

Background: Following AIM2 inflammasome activation, EB1 is required for speck formation.
Results: EB1 regulates AIM2 inflammasome-induced IL-1β secretion via autophagic shedding after speck formation; furthermore, AMPK may regulate this via EB1.
Conclusion: AIM2 inflammasome-induced IL-1β secretion depends on autophagy-based secretory trafficking through EB1 regulation.
Significance: We identify a novel role for EB1 in the non-classical secretion of IL-1β by AIM2 inflammasome complexes.
Keywords: AMP-activated Kinase (AMPK), Autophagy, Inflammation, Interleukin, Microtubule, EB1, IL-1b, Inflammasome
Abstract
Inflammasomes are multi-protein complexes that regulate chronic inflammation-associated diseases by inducing interleukin-1 β (IL-1β) secretion. Numerous components involved in inflammasome activation have been identified, but the mechanisms of inflammasome-mediated IL-1β secretion have not yet been fully explored. Here, we demonstrate that end-binding protein 1 (EB1), which is required for activation of AIM2 inflammasome complex, links the AIM2 inflammasome to autophagy-dependent secretion. Imaging studies revealed that AIM2 inflammasomes colocalize with microtubule organizing centers and autophagosomes. Biochemical analyses showed that poly(dA-dT)-activated AIM2 inflammasomes induce autophagy and IL-1β secretion in an LC3-dependent fashion. Furthermore, depletion of EB1 decreases autophagic shedding and intracellular trafficking. Finally, we found that the 5′-AMP activated protein kinase may regulate this EB1-mediated autophagy-based inflammasome-induced secretion of IL-1β. These findings reveal a novel EB1-mediated pathway for the secretion of IL-1β.
Introduction
Inflammasomes are intracellular protein complexes that sense various pathogenic infections and cause inflammation by inducing the secretion of interleukin 1 β (IL-1β) or interleukin 18 (IL-18) (1). Numerous studies have revealed that inflammasomes participate in various diseases (e.g. cancers, diabetes, and Alzheimer disease) by regulating IL-1β secretion (2–4). Inflammasomes are multi-protein complexes that comprise receptors (e.g. NLPR3, AIM2, and NLRC4), an adaptive protein ASC, and procaspase 1. The receptors activate procaspase 1 via ASC (apoptosis-associated speck-like protein containing a CARD)-dependent or -independent mechanisms, and the activated caspase 1 subsequently triggers IL-1β secretion (1). The typical mechanism of IL-1β secretion requires two signals: (i) transcriptional regulation of pro-IL-1β and (ii) inflammasome activation-mediated secretion of IL-1β. An activated inflammasome is seen as a single perinuclear complex in cells; this is called a “speck-like particle” (5). Interestingly, inflammasomes not only sense pathogenic infections, they also detect cellular damage signals (e.g. reactive oxygen species production and ATP release) (6, 7). Such innate immune responses are seen in immune cells and other cell types (2, 8), indicating that inflammasome activation is a topic of general interest. However, the underlying biological processes are not yet fully understood.
We previously reported that the NLRP3, AIM2, and RIG-I inflammasomes are activated in nasopharyngeal carcinoma (NPC)2 (2) and that end-binding protein 1 (EB1) is a novel component of these activated inflammasome complexes (9). EB1, which is a plus-end tracking protein (+TIP) that regulates microtubule polymerization by recruiting other +TIPs (10), is involved in various biological processes, including mitosis, migration, and signal transduction (10, 11). In our recent report, we showed that EB1 is critical for speck-like particle formation by AIM2 inflammasomes following the perception of double-stranded DNA (9). Furthermore, the perinuclear location of the inflammasome speck is known to be crucial for proper inflammasome activity and IL-1β secretion. However, the mechanisms underlying EB1-mediated speck formation and localization have not yet been thoroughly elucidated.
Autophagy is cellular vacuoles that governs cellular homeostasis and responds to cellular stress (12). Mechanistically, autophagy begins with the nucleation of proautophagic vacuoles at the microtubule organizing center (MTOC) (13). The autophagic vacuoles then mature; this may be tracked by the processing of LC3-I to LC3-II via upstream regulators (12). The mature autophagic vesicles then fuse with lysosomes, enabling the enzymatic digestion of cellular components. This supports cellular homeostasis and the clearance of pathogens (12). The intracellular trafficking of autophagic vacuoles has been shown to require microtubules (14), suggesting that microtubule polymerization is important for autophagic movement. Interestingly, autophagy not only prevents cellular damage by clearing proteins and pathogens, it also participates in the unconventional secretion of certain factors (15, 16). Given that autophagy can be triggered by ubiquitinated inflammasomes (17) and the secretion of IL-1β is known to require vesicle shedding (18, 19), we hypothesized that autophagy may be involved in the secretion of IL-1β.
Here, we demonstrate that EB1-activated AIM2 inflammasome specks colocalize with MTOCs and are engulfed in autophagic vacuoles. We show that AIM2 inflammasome-induced IL-1β secretion depends on autophagy and can be inhibited by silencing of the autophagic marker, LC3. In addition, we report that depletion of EB1 decreases autophagy and IL-1β shedding after AIM2 inflammasome activation. Finally, we show that 5′-AMP-activated protein kinase (AMPK) may regulate EB1-mediated AIM2 inflammasome activation and autophagy-based IL-1β secretion. Conclusively, we performed the third signal of inflammasome-mediated IL-1β secretion via autophagy, which was elicited by EB1-depended MTOCs location of AIM2 inflammasome speck and autophagic shedding.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following antibodies were utilized: anti-caspase 1, anti-EB1 (for immunoprecipitation), anti-LAMP1 and anti-α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA); anti-FLAG, anti-γ-tubulin, anti-LC3, anti-mouse IgG (10 nm of colloidal gold) and anti-FLAG M2 affinity gel (Sigma-Aldrich); anti-ASC (MBL, Woburn, MA); anti-GFP (GeneTex, Irvine, CA); anti-EB1 (for Western blot analysis; BD Transduction Laboratories); anti-AMPKα (Epitomic, Burlingame, CA); and anti-Thr(P)-172-AMPKα (Cell Signaling Technology, Danvers, MA). The poly(dA-dT), PMA, AICAR, rapamycin, chloroquine, and compound C were purchased from Sigma-Aldrich. Protein G Sepharose 4 Fast Flow was purchased from GE Healthcare. Finally, the Amicon Ultra 2-ml centrifugal filters 3K were purchased from Merck Millipore.
