolution of 0.6 s/frame. Super-resolution images were recorded every 10 s for 15 min for the time lapse observation.
2′,7′-Dichlorofluorescein (DCF) Staining
Intracellular ROS levels were monitored by DCF fluorescence (Invitrogen). H9c2 cells were loaded with DCF according to the manufacturer's protocol. DCF-loaded cells underwent 120-min hypoxia/30-min reoxygenation using hypoxic GasPak pouches (BD Biosciences) or exposure to antimycin A (50 μmol/liter) for 1 h in the presence of LiCl or a vehicle. Treatment with LiCl or the vehicle was started 30 min before hypoxia/reoxygenation or antimycin A exposure and continued until the end of the experiments. DCF fluorescence was recorded by a FLoid Cell Imaging Station (Invitrogen) at 1 h after exposure to antimycin A or at 30 min after reoxygenation.
Detection of Superoxide in GFP-GSK-3β-expressing Cells
To determine the level of superoxide production in cells expressing GFP-tagged GSK-3β, a Total ROS/Superoxide Detection kit (Enzo Life Sciences) was used according to the manufacturer's instructions. Cells were loaded with Superoxide Detection Solution and then incubated for 30 min. After washing the cells twice with wash buffer, the cells were observed by fluorescence microscopy. Fluorescence images of orange fluorescence, indicating superoxide, in GFP-positive cells at a magnification of 400× were analyzed by counting positive pixels per cell by image analysis software (Photoshop CS6, Adobe).
Cell Death Assay
The LDH activity of the culture medium was assayed with CytoTox96 (Promega) according to the manufacturer's instructions. LDH activity in the medium as a percentage of total cellular LDH activity in the well was used as an index of cell necrosis.
Calcein Assay
Mitochondrial permeability was monitored by calcein using an Image-iTTM LIVE Mitochondrial Transition Pore Assay kit (Molecular Probes) according to the manufacturer's instructions with slight modification. Cells were incubated with 0.25 μmol/liter calcein and MitoTracker Red for 15 min, and the medium was changed to calcein-free medium containing 4 mmol/liter cobalt chloride (CoCl2). CoCl2 does not reach inside mitochondria and quenches cytosolic calcein fluorescence, resulting in selective mitochondrial accumulation of calcein as long as the mPTPs are closed. After 15-min incubation, cells were pretreated with 30 mmol/liter LiCl or a vehicle for 60 min before treatment with 100 μmol/liter H2O2 for 2 h. The calcein-stained area overlapped with the MitoTracker Red-stained area was used as an index of mitochondria with closed mPTPs, and the extent of mPTP opening was determined by the decline in the ratio of the overlapped area to the MitoTracker Red-stained area (calcein-positive mitochondria/total mitochondria).
Two-dimensional Electrophoresis
Whole cell lysates of H9c2 cells exposed to H2O2 or a vehicle were harvested using CellLytic-M Mammalian Cell Lysis/Extraction agent (Sigma-Aldrich) and stored at −80 °C for later use. The samples were immunoprecipitated with a GSK-3β antibody (Abcam) as reported previously (18). The immunoprecipitates were then separated by two-dimensional electrophoresis using a ReadyPrepTM 2-D Starter kit (Bio-Rad) according to the manufacturer's instructions as follows. The protein samples were mixed with Rehydration Buffer (Bio-Rad) for isoelectrical focusing. The immobilized pH gradient gel strips (pH range, 3–10; 7 cm; Bio-Rad) were rehydrated in 125 μl of samples diluted with ReadyPrep Rehydration/Sample Buffer with a final protein concentration of 0.6 g/μl for 12 h at 20 °C. Isoelectric focusing was performed on the pre-rehydrated immobilized pH gradient gel strips at 20 °C in four steps: (i) 250 V for 15 min and (ii) 4000 V for 1 h in a linear mode where voltage increases are in a linear fashion to the set voltage followed by (iii) 4000 V for 3 h and (iv) 500 V for 24 h in a rapid mode where voltage is limited by the set current value using a Protean isoelectric focusing cell (Bio-Rad). When isoelectrical focusing was completed, the focused immobilized pH gradient strips were incubated in Equilibration Buffer I (6 m urea, 2% SDS, 0.375 m Tris-HCl (pH 8.8), 20% glycerol, and 2% (w/v) DTT) from the ReadyPrep 2-D Starter kit for 20 min with gentle shaking. Then Equilibration Buffer I was discarded, and Equilibration Buffer II (containing iodoacetamide) was added to each strip with gentle shaking for 10 min. Strips were then mounted on top of 12% gels (Mini-Protean TGX Precast Gels, Bio-Rad) in wells filled with warm agarose (0.5% low melting point agarose in 25 mm Tris, 192 mm glycine, 0.1% SDS, and a trace of bromphenol blue). The gel box apparatus was filled with running buffer, and the gels were run at 150 V for 1 h. After second dimension electrophoresis, the gels were stained with Coomassie Blue at 4 °C for 24 h, scanned by a laser scanner (GT-X970, Epson), and then analyzed using PDQuest 2-D Analysis Software. A total of eight gels, four for vehicle-treated cells and four for H2O2-exposed cells, were analyzed under the same experimental conditions. Nine protein spots with an increase in stain density of 3-fold or more in response to H2O2 stimulation were excised and analyzed by MALDI-TOF/MS.
Mass Spectrometry
Spots of interest in the Coomassie Blue-stained gels were excised in small cubes of ∼1 mm square in size. After reduction and alkylation, in-gel digestion was performed with trypsin overnight at 37 °C. Peptides were subsequently separated on a HiQsilC18W-3 column (100-μm inner diameter × 100 mm; KYA Technology, Tokyo, Japan). 0.1% trifluoroacetic acid (TFA; solvent A) and 0.1% TFA in 70% acetonitrile (solvent B) were used for elution. The gradient was 5–50% for solvent B over 50 min at a flow rate of 300 nl/min. Separated peptides were spotted onto 384-well AB OptiTOF MALDI plate inserts (AB Sciex). Peptide fractions were collected every 30 s, and 150-nl peptide fractions were overlaid with 700 nl of α-cyano-4-hydroxycinnamic acid (Sigma) in 80 mg/ml ammonium citrate, 70% acetonitrile, and 0.1% TFA. Mass spectrometric analyses were performed on a 4800 Plus MALDI-TOF/TOF Analyzer (AB Sciex) with 4000 Series Explorer version 3.5 software (AB Sciex). A tryptic BSA standard (KYA Technology) and a six-peptide mixture (AB Sciex) were used to calibrate the mass accuracy. MS/MS acquisitions of MS spectra with a signal to noise ratio over 100 were carried out using air as the collision gas and a collision energy of 1 kV. Data were processed by Protein Pilot versions 2.0 and 3.0 (AB Sciex) using the Paragon search algorithm (23). TOF/MS data were searched against the human International Protein Index database (version 3.63).
Statistical Analysis
Group mean data are shown as means ± S.E. One-way analysis of variance was used for testing differences among group mean data. Differences between groups in response to oxidative stress were examined by two-way repeated measures analysis of variance. When analysis of variance indicated a significant overall difference, multiple comparisons were performed using the Student-Newman-Keul post hoc test. p < 0.05 was considered statistically significant.
RESULTS
To explore the mechanism of GSK-3β translocation to mitochondria, we first examined the relationship between kinase activity of GSK-3β and extent of mitochondrial translocation of GSK-3β in response to oxidative stress. We prepared GFP-tagged wild-type GSK-3β (WT) and its site-directed mutants, constitutively active GSK-3β (S9A) and kinase-deficient GSK-3β (K85R). H9c2 cells were transfected with WT, S9A, or K85R and stained with MitoTracker Red, and time lapse observation by fluorescence microscopy was performed during 15-min exposure to H2O2 (10 μmol/liter). WT gradually translocated from the cytosol to the mitochondria as indicated by an increase in overlap of green fluorescence and red fluorescence (Fig. 1, A and B). The mitochondrial translocation of WT was followed by cell death as revealed by plasma membrane disruption and severe cell deformation. S9A signals appeared to be present in some of the mitochondria even before H2O2 challenge, and S9A translocated to the mitochondria more promptly than did WT, leading to cell death (Figs. 1, A and B, and 2 and supplemental Movie 1A). In contrast, mitochondrial translocation of K85R was much slower than that of WT and was accompanied by a lower prevalence of cell death compared with that of WT or S9A (Figs. 1 and 2 and supplemental Movie 1B). These findings indicate that mitochondrial translocation of GSK-3β is dependent on its kinase activity, although involvement of a factor besides GSK-3β activity is indicated by a low but significant level of co-localization of K85R with mitochondria. This notion was further supported by the finding that treatment with LiCl, which reduces GSK-3β activity both directly and by increasing inhibitory Ser-9 phosphorylation (24), reduced mitochondrial localization of WT both before and after exposure to H2O2 (Fig. 1, A and B). The GFP control that was not tagged with GSK-3β remained only in the cytoplasm throughout the experiments (Fig. 1, A and B), indicating that fusion with GFP per se did not induce translocation of GSK-3β. Although GFP-tagged GSK-3β might not have behaved exactly the same as endogenous GSK-3β because of its large molecular size (32.7 kDa) and the biophysical property of GFP, mitochondrial translocation of endogenous GSK-3β induced by H2O2 was confirmed by Western blotting for mitochondrial and cytosolic fractions of H9c2 cells (Fig. 1C) as was GSK-3β in the rat myocardium (18). Furthermore, the amount of GSK-3β co-immunoprecipitated with cyclophilin D, a matrix protein, and with Rieske, a subunit of complex III, increased after H2O2 treatment (Fig. 1D), suggesting that GSK-3β resides in the matrix side of the inner membrane after the translocation.
FIGURE 1.

Kinase activity-dependent mitochondrial translocation of GSK-3β under the condition of oxidative stress. A, fluorescence images obtained from time lapse observation. H9c2 cells were transfected with GFP-tagged GSK-3β (WT), constitutively active mutant GSK-3β (S9A), kinase-inactive mutant GSK-3β (K85R), or GFP alone (GFP control) and then stained with MitoTracker Red. Photos at 3 min after exposure of cells to H2O2 (10 μmol/liter) in the presence or absence of LiCl (30 mmol/liter) are shown. B, quantification of mitochondrial (mito) localization of each plasmid. MitoTracker-stained area overlapped with GFP signal was expressed as a percentage of total MitoTracker-stained area. *, p < 0.05 versus cells transfected with the same plasmid and treated with a vehicle; †, p < 0.05 versus cells transfected with WT and treated with a vehicle; ‡, p < 0.05 versus cells transfected with WT and exposed to H2O2. C, representative immunoblotting for total GSK-3β in mitochondrial fractions (Mito) and cytosolic fractions (Cyto). Prohibitin and β-actin were used as markers of the mitochondria and cytosol, respectively. Three separate experiments showed similar results. D, interaction of GSK-3β with mitochondrial proteins. Immunoblots (IB) for GSK-3β co-immunoprecipitated (IP) with cyclophilin D (CyD) and Rieske co-immunoprecipitated with GSK-3β with or without exposure to H2O2 (100 μmol/liter; 4 h) are shown. Error bars represent S.E.
FIGURE 2.
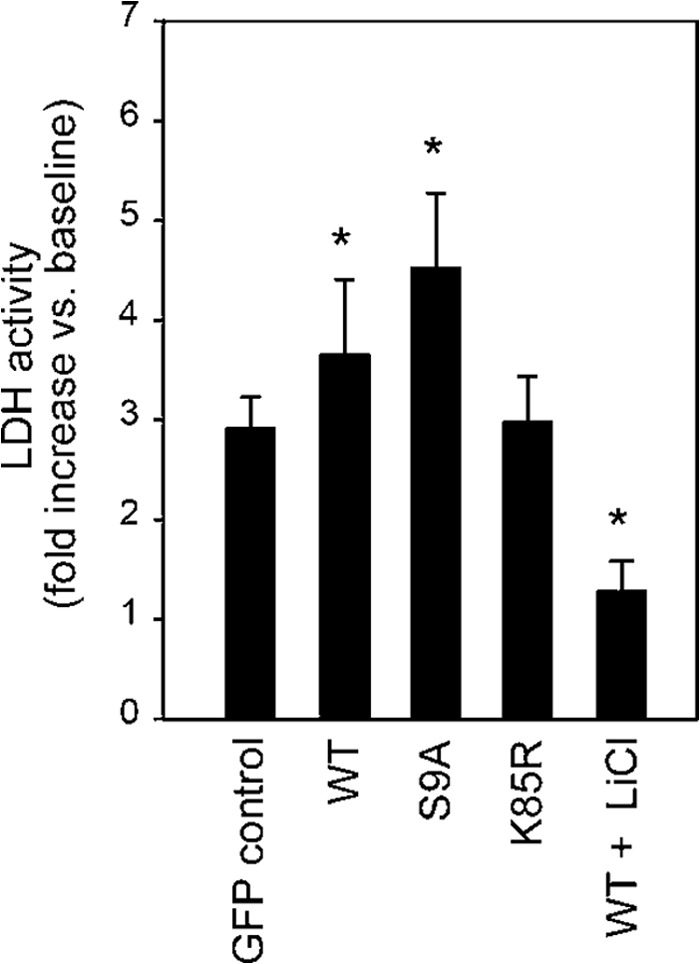
Activity of GSK-3β is associated with cell death. LDH activity in the culture medium was measured at 4 h after addition of H2O2 (100 μmol/liter). -Fold increases in LDH activity compared with the baseline value are shown. *, p < 0.05 versus GFP control. Error bars represent S.E.
Results of quantitative analyses of cell death indicated an association of cell death with mitochondrial translocation of GSK-3β. LDH activity in the culture medium during 4-h treatment with H2O2 (100 μmol/liter), a dose and duration that induced cell death in 12% of H9c2 cells in preliminary experiments, was significantly increased by transfection with WT or S9A, but not by transfection with K85R, compared with GFP control (Fig. 2). As expected, inhibition of GSK-3β by LiCl significantly inhibited the LDH release. The insignificant effect of transfection with K85R on LDH release is most likely explained by low transfection efficiency (∼10%) by nucleofection.