Plasmid Construction
The wild-type EB1 construct was PCR amplified from HK1 cDNA and cloned into HindIII/BamHI-treated pFLAG-CMV2 vectors (Clontech). Wild-type ASC was PCR amplified from HK1 cDNA and cloned into HindIII/BamHI-treated pEGFP-N1 vectors (Clontech).
Cell Culture
HeLa, NPC TW02, and HK1 cells were cultured as described previously (9), and THP1 cells were cultured in RPMI medium (9). Lentiviruses were established following the protocol of the RNAi Core of Taiwan and used to generate HK1 cells expressing mature human IL-1β. The cells were maintained with 500 μg/ml G418 (Invitrogen). For transfection assays, cells were transfected with poly(dA-dT) using Lipofectamine 2000 (Invitrogen).
Preparing the Anti-AIM2 Antibody
The sequence encoding the full-length human AIM2 cDNA was cloned into NdeI/BamHI-treated pET-15b vectors (Novagen). His-AIM2 was expressed in vector-transformed Rosetta competent cells treated with 0.5 mm isopropyl β-d-1-thiogalactopyranoside for 1.5 h, and the expressed proteins were purified using a nickel Sepharose High Performance system (GE Healthcare). Two American Rabbit Breeders Association (New Zealand) rabbits were immunized twice (interval, 1 month) with 1 mg of purified His-AIM2 in Freund's complete adjuvant. At 10–14 days after the second (boost) injection, anti-AIM2 sera were obtained from the immunized rabbits and purified.
Immunoprecipitation
Cells were treated with poly(dA-dT) for 6 h. Cell extracts (1 mg) were collected using radioimmune precipitation assay buffer (50 mm Tris-Cl, pH 7.5, 150 mm NaCl, 10 mm MgCl2, 1 mm ethylenediamine tetraacetate, pH 8.0, 1% Nonidet P-40, 100 mm sodium fluoride, and 1 mm phenylmethylenesulfonyl fluoride) and immunoprecipitated with specific antibodies (1 μg) or anti-FLAG M2 affinity gel for 24 h. The antibody-bound samples were precipitated with Protein G Sepharose 4 Fast Flow for 1 h at 4 °C, and the immunoprecipitated products were collected for Western blot analysis.
RNA Interference
Stealth RNAi reagents were purchased from Invitrogen. These included three 25-bp RNA duplexes for each of three targets, as follows: AIM2 (5′-UAUGGUGCUAUGAACUCCAGAUGUC-3′; 5′-GACAUCUGGAGUUCAUAGCACCAUA-3′; 5′-AAACCAUUCACAAUUGUUCCAAGGG-3′), EB1 (5′-CCGAAGAAACCUCUCACUUCUAGCA-3′; 5′-GGAUCAAUGAGUCUCUGCAGUUGAA-3′; 5′-UCAACGUAUUGAAACUUACUGUUGA-3′), LC3 (5′-CGGACCAUGUCAACAUGAGCGAGUU-3′; 5′-UCGCGGACAUCUACGAGCAGGAGAA-3′; 5′-GGCUUCCUCUAUAUGGUCUACGCCU-3′), and AMPKα (5′-CCCAUCCUGAAAGAGUACCAUUCUU-3′; 5′-CCCUCAAUAUUUAAAUCCUUCUGUG-3′; 5′-ACCAUGAUUGAUGAUGAAGCCUUAA-3′). The negative control siRNA was synthesized by Research Biolabs (Singapore). Cells were transfected with 75 pmol of dsRNA duplexes using Lipofectamine 2000 according to the manufacturer's instructions. After incubation for 6 h, the dsRNA complexes were removed, and the cells were replated in 5 ml of fresh medium.
IL-1β ELISA
Cell culture supernatants were assayed for human IL-1β using the human IL-1β ELISA Ready-SET-Go kit (eBioscience, San Diego, CA) according to the manufacturer's instructions.
Western Blot Analysis
Whole-cell extracts were homogenized and lysed in a buffer containing 50 mm Tris-Cl (pH 7.5), 150 mm NaCl, 10 mm MgCl2, 1 mm ethylenediamine tetraacetate (pH 8.0), 1% Nonidet P-40, 100 mm sodium fluoride, 1 mm phenylmethylenesulfonyl fluoride, and 2 μl/ml protease inhibitor mixture (Sigma-Aldrich). Protein concentrations were determined using a protein assay reagent (Bio-Rad), and equal protein amounts (30–50 μg/lane) were resolved by 12% SDS-PAGE. For the concentrated samples, the culture media were collected and concentrated with Amicon Ultra 2-ml centrifugal filters. After electrophoresis, the proteins were electrotransferred onto nitrocellulose membranes (Amersham Biosciences). The blots were blocked with 5% nonfat powdered milk in TBS, incubated overnight with the respective primary antibodies at 4 °C, and then incubated with HRP-conjugated rabbit/goat/mouse anti-IgG (Invitrogen) for 1 h at room temperature. Protein bands were detected by enhanced chemiluminescence (Pierce ECL, Thermo Scientific) on Fuji SuperRx films.
Immunofluorescence Microscopy
Cells grown on coverslips were fixed with 3.7% formaldehyde, permeabilized, and blocked overnight at 4 °C with PBS containing 1% BSA. The coverslips were incubated with the indicated primary antibodies for 2 h and then with the appropriate fluorophore-conjugated secondary antibodies (Invitrogen) for 1 h at room temperature. Nuclei were stained with 4′-6-diamidino-2-phenylindole (Sigma-Aldrich). All coverslips were mounted with the Vectashield reagent (Vector Laboratories) and visualized by confocal microscopy (Zeiss LSM510 META laser-scanning microscope; Carl Zeiss, Germany) with a 63 × 1.32 or 100 × 1.32 NA oil immersion objective.