Next, we examined whether production of ROS under the condition of pathological stress is regulated by GSK-3β activity. We used two different stimuli to induce ROS production in mitochondria, namely hypoxia/reoxygenation and antimycin A, an inhibitor of mitochondrial complex III (25, 26), and DCF was used as a probe for ROS. The amount of ROS produced after hypoxia/reoxygenation was significantly increased, but ROS production was almost abolished by LiCl (Fig. 3A). Similarly, LiCl attenuated antimycin A-induced ROS production (Fig. 3B). These findings indicate that activity of GSK-3β contributes not only to its translocation to mitochondria but also to production of ROS from mitochondria.
FIGURE 3.
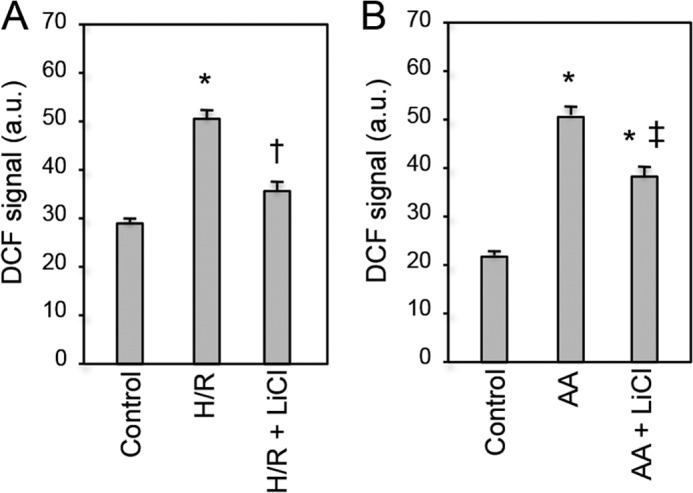
Effects of GSK-3β inhibition on ROS production by oxidative stress. Production of ROS after hypoxia/reoxygenation (A) or exposure to 50 μmol/liter antimycin A (B) was monitored by DCF staining. GSK-3β was inhibited by lithium chloride. Signal intensities of DCF staining are shown. *, p < 0.05 versus control; †, p < 0.05 versus hypoxia/reoxygenation (H/R); ‡, p < 0.05 versus antimycin A (AA). Error bars represent S.E. a.u., arbitrary units.
Having found that activity of GSK-3β is required for mitochondrial translocation and ROS production under the condition of oxidative stress, we investigated whether oxidative stress enhances the activity of GSK-3β, thereby inducing its translocation. However, Ser-9 phosphorylation of GSK-3β, Tyr-216 phosphorylation of GSK-3β, and phosphorylation of glycogen synthase were not affected by H2O2 challenge (Fig. 4A, left panels). This was also the case when phosphorylation status of mitochondrial GSK-3β was assessed (Fig. 4A, right panels). These results exclude the possibility that alteration in phosphorylation or kinase activity of GSK-3β contributes to the translocation induced by H2O2.
FIGURE 4.

Effects of H2O2 on GSK-3β activity and its interaction with other proteins. A, H9c2 cells were exposed to 100 μmol/liter H2O2 or a vehicle (Ve) for 4 h and harvested. Results of Western blotting for total GSK-3β, Ser-9-phospho-GSK-3β, Tyr-216-phospho-GSK-3β, total glycogen synthase, and phospho-glycogen synthase in the total homogenate (left panels) and total GSK-3β and Ser-9-phospho-GSK-3β in the mitochondrial fraction (right panels) are shown. β-Actin and prohibitin serve as loading controls. B, representative two-dimensional gels stained by Coomassie Blue. Samples were obtained from GSK-3β immunoprecipitates of H9c2 cells that were treated with a vehicle or H202 (100 μmol/liter) for 3 h. Results of four experiments for vehicle-treated cells and four experiments for H2O2-exposed cells were similar. Spots that exhibited increases in the density by 3-fold or more after exposure to H2O2 are labeled as 1–9. C, HEK293 cells transfected with FLAG-VDAC2 were exposed to H2O2 (100 μmol/liter; 4 h) in the presence or absence of LiCl (30 mmol/liter). Immunoblots (IB) for FLAG and GSK-3β co-immunoprecipitated (IP) with FLAG-VDAC2 are shown.
We next examined whether oxidative stress-induced interaction of GSK-3β with other proteins plays a role in the mitochondrial translocation of GSK-3β. H9c2 cells exposed to H2O2 or a vehicle were harvested and immunoprecipitated by an anti-GSK-3β antibody. LDH release into the culture medium at the time of cell harvest under this condition was only 12% of the total LDH, and absence of electroporation before H2O2 challenge and higher confluence are possible explanations for the much smaller rate of cell death than that in experiments using cells transfected with plasmids. The anti-GSK-3β immunoprecipitates were then separated by two-dimensional gel electrophoresis and stained with Coomassie Blue, which showed that densities of nine spots were increased after H2O2 treatment by more than 3-fold as labeled in Fig. 4B. We excised the nine spots and performed mass spectrometry analyses.
Among the proteins identified (Table 1), we focused on VDAC2, which was present in spot number 2 in Fig. 4B. Oxidative stress-induced interaction between GSK-3β and VDAC2 was further confirmed by immunoprecipitation experiments in which FLAG-tagged VDAC2 was co-immunoprecipitated with GSK-3β under an oxidant stress condition (Fig. 4C). The GSK-3β/VDAC2 interaction was also dependent on the kinase activity as it was inhibited by pretreatment with LiCl.
TABLE 1.
Proteins predicted from the peptides identified by mass spectrometric analysis
Peptide (95%), number of peptides observed in each sample with a probability of >95% by PARAGON algorithm; Coverage, percentage of the sequence of corresponding protein covered by identified peptides (%); N/A, not available.
| Spot | Protein name | Accession no. | Peptide (95%) | Coverage |
|---|---|---|---|---|
| % | ||||
| 1 | α-Actin 3 | P63269 | 32 | 60.1 |
| Major β-hemoglobin | AAA41309.1 | 3 | 34.0 | |
| β-Actin | P60711 | 59 | 65.3 | |
| Phosphatidylinositol-4,5-bisphosphate 5-phosphatase A | Q9JMC1.1 | 1 | 4.2 | |
| Pretrypsinogen 1 | P00762 | 1 | 8.1 | |
| α-Cardiac actin | P68035.1 | 37 | 55.7 | |
| 2 | Latent transforming growth factor β-binding protein 3 | XP_341998.1 | 1 | 2.9 |
| Voltage-dependent anion-selective channel protein 2 | P81155 | 1 | 6.4 | |
| α2-Globin chain | Q91V15 | 1 | 10.6 | |
| ATP-binding cassette protein B1b | Q8R427 | 1 | 1.5 | |
| 3 | N/A | |||
| 4 | Albumin | Q5U3X3 | 5 | 10.2 |
| α-Actin 2 | P62738 | 11 | 40.3 | |
| DiGeorge syndrome critical region 6 homolog | NP_001100550 | 1 | 10.0 | |
| Mosaic serine protease | XP_236201.4 | 1 | 4.4 | |
| α-Fetoglobulin | P02773.1 | 1 | 1.1 | |
| 5 | Protein-disulfide isomerase A3 | P11598.2 | 1 | 6.9 |
| 6 | Heterogenous nuclear ribonucleoprotein H1 | EDM04273.1 | 1 | 5.1 |
| 7 | α-Tropomyosin | Q91XN7 | 5 | 33 |
| Angiotensinogen | P01015 | 1 | 4.6 | |
| Unconventional myosin-9b | Q63358.1 | 1 | 1.2 | |
| 8 | Tropomyosin-4 | P09495.3 | 20 | 50.4 |
| 9 | β-Tropomyosin | P58775.1 | 2 | 23.6 |
To explore the functional significance of VDAC2 in the mitochondrial translocation of GSK-3β, H9c2 cells were transfected with VDAC2 siRNA, which suppressed the expression of mRNA (Fig. 5A) and protein (Fig. 5B) of VDAC2 but not the mRNA or protein expression of other VDAC isoforms VDAC1 and VDAC3. When VDAC2 was knocked down by siRNA, mitochondrial localization of WT was significantly reduced at baseline, and its mitochondrial translocation under the condition of oxidative stress was almost abolished (Fig. 5C and supplemental Movie 2B) compared with mitochondrial localization and translocation in cells transfected with control siRNA (supplemental Movie 2A). Knockdown of other isoforms VDAC1 and VDAC3 did not affect GSK-3β translocation (Fig. 5D), indicating an isoform-specific role of VDAC2 in translocation of GSK-3β. We next examined whether GSK-3β activity-associated ROS production (Fig. 3) is mechanistically linked to the VDAC2-mediated mitochondrial translocation of GSK-3β (Fig. 5C). Mitochondrial superoxide production was correlated with the extent of mitochondrial translocation of GFP-GSK-3β constructs; the level of superoxide was higher in S9A-transfected cells and lower in K85R-transfected cells than in WT-transfected cells. Knockdown of VDAC2 markedly suppressed superoxide production in cells transfected with WT or S9A (Fig. 5, E and F).
FIGURE 5.
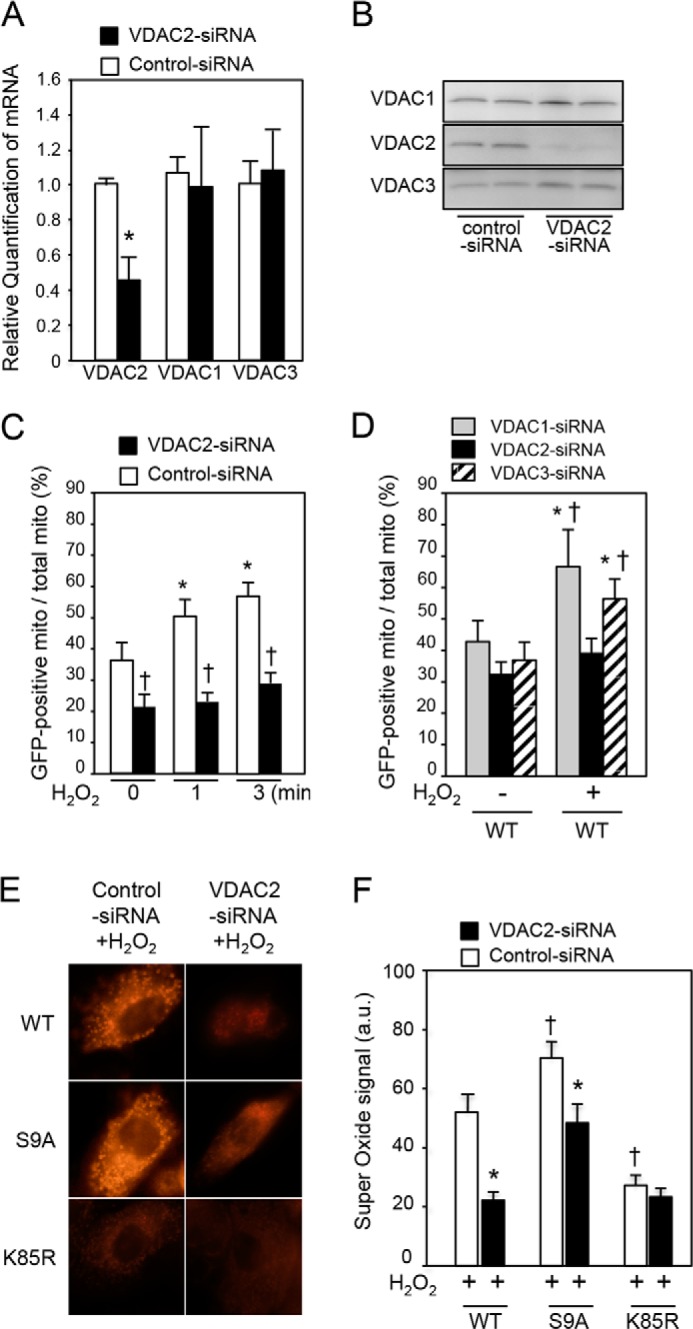
VDAC2-dependent mitochondrial translocation of GSK-3β and mitochondrial production of superoxide. A and B, mRNA levels in H9c2 cells (A) and protein levels in HEK293 cells (B) of VDAC1, VDAC2, and VDAC3 at 48 h after transfection of VDAC2 siRNA. *, p < 0.05 versus control siRNA. C, mitochondrial localization of GFP-tagged GSK-3β (WT) expressed as a percentage of GFP-positive mitochondria among total mitochondria at baseline and at 1 and 3 min after exposure to H2O2 in H9c2 cells. *, p < 0.05 versus baseline; †, p < 0.05 versus control siRNA. D, HEK293 cells were transfected with VDAC1 siRNA, VDAC2 siRNA, or VDAC3 siRNA. Mitochondrial localization of GFP-tagged GSK-3β (WT) expressed as a percentage of GFP-positive mitochondria (mito) among total mitochondria with or without exposure to H2O2 (10 μmol/liter; 3 min) is shown. *, p < 0.05 versus baseline; †, p < 0.05 versus VDAC2 siRNA. E, effects of VDAC2 and kinase activity of GSK-3β on generation of superoxide. Superoxide signal after exposure to H2O2 in H9c2 cells transfected with WT, S9A, or K85R is shown. F, quantitative analysis of superoxide-positive pixels per cell is shown. *, p < 0.05 versus control siRNA; †, p < 0.05 versus WT. Error bars represent S.E. a.u., arbitrary units.
To examine whether knockdown of VDAC2 attenuates mPTP opening, we observed morphology of mitochondria by super-resolution microscopy (N-SIM, Nikon). In cells transfected with control siRNA, intermittent exposure to a laser for time lapse observation induced mitochondrial deformation from a long and tubular shape to a swollen and round shape with occasional fragmentation (Fig. 6A, arrows, and supplemental Movie 3), indicating opening of the mPTP and mitochondrial destruction. The mitochondrial deformation/fragmentation were significantly suppressed by siRNA-mediated knockdown of VDAC2 (Fig. 6, A and B). Because outer membrane permeabilization could have been involved in the mitochondrial deformation in response to exposure to the laser, we next examined the protein levels of mitochondrial BAX and BAK, members of the multidomain subfamily of proapoptotic Bcl-2 proteins. BAX, but not BAK, in mitochondrial fractions increased in response to H2O2 (Fig. 6C) most likely due to translocation from the cytoplasm as reported previously (22), possibly inducing outer membrane permeabilization together with BAK. However, mitochondrial levels of the proteins were not affected by VDAC2 knockdown (Fig. 6C).