Immunoelectron Microscopy
Cells were fixed in a mixture of 1% glutaraldehyde and 2% paraformaldehyde in 0.1 m sodium cacodylate buffer for 2 h and then embedded in 12% gelatin. The blocks were trimmed to size, immersed overnight at 4 °C in PVP/sucrose (15%/1.7 M), mounted on aluminum pins in the proper orientation, and stored in liquid nitrogen. Frozen sections were prepared with an ultracryomicrotome (Reichert Ultracut S/Reichert FCS; Leica, Vienna, Austria). To obtain ultrathin sections, 0.5-μm sections were first cut at −70 °C, stained with 0.1% Toluidine Blue in 1% boric acid, and examined under light microscopy. When the cells were reached within each block, the cutting temperature was lowered to −100 °C, and the section thickness was decreased to 55 nm. Ultrathin sections were picked up with a drop of an equal volume mixture of methylcellulose (2%)/sucrose (2.3 m). Each section was stained with anti-GFP antibodies in 0.1% BSA buffer for 30 min, followed by anti-IgG gold antibodies for 25 min at room temperature. The samples were finally mounted with methylcellulose/uranyl acetate buffer and viewed under a JOEL JEM-1230 transmission electron microscope.
RESULTS
Activated AIM2 Inflammasomes Colocalize with MTOCs
We previously reported that EB1 is a component of the NLRP3, AIM2, and RIG-I inflammasomes in NPC cells and that it is required for the formation of activated inflammasome specks (9). To investigate the biological function of EB1 in speck formation, we monitored specks (i.e. activated inflammasomes (5)) using fluorescence-tagged ASC. Following poly(dA-dT)-induced AIM2 inflammasome activation, GFP-ASC-labeled specks showed perinuclear localization (Fig. 1A). Together with our previous work (9), this finding indicates that AIM2 inflammasome specks show EB1-dependent perinuclear localization. Notably, we observed only one visible inflammasome speck per poly(dA-dT)-treated cell. Because EB1 regulates microtubule polymerization at the plus end of the microtubule (10), we speculated that EB1 may recruit the AIM2 inflammasome to the origin of the microtubule or to the MTOC. Immunofluorescence microscopy showed that the perinuclear location of the GFP-ASC specks in poly(dA-dT)-treated cells was specific to the central cytoskeletal network, as indicated by staining with anti-α-tubulin antibodies (Fig. 1A). Because γ-tubulin is an essential component of the MTOC, we used anti-γ-tubulin antibodies to identify the position of the MTOC in our system. Our results revealed that the GFP-ASC specks in poly(dA-dT)-treated cells colocalized with the MTOCs (Fig. 1A). Interestingly, this phenomenon has also been observed for NLRP7 inflammasomes (20), suggesting that inflammasome specks may commonly localize to the MTOC upon activation.
FIGURE 1.

AIM2 inflammasomes localize with MTOCs and induce IL-1β secretion via autophagy. A, NPC TW02 cells were transfected with GFP-ASC for 48 h and then treated with poly(dA-dT) for 6 h. The locations of GFP-ASC, α-tubulin, or γ-tubulin were observed by confocal fluorescence microscopy. Scale bar, 20 μm. B, NPC TW02 cells were pretreated with TNFα for 48 h. Then, NPC TW02 and HK1 cells were treated with rapamycin (30 nm) or chloroquine (50 μm) for 2 h, followed by poly(dA-dT) for 12 h. Culture media were collected and subjected to IL-1β ELISA. C, At the indicated time point, HK1 cell extracts were subjected to Western blot analysis. *, p < 0.05; **, p < 0.01. D, HK1 cells were treated with control or LC3-specific siRNA for 48 h and then treated with poly(dA-dT) for 12 h. Culture media and cell extracts were collected for IL-1β ELISA and Western blot analysis, respectively. Symbols: *, p < 0.05; and **, p < 0.01. E, NPC TW02 cells were transfected with Flag-EB1 for 48 h and then treated with poly(dA-dT) for 6 h. Cell extracts were collected for immunoprecipitation with an anti-FLAG matrix and subjected to Western blot analysis.
The colocalization of activated inflammasome specks with the MTOC suggests that the microtubule network may be involved in inflammasome activation and/or IL-1β secretion. For the past few decades, researchers have examined cellular cargo trafficking (21), including the secretion of cytokines or growth factors. IL-1β secretion has been shown to depend on vesicular transport (18, 19), and prior reports have shown that (i) autophagy acts on ubiquitinated inflammasomes (17); (ii) the intracellular trafficking of autophagosomes depends on the microtubule network; and (iii) the shedding of these vacuoles begins at the MTOC (14). Based on our present findings, we therefore hypothesize that in addition to being required for speck formation, EB1 also participates in inflammasome activation/IL-1β secretion by recruiting inflammasomes to the MTOC and regulating autophagy-dependent secretion.
AIM2 Inflammasome-induced IL-1β Secretion Requires Autophagy
To examine whether EB1 is involved in the secretion of IL-1β, we used NPC cells that stably expressed mature IL-1β (HK1/mIL1b) and constitutively secreted IL-1β regardless of inflammasome stimulation (Fig. 2A) (2). To examine whether EB1-mediated inflammasome activation was required for IL-1β secretion, we used the EB1-silenced cells generated in our previous work (9). Our results revealed that EB1 depletion reduced the secretion of mature IL-1β from poly(dA-dT)-treated HK1/mIL1b cells (Fig. 2A), indicating that EB1 is involved in both inflammasome activation and IL-1β secretion. To test whether autophagy was also involved in these processes, we treated two NPC cell lines (HK1 and NPC TW02 cells) with rapamycin (which induces autophagy by targeting mTOR) or chloroquine (which inhibits autophagy by blocking the fusion of the vesicle with the cell membrane) and then subjected the cells to AIM2 inflammasome activation. We found that chloroquine pretreatment significantly decreased IL-1β secretion following poly(dA-dT) treatment in both NPC cell lines, but rapamycin did not have a significant opposite effect (Fig. 1B). Because the processing of LC3-I to LC3-II reflects the maturation of autophagy (12), we used Western blot analysis to monitor autophagic maturation. We found that poly(dA-dT) treatment increased the levels of LC3-II by 2.2-fold (Fig. 1C; 12 h), suggesting that the activation of autophagy was subsequent to that of AIM2 inflammasomes in our system. Notably, HK1 cells expressed a basal level of LC3-II in the absence of poly(dA-dT) treatment (Fig. 1C, 0 h). This may explain the low level secretion of IL-1β previously observed from these cells in the absence of inflammasome stimulation (9), and the less significant induction of IL-1β secretion following rapamycin treatment (Fig. 1B). Knockdown of autophagy by silencing of LC3 significantly decreased IL-1β secretion in poly(dA-dT)-treated HK1 and HK1/mIL1b cells (Figs. 1D and 2B), as well as in a monocyte cell line (THP1) (Fig. 3A). Immunoprecipitation of FLAG-EB1 in NPC TW02 cells revealed that the levels of LC3-II and ASC increased after poly(dA-dT) treatment (Fig. 1E), indicating that mature autophagosomes were associated with the activated AIM2 inflammasomes. A previous study showed that the extracellular delivery of IL-1β requires the autophagy-based secretory pathway (15). Here, we show for the first time that both EB1 and autophagy are involved in AIM2 inflammasome-induced IL-1β secretion. This is likely to be an innate and common biological process that occurs in the various cell types that undergo inflammasome-associated IL-1β secretion.
FIGURE 2.
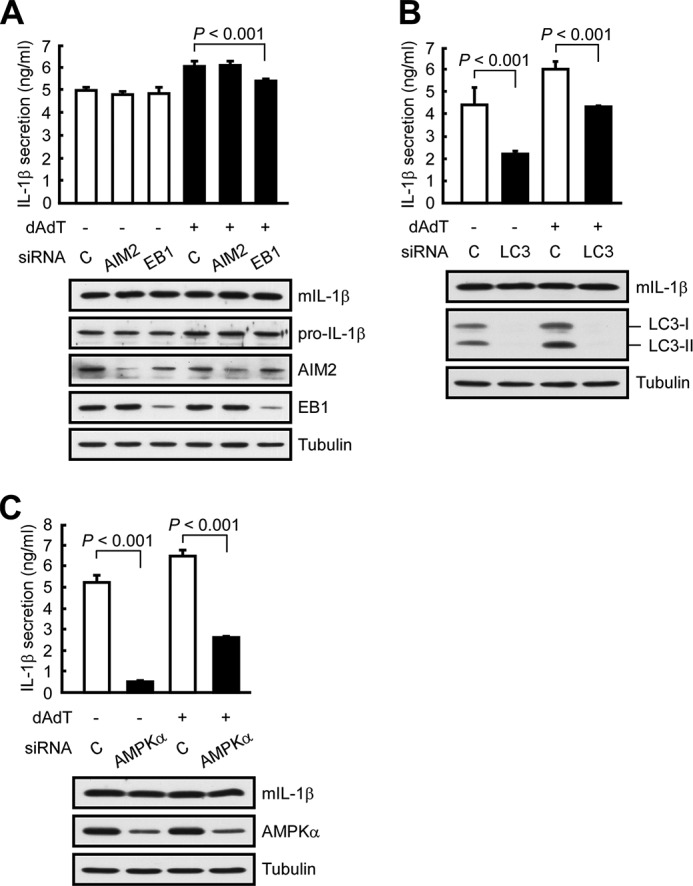
EB1, autophagy, and AMPK are all involved in IL-1β secretion. A, HK1/mIL1b cells were pretreated with control siRNA, AIM2 siRNA, or EB1 siRNA for 48 h and then treated with/without poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis, respectively. B, HK1/mIL1b cells were treated with control siRNA or LC3 siRNA for 48 h and then treated with/without poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis, respectively. C, HK1/mIL1b cells were pretreated with control siRNA or AMPKα siRNA for 48 h and then treated with/without poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis, respectively.
FIGURE 3.

In THP1 cells, EB1 regulates poly(dA-dT)-induced autophagy and AIM2 inflammasome shedding, and AMPK is involved in poly(dA-dT)-induced IL-1β secretion. A, THP1 cells were pretreated with PMA (2 μm for 6 h); treated with control siRNA, AIM2 siRNA, EB1 siRNA, or LC3 siRNA for 48 h; and treated with/without poly(dA-dT) for 4 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis, respectively. B, THP1 cells were pretreated with PMA (2 μm for 6 h), treated with control siRNA or AMPKα siRNA for 48 h, and then treated with/without poly(dA-dT) for 4 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis, respectively. **, p < 0.01; ***, p < 0.001.
GFP-ASC Is Engulfed in Autophagic Vacuoles Following AIM2 Inflammasome Activation
Because poly(dA-dT) treatment triggers EB1-mediated AIM2 inflammasome speck formation (9), followed by autophagy-dependent IL-1β secretion in this paper, we next used immunoelectron microscopy to examine whether AIM2 inflammasome specks were engulfed in autophagosomes. Consistent with the data from our Western blot analysis, the microscopic images revealed that poly(dA-dT) treatment of NPC TW02 cells increased the number of autophagosomes (Fig. 4, A and C). As indicated by gold particles coupled to anti-GFP antibodies in GFP-ASC-overexpressing NPC TW02 cells, the aggregated GFP-ASC was engulfed within autophagosomes in poly(dA-dT)-treated cells but not in untreated controls (Fig. 4, B, D, and E). Furthermore, extracellular GFP-ASC was released from the autophagosomes (Fig. 4F). These findings suggest that AIM2 inflammasome complexes and IL-1β enter the secretory pathway via autophagy, which is required for the inflammasome-mediated secretion of IL-1β.
FIGURE 4.