FIGURE 6.

Effects of VDAC2 knockdown on mitochondrial deformation, mPTP opening, and cell necrosis by oxidant stress. A, photos of mitochondria obtained from time lapse observation by super-resolution microscopy (N-SIM). H9c2 cells were transfected with control siRNA or VDAC2 siRNA and then stained with MitoTracker Red before observation by N-SIM. Arrows indicate swollen mitochondria after intermittent laser scanning for time lapse observation. B, percentage of swollen or fragmented mitochondria after 4-min observation by N-SIM. *, p < 0.05 versus control siRNA. C, immunoblots for BAK and BAX in the mitochondrial fraction are shown. Prohibitin served as a loading control. Successful mitochondrial fractionation is indicated by clear prohibitin bands and barely detectable β-actin bands. D, open-closed status of mPTPs determined by calcein assay in HEK293 cells. Images of calcein-stained cells transfected with VDAC1 siRNA, VDAC2 siRNA, or VDAC3 siRNA in the presence or absence of H2O2 and/or LiCl are shown. E, the ratio of calcein-positive area to MitoTracker-positive area is shown as an index for mitochondria (mito) that were not subjected to opening of mPTPs. *, p < 0.05 versus baseline (without H2O2 and LiCl); †, p < 0.05 versus VDAC2 siRNA. F, LDH activity in the culture medium was measured at 4 h after addition of H2O2 (100 μmol/liter). -Fold increases in LDH activity compared with the baseline value are shown. G, Western blotting for GSK-3β in H9c2 cells transfected with GFP-GSK-3β construct (WT, S9A, or K85R) or GFP control. Endogenous GSK-3β and GFP-tagged GSK-3β constructs were detected around 46 and 79 kDa, respectively. Error bars represent S.E.
To further confirm the suppression of mPTP opening by selective knockdown of VDAC2, a calcein assay was performed. As shown in Fig. 6D, calcein preloaded in mitochondria leaked into the cytoplasm after exposure to H2O2, indicating opening of mPTPs, in VDAC1- or VDAC3-knocked down cells. Opening of mPTPs was significantly attenuated in those cells by treatment with LiCl (Fig. 6, D and E). In contrast, knockdown of VDAC2 alone significantly suppressed opening of mPTPs (Fig. 6, D and E). As expected from its effect on the mPTP (Fig. 6, D and E) and superoxide production (Fig. 5E), cell necrosis after H2O2 exposure was also suppressed by knockdown of VDAC2 (Fig. 6F). Significant reduction in LDH release by VDAC2 siRNA in the K85R group can be explained by reduction of interaction between VDAC2 and endogenous GSK-3β in the K85R-transfected cell group (Fig. 6G).
Co-localization of GSK-3β with mitochondria (Fig. 1) and co-immunoprecipitation of GSK-3β with cyclophilin D and Rieske in the present study (Fig. 1D) and adenine nucleotide translocase in previous studies (17, 18) indicate an intramitochondrial pool of GSK-3β. However, to our knowledge, an MTS by which nucleus-encoded mitochondrial proteins are directed to the mitochondria (27–30) has not been identified in GSK-3β. MTS consists of 15–40 N-terminal amino acid residues dotted with a few positively charged amino acid residues, arginine (Arg) or lysine (Lys) (30). Because MitoProt II, an MTS-predicting algorithm (31), indicated moderate probability of the N-terminal domain of GSK-3β functioning as an MTS, we examined whether this mechanism indeed operates in GSK-3β. A positively charged amino acid, namely Arg-4, Arg-6, or Lys-15, in WT was replaced with alanine by site-directed mutagenesis. H9c2 cells were transfected with the mutants (R4A, R6A, and K15A) and exposed to H2O2. Unexpectedly, R4A translocated to the mitochondria more promptly than did WT in response to H2O2 challenge (Fig. 7). Conversely, mitochondrial translocation of R6A tended to be attenuated, and that of K15A was significantly suppressed compared with that of WT. These findings suggest that the N-terminal domain of GSK-3β serves as a functional MTS, although the inhibitory effect of the site-directed mutation was much less prominent than the effect of VDAC2 knockdown.
FIGURE 7.
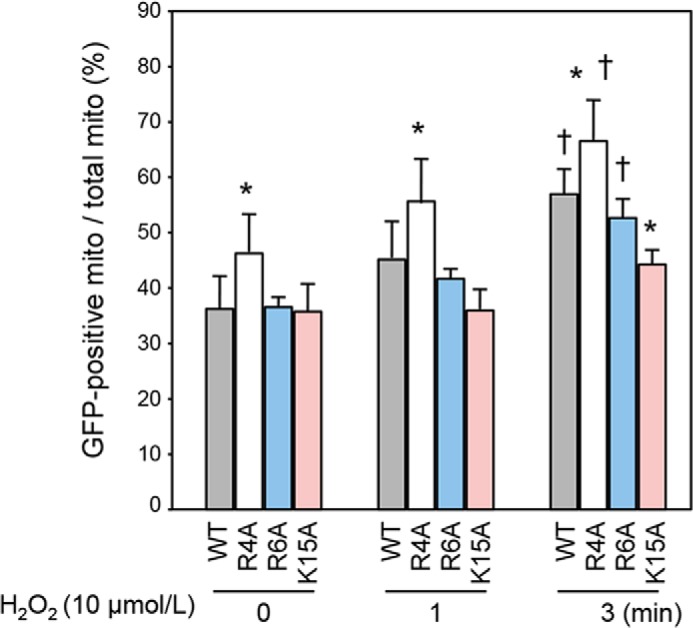
Effects of mutations in the N-terminal domain of GSK-3β on mitochondrial translocation. Percentages of GFP-positive mitochondria (mito) among total mitochondria at baseline and at 1 and 3 min after H2O2 exposure are shown. *, p < 0.05 versus WT; †, p < 0.05 versus baseline. Error bars represent S.E.
DISCUSSION
In this study, we demonstrated that kinase activity of GSK-3β and interaction with VDAC2 are major determinants of GSK-3β translocation to mitochondria in response to ROS and that GSK-3β activity enhances GSK-3β/VDAC2 interaction in H9c2 cardiomyocytes. Increased ROS production from the mitochondria by hypoxia/reoxygenation or inhibition of complex III is also dependent on the activity of GSK-3β (Fig. 3). The findings are consistent with an earlier finding by King et al. (32) that transfection of unregulated GSK-3β fused to an MTS of subunit VIII of cytochrome oxidase significantly increased ROS production in SH-SY5Y cells. An association between mitochondrial translocation of GSK-3β and mPTP-mediated cell death has been observed in various cell and tissue preparations (33–35) and was also confirmed in this study (Figs. 1, 2, and 6). Thus, kinase activity of GSK-3β seems to play dual roles: it is required both for cytosolic GSK-3β to translocate to the mitochondria and for mitochondrial GSK-3β to induce ROS production, leading to mPTP opening.