GFP-ASC localizes to autophagic vacuoles following AIM2 inflammasome activation. A, NPC TW02 cells were transfected with GFP-ASC for 48 h and then treated with/without poly(dA-dT). Ultrathin sections were stained with gold particles targeting the anti-GFP antibodies and observed by electron microscopy. Scale bar, 2 μm. B, image magnified from A. Scale bar, 500 nm. C, NPC TW02 cells were transfected with GFP-ASC for 48 h, treated with poly(dA-dT) for 4 h, and then examined as described in A. Scale bar, 2 μm. D–F, images magnified from C. Scale bar, 500 nm. The arrowhead indicates the gold particles targeting GFP-ASC. N indicates the nucleus.
EB1 Is Required for the Shedding of AIM2 Inflammasomes and Autophagic Vesicles
To further show that EB1 regulates IL-1β secretion by eliciting the shedding of inflammasome complex-containing autophagic vesicles, we investigated the maturation and shedding of autophagic vesicles in EB1 knockdown cells. Immunofluorescence analyses revealed that cytosolic LC3 puncta (representing activated autophagy) could be observed in control, AIM2 knockdown, and EB1 knockdown cells exposed to poly(dA-dT) treatment for 6 h (Fig. 5A). Interestingly, LC3 punctation was reduced at 12 h post-treatment in control and AIM2 knockdown cells, but not in EB1 knockdown cells (Fig. 5A). This suggests that EB1 depletion may interfere with autophagosome trafficking or lead to constitutive activation of autophagy. Indeed, the levels of LC3-II were >2-fold higher in cultured media from control and AIM2-depleted cells treated with poly(dA-dT) (2.21- and 2.54-fold, respectively) but not that from the corresponding EB1-depleted cells (0.69-fold) (Fig. 5B). AIM2 was detected in the culture medium, and poly(dA-dT) treatment enhanced AIM2 secretion in control knockdown cells. Importantly, EB1 depletion decreased poly(dA-dT)-induced autophagy (LC3-II) and AIM2 secretion, compared with that seen in control siRNA-treated cells (Fig. 5B). Similar results were obtained in THP1 cells: EB1 depletion affected poly(dA-dT)-induced autophagy and AIM2 inflammasome secretion, as monitored by the levels of LC3-II, AIM2, and caspase 1 in the culture medium, compared with the knockdown controls (Fig. 3A). Furthermore, LC3 depletion reduced poly(dA-dT)-induced AIM2 inflammasome secretion in THP1 cells (Fig. 3A). These results collectively indicate that AIM2-inflammasome-mediated IL-1β secretion is dependent on the EB1-directed shedding of autophagosomes.
FIGURE 5.

EB1 knockdown reduces autophagy and AIM2 shedding. A, NPC TW02 cells were pretreated with control siRNA, AIM2 siRNA, or EB1 siRNA for 48 h and then treated with poly(dA-dT) for the indicated durations. The cells were observed by confocal fluorescence microscopy. Scale bar, 20 μm. B, NPC TW02 cells were treated with control siRNA, AIM2 siRNA, or EB1 siRNA for 48 h and then treated with/without poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to Western blot analysis.
We also examined whether EB1 regulates the shedding of autophagic vesicles by altering their intracellular trafficking. Autophagosomes can fuse with lysosomes to form autolysosomes (12), which digest toxic or renewable components. Here, we used immunofluorescence microscopy to observe the colocalization of autophagosomes and lysosomes, which were visualized with anti-LC3 and anti-LAMP1 antibodies, respectively. Our results revealed that the colocalization of LC3 and LAMP1 was reduced in poly(dA-dT)-treated NPC and HeLa cells subjected to EB1 knockdown but not in those transfected with control siRNA (Fig. 6). This suggests that EB1 regulates the shedding of autophagic vesicles through intracellular trafficking. Collectively, these results indicate that EB1 regulates inflammasome release by eliciting the shedding of autophagic vesicles, which is required for the inflammasome-mediated secretion of inflammasomes and IL-1β.
FIGURE 6.

EB1 knockdown reduces the fusion of autophagic vesicles and lysosomes. A, NPC TW02 cells were pretreated with control siRNA (siC) or EB1 siRNA for 48 h and then treated with/without poly(dA-dT) for 6 h. The cells were fixed and stained with anti-LC3 and anti-LAMP1 antibodies and then observed under confocal fluorescence microscopy. B, HeLa cells were prepared and observed as described in A. Scale bar, 20 μm.
AMPK May Regulate AIM2 Inflammasome Activation by Targeting EB1
Finally, we sought to identify potential regulators of the EB1-mediated secretions of inflammasomes and IL-1β. AMPK has been reported to control microtubule polymerization via the post-translational regulation of microtubule plus-end proteins (22) and appears to be involved in NLRP1b inflammasome-derived IL-1β secretion (23). Based on these previous results, we hypothesized that AMPK may be an upstream regulator of EB1. To test this possibility, we first pretreated cells with the AMPK activator, AICAR (24), or its inhibitor, compound C (25), and examined poly(dA-dT)-induced IL-1β secretion. As shown in Fig. 7A, AMPK phosphorylated at threonine 172 (Thr(P)-172; which was taken as indicating activated AMPK) was detected in control cells regardless of poly(dA-dT) treatment. Cells treated with compound C (5 or 10 mm) showed far less AMPK activation, whereas AICAR had no significant effect (Fig. 7A). Importantly, pretreatment with compound C significantly and dose-dependently blocked poly(dA-dT)-induced IL-1β secretion, whereas this secretion was potently induced by AICAR (Fig. 7A).
FIGURE 7.