Interaction of GSK-3β with cyclophilin D, a mitochondrial matrix protein, and with Rieske, a subunit of complex III, was increased upon oxidative stress (Fig. 1D). These findings are consistent with results of studies showing that GSK-3β interacts with cyclophilin D in SAOS-2 cells (36) and that cyclophilin D interacts with F0F1 ATPase, a protein in the inner membrane, in bovine heart mitochondria (37). Together with previous findings, the present results indicate that GSK-3β is localized in the mitochondrial inner membrane where a part of its molecule is facing the matrix. Furthermore, interaction of VDAC2 and GSK-3β (Fig. 4, B and C) and significant suppression of mitochondrial translocation of GSK-3β by VDAC2 knockdown (Fig. 5, C and D) suggest that transport of GSK-3β into the inner membrane via translocase of the outer membrane (TOM) complex is promoted by its interaction with VDAC2 initially in the cytoplasm or on the outer membrane. However, there is also the possibility that VDAC2 and GSK-3β form a complex at the contact site between the outer and inner membranes (38).
VDAC2 has been identified as an important substrate of GSK-3β under the condition of ischemia/reperfusion in cardiomyocytes (39). Das et al. (39) showed that inhibition of GSK-3β slowed ATP consumption in cardiomyocytes under an anoxic condition. They performed Western blotting and one-dimensional/two-dimensional gel phosphorylation site analysis and detected proteins that were less phosphorylated in hearts treated with GSK-3β inhibitors. One of the less phosphorylated proteins was identified as VDAC2 by MALDI-TOF/MS analysis. Collectively, their findings suggest that VDAC2 phosphorylated by GSK-3β on the mitochondrial outer membrane accelerates ATP depletion, lowering the threshold for mPTP opening. The present study showed a novel role for VADC2 in mPTP regulation as a promoter of GSK-3β import into mitochondria in response to oxidant stress.
Cheng et al. (40) demonstrated previously that VDAC2 interacts with BAK and maintains its inactive conformation. Conversely, tBID, BIM, or BAD activated by death signals displaces VDAC2 from BAK and induces apoptosis via homo-oligomerization of BAK. In fact, cells lacking VDAC2 exhibited enhanced BAK oligomerization and were susceptible to apoptotic death induced by TNFα, cycloheximide, or etoposide, whereas overexpression of VDAC2 selectively prevented BAK activation and inhibited the mitochondrial apoptotic pathway (40). However, in the present study, knockdown of VDAC2 did not modify levels of BAX and BAK in mitochondria before and after exposure to oxidant stress (Fig. 6C) but rather protected H9c2 cells. Knockdown of VDAC2 significantly suppressed translocation of GSK-3β to mitochondria (Fig. 5, C and D), mPTP opening (Fig. 6, A, B, D, and E), ROS production (Fig. 5, E and F), and cell necrosis (Fig. 6F) after oxidant stress in H9c2 and/or HEK293 cells. These results indicate that VDAC2 plays distinct roles in cell death depending on the mode of cell death and/or stimuli that trigger cell death: promotion of cell necrosis by interaction with GSK-3β versus protection via interaction with BAK against apoptosis triggered by inflammation or DNA damage.
There has been solid genetic evidence that VDAC protein is not an essential structural component of the mPTP. Baines et al. (41) demonstrated that isolated mitochondria from VDAC1-, VDAC2-, or VDAC3-null mice exhibited Ca2+- and oxidative stress-induced mPTP opening that was indistinguishable from wild-type mitochondria. Similarly, Ca2+- and oxidative stress-induced mPTP opening was unaltered in fibroblasts lacking VDAC1, VDAC2, or VDAC3 (41). However, these results are basically derived from isolated mitochondria and do not exclude the possibility that VDAC2 plays a regulatory role in opening/closure of the mPTP in some cell types.
Several lines of evidence indicate that the role of VDAC2 in determination of cell fate under pathological conditions is not limited to cardiomyocytes. Huge VDAC accumulation has been observed in the dystrophic neurites of β-amyloid plaques in Alzheimer disease patients and in a related transgenic mouse model (42). In addition, post-mortem analysis of Alzheimer disease patient brains revealed that VDAC2 was significantly increased in the temporal cortex (43), a region frequently affected in Alzheimer disease pathology. Transcript levels of the VDAC1–3 isoforms in rat hepatoma cells were found to be significantly higher than those in normal rat liver tissue (44). More interestingly, erastin, a selective anticancer agent discovered in screening by Yagoda et al. (45), has been shown to bind to VDAC2, which causes mitochondrial damage via ROS production, inducing non-apoptotic cell death.
Our data showed that the each site-directed mutant at the N terminus had a different impact on mitochondrial translocation of GSK-3β: R4A translocated to the mitochondria more promptly than did WT, whereas mitochondrial translocation of R6A and that of K15A were slightly attenuated and significantly suppressed, respectively. A possible explanation for the differences depending on the site of Arg- or Lys-to-Ala mutation is co-modification of interaction with Akt/PKB. RXRXX(S/T) was found to be a consensus phosphorylation motif for Akt/PKB (46), a major upstream kinase that phosphorylates GSK-3β on Ser-9, leading to inactivation. In fact, the levels of Ser-9 phosphorylation of R4A and R6A were much lower, indicating higher kinase activities, than that of WT or K15A (data not shown). The up-regulation of kinase activities in R4A and R6A might have canceled the effect of MTS elimination in these mutants. Another possibility is that the N-terminal domain functions as a binding site for VDAC2 as is the case with 14-3-3 or p53 (47), resulting in different levels of interaction with VDAC2 depending on mutated amino acids.
Even K15A yielded only a small change in the mitochondrial translocation. Thus, site-directed mutation of multiple amino acids, but not a single amino acid, might have been necessary to fully disable the function of MTS. Alternatively, the role MTS plays in mitochondrial translocation might be relatively small compared with that of kinase activity of GSK-3β and interaction with VDAC2.
In the present study, 30 mmol/liter LiCl was used for inhibiting GSK-3β. This dose significantly increased the Ser-9 phosphorylation of GSK-3β and decreased the phosphorylation of glycogen synthase in H9c2 cells (data not shown). LiCl is not specific to GSK-3β and has other targets including voltage-dependent sodium channels (48), Na/K-ATPase (49), and inositol monophosphate (50). However, LiCl significantly suppressed H2O2-induced mitochondrial translocation of WT (Fig. 1, A and B), and such an effect was not observed for constitutively active S9A or kinase-deficient K85R (data not shown). Thus, it is unlikely that the effect of LiCl on a target other than GSK-3β activity was responsible for the attenuation of mitochondrial translocation.
As a technical problem, cells stained with MitoTracker Red and/or transfected with GFP-GSK-3β constructs are more vulnerable to exposure to H2O2; in those cells, H2O2 exposure for a much shorter duration at a lower concentration was sufficient to induce cell death during observation by fluorescence microscopy. This is most likely due to light-induced photoexcitation of GFP and MitoTracker Red. Thus, we performed preliminary experiments to optimize the duration and concentration of H2O2 in each series of experiments in this study. We used a concentration of H2O2 as low as 10 μmol/liter for 3-min exposure to the laser to induce mitochondrial translocation of GSK-3β and under fluorescence observation in H9c2 cells that were transfected with a GFP-GSK-3β construct and stained with MitoTracker Red. In contrast, it took 4 h for 100 μmol/liter H2O2 to induce a similar level of cell death in cells that were not stained by MitoTracker Red, transfected with GFP constructs, or observed by fluorescence microscopy.
Limitation of myocardial infarct size after ischemia/reperfusion by inhibition of mPTP opening has been demonstrated not only in animal experiments (51–54) but also in a small clinical trial (55). However, it is difficult to use currently available mPTP inhibitors in patients because of their various side effects. Selective inhibition of GSK-3β translocation to mitochondria would be another option to suppress mPTP opening upon reperfusion. This approach would be superior to the use of GSK-3β inhibitors, which can unfavorably affect the physiological function of GSK-3β (56). The results of the present study indicate that inhibition of GSK-3β/VDAC2 interaction and/or function of MTS in GSK-3β is a promising and novel approach to cardioprotection from lethal reperfusion injury.
Supplementary Material
Acknowledgment
We are grateful to the Nikon Imaging Center at Hokkaido University for technical assistance with the time lapse study.
This work was supported by Japanese Society for the Promotion of Science Grants-in-aid for Scientific Research 23501086 and 26461133.

This article contains supplemental Movies 1–3.
- mPTP
- mitochondrial permeability transition pore
- GSK-3β
- glycogen synthase kinase-3β
- VDAC
- voltage-dependent anion channel
- MTS
- mitochondrial targeting sequence
- DCF
- 2′,7′-dichlorofluorescein
- ROS
- reactive oxygen species
- LDH
- lactate dehydrogenase.
REFERENCES
- 1. Miura T., Tanno M. (2012) The mPTP and its regulatory proteins: final common targets of signalling pathways for protection against necrosis. Cardiovasc. Res. 94, 181–189 [DOI] [PubMed] [Google Scholar]
- 2. Weiss J. N., Korge P., Honda H. M., Ping P. (2003) Role of the mitochondrial permeability transition in myocardial disease. Circ. Res. 93, 292–301 [DOI] [PubMed] [Google Scholar]
- 3. Halestrap A. P. (2009) What is the mitochondrial permeability transition pore? J Mol. Cell. Cardiol. 46, 821–831 [DOI] [PubMed] [Google Scholar]
- 4. Miura T., Tanno M., Sato T. (2010) Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc. Res. 88, 7–15 [DOI] [PubMed] [Google Scholar]
- 5. Halestrap A. P. (2010) A pore way to die: the role of mitochondria in reperfusion injury and cardioprotection. Biochem. Soc. Trans. 38, 841–860 [DOI] [PubMed] [Google Scholar]
- 6. Di Lisa F., Bernardi P. (2006) Mitochondria and ischemia-reperfusion injury of the heart: fixing a hole. Cardiovasc. Res. 70, 191–199 [DOI] [PubMed] [Google Scholar]
- 7. Suh D. H., Kim M. K., Kim H. S., Chung H. H., Song Y. S. (2013) Mitochondrial permeability transition pore as a selective target for anti-cancer therapy. Front. Oncol. 3, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eckert G. P., Renner K., Eckert S. H., Eckmann J., Hagl S., Abdel-Kader R. M., Kurz C., Leuner K., Muller W. E. (2012) Mitochondrial dysfunction—a pharmacological target in Alzheimer's disease. Mol. Neurobiol. 46, 136–150 [DOI] [PubMed] [Google Scholar]
- 9. Guo L., Du H., Yan S., Wu X., McKhann G. M., Chen J. X., Yan S. S. (2013) Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer's neurons. PLoS One 8, e54914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen B., Xu M., Zhang H., Wang J. X., Zheng P., Gong L., Wu G. J., Dai T. (2013) Cisplatin-induced non-apoptotic death of pancreatic cancer cells requires mitochondrial cyclophilin-D-p53 signaling. Biochem. Biophys. Res. Commun. 437, 526–531 [DOI] [PubMed] [Google Scholar]
- 11. Juhaszova M., Zorov D. B., Yaniv Y., Nuss H. B., Wang S., Sollott S. J. (2009) Role of glycogen synthase kinase-3β in cardioprotection. Circ. Res. 104, 1240–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liao R., Force T. (2007) Not all hypertrophy is created equal. Circ. Res. 101, 1069–1072 [DOI] [PubMed] [Google Scholar]
- 13. Dajani R., Fraser E., Roe S. M., Young N., Good V., Dale T. C., Pearl L. H. (2001) Crystal structure of glycogen synthase kinase 3β: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105, 721–732 [DOI] [PubMed] [Google Scholar]
- 14. Eom T. Y., Jope R. S. (2009) GSK3β N-terminus binding to p53 promotes its acetylation. Mol. Cancer 8, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pedretti S., Raddatz E. (2011) STAT3α interacts with nuclear GSK3β and cytoplasmic RISK pathway and stabilizes rhythm in the anoxic-reoxygenated embryonic heart. Basic Res. Cardiol. 106, 355–369 [DOI] [PubMed] [Google Scholar]
- 16. Jope R. S., Johnson G. V. (2004) The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 29, 95–102 [DOI] [PubMed] [Google Scholar]
- 17. Juhaszova M., Zorov D. B., Kim S. H., Pepe S., Fu Q., Fishbein K. W., Ziman B. D., Wang S., Ytrehus K., Antos C. L., Olson E. N., Sollott S. J. (2004) Glycogen synthase kinase-3β mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J. Clin. Investig. 113, 1535–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nishihara M., Miura T., Miki T., Tanno M., Yano T., Naitoh K., Ohori K., Hotta H., Terashima Y., Shimamoto K. (2007) Modulation of the mitochondrial permeability transition pore complex in GSK-3β-mediated myocardial protection. J. Mol. Cell. Cardiol. 43, 564–570 [DOI] [PubMed] [Google Scholar]
- 19. Miki T., Miura T., Hotta H., Tanno M., Yano T., Sato T., Terashima Y., Takada A., Ishikawa S., Shimamoto K. (2009) Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3β-mediated suppression of mitochondrial permeability transition. Diabetes 58, 2863–2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yano T., Miki T., Tanno M., Kuno A., Itoh T., Takada A., Sato T., Kouzu H., Shimamoto K., Miura T. (2011) Hypertensive hypertrophied myocardium is vulnerable to infarction and refractory to erythropoietin-induced protection. Hypertension 57, 110–115 [DOI] [PubMed] [Google Scholar]
- 21. Hoeflich K. P., Luo J., Rubie E. A., Tsao M. S., Jin O., Woodgett J. R. (2000) Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 406, 86–90 [DOI] [PubMed] [Google Scholar]
- 22. Ohori K., Miura T., Tanno M., Miki T., Sato T., Ishikawa S., Horio Y., Shimamoto K. (2008) Ser9 phosphorylation of mitochondrial GSK-3β is a primary mechanism of cardiomyocyte protection by erythropoietin against oxidant-induced apoptosis. Am. J. Physiol. Heart Circ. Physiol. 295, H2079–H2086 [DOI] [PubMed] [Google Scholar]
- 23. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 24. Jope R. S. (2003) Lithium and GSK-3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol. Sci. 24, 441–443 [DOI] [PubMed] [Google Scholar]
- 25. Muller F. L., Liu Y., Van Remmen H. (2004) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 279, 49064–49073 [DOI] [PubMed] [Google Scholar]
- 26. Lenaz G. (2001) The mitochondrial production of reactive oxygen species: mechanisms and implications in human pathology. IUBMB Life 52, 159–164 [DOI] [PubMed] [Google Scholar]