AMPK may regulate EB1. A, HK1 cells were pretreated with the indicated doses of compound C or AICAR for 6 h, and then treated with poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis, respectively. *, p < 0.05; **, p < 0.01. B, HK1 cells were pretreated with control siRNA or AMPKα siRNA for 48 h and then treated with poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to IL-1β ELISA and Western blot analysis. *, p < 0.05; **, p < 0.01. C, NPC TW02 cells were treated with poly(dA-dT) for the indicated durations, and cell extracts were subjected to immunoprecipitation. D, NPC TW02 cells were treated with control siRNA or AMPKα siRNA for 48 h, treated with poly(dA-dT) for 6 h, extracted, and subjected to immunoprecipitation. E, NPC TW02 cells were treated with control siRNA or AMPKα siRNA for 48 h and then treated with/without poly(dA-dT) for 12 h. Culture media and cell extracts were subjected to Western blot analysis. F, the time course of poly(dA-dT) treatment-induced EB1-mediated AIM2 inflammasome delivery via autophagic vacuole shedding, which occurs as follows: 1) AMPK regulates EB1; 2) EB1 regulates the MTOC localization of the AIM2 inflammasome speck; and 3) EB1 elicits AIM2 inflammasome release via autophagic vacuole shedding.
Because AMPKα is critical for AMPK kinase activity in the AMPK protein complex, we tested whether depletion of AMPKα specifically disrupted the function of AMPK. Similar to the results from our EB1-depletion experiments, AMPKα depletion reduced IL-1β secretion in poly(dA-dT)-treated HK1 and THP1 cells, whereas control siRNAs had no such effect (Figs. 3B and 7B). Depletion of AMPKα also decreased the secretion of mature IL-1β from HK1/mIL1b cells (Fig. 2C), indicating that AMPK participates in the secretions of inflammasomes and IL-1β. Co-immunoprecipitation assays with anti-AMPKα antibodies confirmed that EB1 was associated with AMPK in poly(dA-dT)-treated cells (Fig. 7C). Notably, the association of EB1 and AMPK occurred at 1 to 3 h after poly(dA-dT) treatment. This was earlier than the timing of poly(dA-dT)-induced EB1/AIM2 inflammasome speck formation (4 to 8 h) (9), suggesting that AMPK may regulate EB1-mediated AIM2 inflammasome formation and secretion. Depletion of AMPKα abolished the poly(dA-dT)-induced association of EB1 with AIM2 inflammasomes, as visualized in an ASC pulldown assay, and reduced the release of autophagic vesicles and AIM2 to the culture medium (Fig. 7, D and E). Thus, AMPK may interact with EB1, thereby serving as an upstream regulator of EB1-mediated inflammasome secretion.
Taken together, our findings suggest a model for EB1-mediated, autophagy-dependent AIM2 inflammasome and IL-1β secretion (Fig. 7F). In this model, poly(dA-dT) treatment causes AMPK to associate with EB1 upon AIM2 inflammasome activation, whereupon EB1 controls the MTOC localization of AIM2 inflammasome specks, directing them to the autophagic pathway.
DISCUSSION
EB1 mediates various cellular functions by regulating signal transduction via alterations in the microtubule network (11). In the present study, we identify a novel role for EB1, showing for the first time that it serves as a platform for AIM2 inflammasome recruitment (9) and elicits the autophagy-dependent secretions of AIM2 inflammasomes and IL-1β (Fig. 7F). Speck formation has been detected following the activations of multiple inflammasomes (e.g. the NLRP3, RIG-I, and AIM2 inflammasomes (9)) by various stimuli. Our novel finding that EB1 controls the MTOC localization of AIM2 inflammasome specks may suggest that EB1-mediated inflammasome activation/autophagy is a common mechanism for inflammasome-associated IL-1β secretion.
The MTOC is thought to be the site at which autophagosome precursors are initiated via nucleation (13). This may explain why speck formation and localization to the MTOC are required for inflammasome-induced, autophagy-dependent secretion. Autophagy has been shown to negatively regulate inflammasome activation by removing oxidative species or damaged mitochondria (26, 27). Here, we show that autophagy positively regulates AIM2 inflammasome activation and IL-1β secretion through vesicular shedding and that these events are connected via EB1. Thus, autophagy appears to have dual functions with respect to inflammasomes, and future studies are warranted to investigate the maturation of autophagosomes before and after inflammasome activation. Caspase 1 activity, which is involved in the inflammasome-dependent cleavage of pro-IL-1β to mature IL-1β, is reportedly involved in the export of autophagosomes (28). Notably, we found that the maturation of autophagosomes was independent of AIM2 inflammasome activation (Fig. 5A), suggesting that parallel pathways may trigger AIM2 inflammasome activation and autophagy.
The present work also shows for the first time that AMPK may be a potential regulator upstream of EB1-mediated AIM2 inflammasome activation and release. Although a previous report suggested that AMPK regulates the maturation of autophagosomes (29), our results revealed that AMPK depletion did not affect the maturation of autophagosomes but rather reduced their shedding (Fig. 7E). This suggests that AMPK may regulate autophagic secretion via EB1-mediated microtubule regulation. We also found that AMPK associates with EB1 in poly(dA-dT)-treated cells (Fig. 7C). As EB1 contains potential phosphorylation sites (30), further studies are warranted to examine whether AMPK regulates EB1 via post-translational modification. The post-translational modification of EB1-associated proteins may also be important. Recent studies have shown that the EB1-EB3 complex regulates microtubule dynamics via phosphorylation of EB3 (31–33). Notably, phosphopeptides of EB3 were detected by our mass spectrometric analysis of AMPK pulldown complexes obtained from poly(dA-dT)-treated cells (data not shown). Thus, EB-containing complexes may require AMPK to regulate this activity.
Traditionally, IL-1β secretion is believed to require two signals: the transcriptional regulation of pro-IL-1β expression, and the inflammasome-induced, caspase 1-mediated cleavage of pro-IL-1β. Here, we identify a third signal: the autophagy-mediated secretion of inflammasomes and IL-1β (Fig. 8). Our study also uncovers an additional role of EB1 as a key component that connects inflammasome activity with the autophagy-based secretory pathway. Notably, the EB1-autophagy pathway had a stronger effect on IL-1β secretion in NPC cells than in THP1 cells as suppression of this pathway had more modest effects on IL-1β release in the monocyte model (Fig. 3A). This may reflect that additional non-autophagy-based mechanisms, such as caspase 1-mediated pyroptosis (34), are the critical pathways for IL-1β secretion in monocytes.
FIGURE 8.

Model of AIM2 inflammasome-induced IL-1β secretion. Poly(dA-dT)-induced IL-1β secretion depends on a series of signals, including the following: 1) the transcriptional regulation of IL-1β expression; 2) the EB1-mediated recruitment of the activated inflammasome, which induces maturation of IL-1β via caspase 1; and 3) the EB1-mediated delivery of inflammasome-containing autophagic vesicles.
Autophagy is a critical mechanism that protects the host cell from damage, recycles cellular components, and releases cellular components (e.g. insulin) via the vesicular secretory pathway. Thus, autophagy not only functions as a cellular defense machinery; it also acts as a messenger for synapse-like intercellular communication (21). In new preliminary studies, we have found that the releases of additional cytokines are also regulated by the autophagy-dependent secretion system in NPC cell lines following AIM2 inflammasome activation (data not shown). Future work is therefore warranted to broadly investigate whether other proteins (e.g. cytokines and enzymes) are also secreted via autophagic vacuoles and to determine how the autophagic pathway selects its targets for secretion. Interestingly, inflammasome activation is reportedly involved in cellular senescence and physiological aging (35, 36), and activation of autophagy has been observed in aging cells (37). Thus, it would be useful to explore the potential correlation of inflammasome activation and autophagy during systemic aging and in chronic inflammatory diseases. Inflammasome-derived IL-1β secretion participates in various diseases, and this secretion depends on autophagic delivery. Therefore, inflammasome activity, autophagic vesicle maturation, and autophagic vesicle trafficking could all be potential targets for new therapeutic strategies for treating inflammatory diseases. Autophagy-dependent inflammasome/IL-1β secretion is an innate response, and tumoral inflammasome-induced IL-1β secretion is involved in tumor development (2, 38, 39), suggesting that therapeutic drugs capable of targeting cytoskeletal polymerization may help inhibit autophagic vesicle shedding and reduce IL-1β secretion. Here, we add to our previous identification of EB1 in inflammasome complexes by demonstrating a novel pathway in which EB1 elicits a critical step in inflammasome/IL-1β secretion following inflammasome activation.
This work was supported by Grants NSC101-2320-B-182-010-MY3 and MOST 103-2325-B-182-006 (to Y.-S. C.) from the National Science Council of Taiwan; Grants EMRPD1D0031 and 1D0631 (to Y.-S. C.) from the Ministry of Education of Taiwan; and Grant CMRPD1D0101 (to Y.-S. C.) from the Chang Gung Memorial Hospital (Tao-Yuan, Taiwan).
- NPC
- nasopharyngeal carcinoma
- EB1
- end-binding protein 1
- AIM2
- absent in melanoma 2
- MTOC
- microtubule organizing center
- LC3
- microtubule-associated protein 1A/1B-light chain 3
- AMPK
- 5′ AMP-activated protein kinase
- mIL1b
- mature IL-1β.
REFERENCES
- 1. Davis B. K., Wen H., Ting J. P. (2011) The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 29, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen L. C., Wang L. J., Tsang N. M., Ojcius D. M., Chen C. C., Ouyang C. N., Hsueh C., Liang Y., Chang K. P., Chen C. C., Chang Y. S. (2012) Tumour inflammasome-derived IL-1β recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol. Med. 4, 1276–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jourdan T., Godlewski G., Cinar R., Bertola A., Szanda G., Liu J., Tam J., Han T., Mukhopadhyay B., Skarulis M. C., Ju C., Aouadi M., Czech M. P., Kunos G. (2013) Activation of the Nlrp3 inflammasome in infiltrating macrophages by endocannabinoids mediates β cell loss in type 2 diabetes. Nat. Med. 19, 1132–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heneka M. T., Kummer M. P., Stutz A., Delekate A., Schwartz S., Vieira-Saecker A., Griep A., Axt D., Remus A., Tzeng T. C., Gelpi E., Halle A., Korte M., Latz E., Golenbock D. T. (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fernandes-Alnemri T., Wu J., Yu J. W., Datta P., Miller B., Jankowski W., Rosenberg S., Zhang J., Alnemri E. S. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14, 1590–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhou R., Tardivel A., Thorens B., Choi I., Tschopp J. (2010) Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 [DOI] [PubMed] [Google Scholar]
- 7. Duncan J. A., Bergstralh D. T., Wang Y., Willingham S. B., Ye Z., Zimmermann A. G., Ting J. P. (2007) Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. U.S.A. 104, 8041–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reinholz M., Kawakami Y., Salzer S., Kreuter A., Dombrowski Y., Koglin S., Kresse S., Ruzicka T., Schauber J. (2013) HPV16 activates the AIM2 inflammasome in keratinocytes. Arch. Dermatol Res. 305, 723–732 [DOI] [PubMed] [Google Scholar]
- 9. Wang L. J., Hsu C. W., Chen C. C., Liang Y., Chen L. C., Ojcius D. M., Tsang N. M., Hsueh C., Wu C. C., Chang Y. S. (2012) Interactome-wide analysis identifies end-binding protein 1 as a crucial component for the speck-like particle formation of activated absence in melanoma 2 (AIM2) inflammasomes. Mol. Cell Proteomics 11, 1230–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang K., Akhmanova A. (2011) Microtubule tip-interacting proteins: a view from both ends. Curr. Opin. Cell Biol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 11. Martín-Cófreces N. B., Baixauli F., López M. J., Gil D., Monjas A., Alarcón B., Sánchez-Madrid F. (2012) End-binding protein 1 controls signal propagation from the T cell receptor. EMBO J. 31, 4140–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boya P., Reggiori F., Codogno P. (2013) Emerging regulation and functions of autophagy. Nat. Cell Biol. 15, 713–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mackeh R., Perdiz D., Lorin S., Codogno P., Poüs C. (2013) Autophagy and microtubules: new story, old players. J. Cell Sci. 126, 1071–1080 [DOI] [PubMed] [Google Scholar]