- 27. Becker T., Böttinger L., Pfanner N. (2012) Mitochondrial protein import: from transport pathways to an integrated network. Trends Biochem. Sci. 37, 85–91 [DOI] [PubMed] [Google Scholar]
- 28. Chacinska A., Koehler C. M., Milenkovic D., Lithgow T., Pfanner N. (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dolezal P., Likic V., Tachezy J., Lithgow T. (2006) Evolution of the molecular machines for protein import into mitochondria. Science 313, 314–318 [DOI] [PubMed] [Google Scholar]
- 30. Omura T. (1998) Mitochondria-targeting sequence, a multi-role sorting sequence recognized at all steps of protein import into mitochondria. J. Biochem. 123, 1010–1016 [DOI] [PubMed] [Google Scholar]
- 31. Claros M. G., Vincens P. (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241, 779–786 [DOI] [PubMed] [Google Scholar]
- 32. King T. D., Clodfelder-Miller B., Barksdale K. A., Bijur G. N. (2008) Unregulated mitochondrial GSK3β activity results in NADH: ubiquinone oxidoreductase deficiency. Neurotox. Res. 14, 367–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pastorino J. G., Hoek J. B., Shulga N. (2005) Activation of glycogen synthase kinase 3β disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545–10554 [DOI] [PubMed] [Google Scholar]
- 34. Chiara F., Gambalunga A., Sciacovelli M., Nicolli A., Ronconi L., Fregona D., Bernardi P., Rasola A., Trevisan A. (2012) Chemotherapeutic induction of mitochondrial oxidative stress activates GSK-3α/β and Bax, leading to permeability transition pore opening and tumor cell death. Cell Death Dis. 3, e444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Z., Ge Y., Bao H., Dworkin L., Peng A., Gong R. (2013) Redox-sensitive glycogen synthase kinase 3β-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic. Biol. Med. 65, 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rasola A., Sciacovelli M., Chiara F., Pantic B., Brusilow W. S., Bernardi P. (2010) Activation of mitochondrial ERK protects cancer cells from death through inhibition of the permeability transition. Proc. Natl. Acad. Sci. U.S.A. 107, 726–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Giorgio V., Bisetto E., Soriano M. E., Dabbeni-Sala F., Basso E., Petronilli V., Forte M. A., Bernardi P., Lippe G. (2009) Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J. Biol. Chem. 284, 33982–33988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Harner M., Körner C., Walther D., Mokranjac D., Kaesmacher J., Welsch U., Griffith J., Mann M., Reggiori F., Neupert W. (2011) The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 30, 4356–4370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Das S., Wong R., Rajapakse N., Murphy E., Steenbergen C. (2008) Glycogen synthase kinase 3 inhibition slows mitochondrial adenine nucleotide transport and regulates voltage-dependent anion channel phosphorylation. Circ. Res. 103, 983–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cheng E. H., Sheiko T. V., Fisher J. K., Craigen W. J., Korsmeyer S. J. (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301, 513–517 [DOI] [PubMed] [Google Scholar]
- 41. Baines C. P., Kaiser R. A., Sheiko T., Craigen W. J., Molkentin J. D. (2007) Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 9, 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pérez-Gracia E., Torrejón-Escribano B., Ferrer I. (2008) Dystrophic neurites of senile plaques in Alzheimer's disease are deficient in cytochrome c oxidase. Acta Neuropathol. 116, 261–268 [DOI] [PubMed] [Google Scholar]
- 43. Yoo B. C., Fountoulakis M., Cairns N., Lubec G. (2001) Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer's disease and Down syndrome. Electrophoresis 22, 172–179 [DOI] [PubMed] [Google Scholar]
- 44. Shinohara Y., Ishida T., Hino M., Yamazaki N., Baba Y., Terada H. (2000) Characterization of porin isoforms expressed in tumor cells. Eur. J. Biochem. 267, 6067–6073 [DOI] [PubMed] [Google Scholar]
- 45. Yagoda N., von Rechenberg M., Zaganjor E., Bauer A. J., Yang W. S., Fridman D. J., Wolpaw A. J., Smukste I., Peltier J. M., Boniface J. J., Smith R., Lessnick S. L., Sahasrabudhe S., Stockwell B. R. (2007) RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Obata T., Yaffe M. B., Leparc G. G., Piro E. T., Maegawa H., Kashiwagi A., Kikkawa R., Cantley L. C. (2000) Peptide and protein library screening defines optimal substrate motifs for AKT/PKB. J. Biol. Chem. 275, 36108–36115 [DOI] [PubMed] [Google Scholar]
- 47. Goñi-Oliver P., Avila J., Hernández F. (2011) Calpain regulates N-terminal interaction of GSK-3β with 14-3-3ζ, p53 and PKB but not with axin. Neurochem. Int. 59, 97–100 [DOI] [PubMed] [Google Scholar]
- 48. Yanagita T., Maruta T., Uezono Y., Satoh S., Yoshikawa N., Nemoto T., Kobayashi H., Wada A. (2007) Lithium inhibits function of voltage-dependent sodium channels and catecholamine secretion independent of glycogen synthase kinase-3 in adrenal chromaffin cells. Neuropharmacology 53, 881–889 [DOI] [PubMed] [Google Scholar]
- 49. Hermans A. N., Glitsch H. G., Verdonck F. (1997) Activation of the Na+/K+ pump current by intra- and extracellular Li ions in single guinea-pig cardiac cells. Biochim. Biophys. Acta 1330, 83–93 [DOI] [PubMed] [Google Scholar]
- 50. Sarkar S., Floto R. A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L. J., Rubinsztein D. C. (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Argaud L., Gateau-Roesch O., Muntean D., Chalabreysse L., Loufouat J., Robert D., Ovize M. (2005) Specific inhibition of the mitochondrial permeability transition prevents lethal reperfusion injury. J. Mol. Cell. Cardiol. 38, 367–374 [DOI] [PubMed] [Google Scholar]
- 52. Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen M. R., Gottlieb R. A., Dorn G. W., Robbins J., Molkentin J. D. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 [DOI] [PubMed] [Google Scholar]
- 53. Clarke S. J., McStay G. P., Halestrap A. P. (2002) Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Biol. Chem. 277, 34793–34799 [DOI] [PubMed] [Google Scholar]
- 54. Nakagawa T., Shimizu S., Watanabe T., Yamaguchi O., Otsu K., Yamagata H., Inohara H., Kubo T., Tsujimoto Y. (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434, 652–658 [DOI] [PubMed] [Google Scholar]
- 55. Piot C., Croisille P., Staat P., Thibault H., Rioufol G., Mewton N., Elbelghiti R., Cung T. T., Bonnefoy E., Angoulvant D., Macia C., Raczka F., Sportouch C., Gahide G., Finet G., André-Fouët X., Revel D., Kirkorian G., Monassier J. P., Derumeaux G., Ovize M. (2008) Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N. Engl. J. Med. 359, 473–481 [DOI] [PubMed] [Google Scholar]
- 56. Phukan S., Babu V. S., Kannoji A., Hariharan R., Balaji V. N. (2010) GSK3β: role in therapeutic landscape and development of modulators. Br. J. Pharmacol. 160, 1–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.