- 14. Korolchuk V. I., Rubinsztein D. C. (2011) Regulation of autophagy by lysosomal positioning. Autophagy 7, 927–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dupont N., Jiang S., Pilli M., Ornatowski W., Bhattacharya D., Deretic V. (2011) Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 30, 4701–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang S., Dupont N., Castillo E. F., Deretic V. (2013) Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J. Innate Immun. 5, 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shi C. S., Shenderov K., Huang N. N., Kabat J., Abu-Asab M., Fitzgerald K. A., Sher A., Kehrl J. H. (2012) Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 13, 255–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Carta S., Tassi S., Semino C., Fossati G., Mascagni P., Dinarello C. A., Rubartelli A. (2006) Histone deacetylase inhibitors prevent exocytosis of interleukin-1β-containing secretory lysosomes: role of microtubules. Blood 108, 1618–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. MacKenzie A., Wilson H. L., Kiss-Toth E., Dower S. K., North R. A., Surprenant A. (2001) Rapid secretion of interleukin-1β by microvesicle shedding. Immunity 15, 825–835 [DOI] [PubMed] [Google Scholar]
- 20. Messaed C., Akoury E., Djuric U., Zeng J., Saleh M., Gilbert L., Seoud M., Qureshi S., Slim R. (2011) NLRP7, a nucleotide oligomerization domain-like receptor protein, is required for normal cytokine secretion and co-localizes with Golgi and the microtubule-organizing center. J. Biol. Chem. 286, 43313–43323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stow J. L. (2013) Nobel Prize discovery paves the way for immunological traffic. Nat. Rev. Immunol. 13, 839–841 [DOI] [PubMed] [Google Scholar]
- 22. Nakano A., Kato H., Watanabe T., Min K. D., Yamazaki S., Asano Y., Seguchi O., Higo S., Shintani Y., Asanuma H., Asakura M., Minamino T., Kaibuchi K., Mochizuki N., Kitakaze M., Takashima S. (2010) AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat. Cell Biol. 12, 583–590 [DOI] [PubMed] [Google Scholar]
- 23. Liao K. C., Mogridge J. (2013) Activation of the Nlrp1b inflammasome by reduction of cytosolic ATP. Infect Immun. 81, 570–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Giri S., Nath N., Smith B., Viollet B., Singh A. K., Singh I. (2004) 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside inhibits proinflammatory response in glial cells: a possible role of AMP-activated protein kinase. J Neurosci 24, 479–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McCullough L. D., Zeng Z., Li H., Landree L. E., McFadden J., Ronnett G. V. (2005) Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J. Biol. Chem. 280, 20493–20502 [DOI] [PubMed] [Google Scholar]
- 26. Zhou R., Yazdi A. S., Menu P., Tschopp J. (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 [DOI] [PubMed] [Google Scholar]
- 27. Nakahira K., Haspel J. A., Rathinam V. A., Lee S. J., Dolinay T., Lam H. C., Englert J. A., Rabinovitch M., Cernadas M., Kim H. P., Fitzgerald K. A., Ryter S. W., Choi A. M. (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sirois I., Groleau J., Pallet N., Brassard N., Hamelin K., Londono I., Pshezhetsky A. V., Bendayan M., Hébert M. J. (2012) Caspase activation regulates the extracellular export of autophagic vacuoles. Autophagy 8, 927–937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mihaylova M. M., Shaw R. J. (2011) The AMPK signalling pathway coordinates cell growth, autophagy, and metabolism. Nat Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rush J., Moritz A., Lee K. A., Guo A., Goss V. L., Spek E. J., Zhang H., Zha X. M., Polakiewicz R. D., Comb M. J. (2005) Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 23, 94–101 [DOI] [PubMed] [Google Scholar]
- 31. Komarova Y., Lansbergen G., Galjart N., Grosveld F., Borisy G. G., Akhmanova A. (2005) EB1 and EB3 control CLIP dissociation from the ends of growing microtubules. Mol. Biol. Cell 16, 5334–5345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ferreira J. G., Pereira A. J., Akhmanova A., Maiato H. (2013) Aurora B spatially regulates EB3 phosphorylation to coordinate daughter cell adhesion with cytokinesis. J. Cell Biol. 201, 709–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leśniewska K., Warbrick E., Ohkura H. (2014) Peptide aptamers define distinct EB1- and EB3-binding motifs and interfere with microtubule dynamics. Mol. Biol. Cell 25, 1025–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brough D., Rothwell N. J. (2007) Caspase-1-dependent processing of pro-interleukin-1β is cytosolic and precedes cell death. J. Cell Sci. 120, 772–781 [DOI] [PubMed] [Google Scholar]
- 35. Acosta J. C., Banito A., Wuestefeld T., Georgilis A., Janich P., Morton J. P., Athineos D., Kang T. W., Lasitschka F., Andrulis M., Pascual G., Morris K. J., Khan S., Jin H., Dharmalingam G., Snijders A. P., Carroll T., Capper D., Pritchard C., Inman G. J., Longerich T., Sansom O. J., Benitah S. A., Zender L., Gil J. (2013) A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 15, 978–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Youm Y. H., Grant R. W., McCabe L. R., Albarado D. C., Nguyen K. Y., Ravussin A., Pistell P., Newman S., Carter R., Laque A., Munzberg H., Rosen C. J., Ingram D. K., Salbaum J. M., Dixit V. D. (2013) Canonical Nlrp3 inflammasome links systemic low-grade inflammation to functional decline in aging. Cell Metab 18, 519–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Warr M. R., Binnewies M., Flach J., Reynaud D., Garg T., Malhotra R., Debnath J., Passegué E. (2013) FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okamoto M., Liu W., Luo Y., Tanaka A., Cai X., Norris D. A., Dinarello C. A., Fujita M. (2010) Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1β. J. Biol. Chem. 285, 6477–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ponomareva L., Liu H., Duan X., Dickerson E., Shen H., Panchanathan R., Choubey D. (2013) AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol. Cancer Res. 11, 1193–1202 [DOI] [PubMed] [Google Scholar]