

Background: Alveolar macrophages are the primary target of Mycobacterium tuberculosis infection.
Results: Mycobacterial PtpA dephosphorylates host GSK3α Tyr279 resulting in modulation of its activity.
Conclusion: Dephosphorylation of GSK3α decreases apoptosis of the host early in infection promoting survival of the macrophage and the pathogen within it.
Significance: Understanding the mechanisms by which Mycobacterium tuberculosis enables successful infection is essential for understanding the pathogenesis of tuberculosis.
Keywords: Apoptosis, Macrophage, Mycobacterium tuberculosis, Signal Transduction, Protein-tyrosine Phosphatase (Tyrosine Phosphatase)
Abstract
Mycobacterium tuberculosis tyrosine phosphatase PtpA inhibits two key cellular events in macrophages required for the elimination of invading organisms, phagosome acidification, and maturation. Kinome analysis revealed multiple PtpA-dependent changes to the phosphorylation status of macrophage proteins upon M. tuberculosis infection. Among those proteins we show that PtpA dephosphorylates GSK3α on amino acid Tyr279, which leads to modulation of GSK3α anti-apoptotic activity, promoting pathogen survival early during infection.
Introduction
Mycobacterium tuberculosis, one of the most notorious infectious agents of humans, is estimated to have caused 1.3 million deaths in 2012 (WHO Report, 2013). The combination of co-infection with HIV and the emergence of multidrug-resistant strains gives tuberculosis the highest mortality rate of any infectious disease (1).
M. tuberculosis infects the human lung, where circulating alveolar macrophages paradoxically serve as both the first line of defense against microbial infections as well as the bacilli's natural habitat (2). Once engulfed by the macrophage, M. tuberculosis replicates and persists in a secluded organelle named the mycobacterial phagosome. M. tuberculosis inhibits phagosome maturation, a natural macrophage process whereby phagosomes harboring foreign particles fuse with lysosomes (3), and thus prevents proteolytic degradation and the downstream immunological processes required to initiate an adaptive immune response (2). This phenomenon highlights how M. tuberculosis interferes with the macrophage trafficking machinery, a process essential for M. tuberculosis infectivity (3–7).
We have previously shown that the low molecular weight tyrosine phosphatase, PtpA, is needed to block phagosome maturation and is essential for M. tuberculosis pathogenicity within human macrophages (8). PtpA's substrate in the host is the human vesicle trafficking protein vacuolar protein sorting 33B (hVPS33B) (8, 9), which plays a key role in the regulation of membrane fusion in the endocytic pathway (10). Dephosphorylation of hVPS33B by PtpA translates directly into phagosome maturation arrest (8). In parallel, PtpA disrupts the macrophage's V-ATPase pump assembly (11), a protein complex that controls phagosome acidification by transporting protons across membranes (12). During phagosome maturation, the recruitment of the pump to the phagosome generally results in a significant reduction in phagosomal pH (13). However, the binding of PtpA to subunit H of the macrophage V-ATPase pump results in reduction of phagosome acidification (11).
Phosphatases play key roles in signal transduction in different pathways (14). To decipher the multifaceted activity of PtpA on macrophage signaling pathways, we conducted a large scale analysis of signaling networks, termed kinome analysis (15), and we discovered that PtpA affects the phosphorylation pattern of a series of host signaling proteins. Most significantly, we identified human glycogen synthase kinase 3 (GSK3) as another potential substrate for mycobacterial PtpA.
GSK3 is a multifunctional serine/threonine kinase that acts as a regulatory switch for numerous signaling pathways, including the insulin response, glycogen regulation, cell survival, and apoptosis (16). There are two mammalian isoforms of GSK3 encoded by distinct genes, GSK3α (51 kDa) and GSK3β (47 kDa). These two isoforms share a high degree of structural similarity, specifically in their kinase domain (98% identity), but are not functionally identical (17). GSK3α and GSK3β are constitutively active in resting cells and are primarily regulated through the inhibition of their activity via phosphorylation of Ser21 and Ser9, respectively (18). Conversely, the activity of the isoforms is positively regulated by the phosphorylation of a tyrosine residue located in the activation loop, Tyr279 (GSK3α) and Tyr216 (GSK3β), and this phosphorylation is essential for full activity of the enzyme (19). Apoptotic stimuli increase the activity of the isoforms by tyrosine phosphorylation (Tyr279/216) in certain cell lines (20), providing evidence for a role for tyrosine phosphorylation in apoptosis.
In this study, we show that PtpA is capable of interfering with multiple signaling pathways within human macrophages, resulting in observable changes in the phosphorylation pattern of host signaling proteins. Most notably, we reveal that PtpA dephosphorylates GSK3α on Tyr279. We suggest that modulation of GSK3α's activity interferes with apoptosis of the macrophage, the programmed self-destruction process considered to be a defense mechanism utilized by the human host against M. tuberculosis.
EXPERIMENTAL PROCEDURES
Tissue Culture Maintenance and Differentiation
The human monocytic leukemia cell line THP-1 (TIB-202; ATCC) was cultured in RPMI 1640 medium (Sigma) supplemented with 10% FBS (PAA Laboratories Inc.), 1% l-glutamine, 1% penicillin, and 1% streptomycin (StemCell). Cells were seeded in 10-cm (diameter) tissue culture dishes at a density of 7.0 × 106 cells/dish and differentiated into a macrophage-like cell line with 20 ng/ml phorbol myristate acetate (Sigma) in RPMI 1640 medium supplemented with 10% FBS and 1% l-glutamine (incomplete RPMI) at 37 °C in a humidified atmosphere of 5% CO2 for 18 h.
Macrophage Infection
Bacterial cells were washed with Middlebrook 7H9 broth supplemented with 0.05% (v/v) Tween 80 (Sigma). Infection of THP-1 macrophage-like cells was performed using human serum-opsonized M. tuberculosis at a multiplicity of infection of 10:1 in RPMI 1640 medium. After a 3-h incubation at 37 °C and 5% CO2, cells were washed with RPMI 1640 medium to remove noninternalized bacteria and re-incubated at 37 °C and 5% CO2 in incomplete RPMI 1640 medium containing 100 μg/ml gentamicin (Invitrogen) for 4, 18, or 48 h.
Macrophage Cellular Extraction
At defined time points after infection, infected THP-1 macrophage-like cells were washed twice with cold PBS, and cellular extracts were harvested in lysis buffer (20 mm Tris-HCl, 5 mm EDTA, 1% Triton X-100, 1 mm DTT, 1 mm phenylmethylsulfonyl fluoride, 1 mm phosphatase inhibitor mixture (Sigma), pH 7.2) by drawing the solution in and out of a blunt syringe 15–20 times. The cellular extracts were centrifuged for 10 min at 13,000 rpm and passed through a 0.22-μm filter column (Millipore Corp.).
Macrophage RNA Extraction and cDNA Synthesis
Total RNA was extracted from M. tuberculosis-infected THP-1-derived macrophages (7.0 × 106 cells) at defined time points (4, 18, and 48 h) using the RNAspin mini kit according to the manufacturer's instructions (GE Healthcare). RNA was reverse-transcribed to cDNA using the EasyScript cDNA synthesis kit following the manufacturer's protocol (ABM). For each cDNA synthesis, 1 μg of total RNA, measured by an Epoch Microplate Spectrophotometer (BioTek), and 0.5 μm oligo(dT) oligonucleotides primers were used.
Quantitative-PCR (qPCR)2
Primers specific for GSK3α and caspase-3 mRNA were designed using Primer-BLAST (National Center for Biotechnology Information) (Table 1). Control PCR amplifications for the expressions of the gene-specific mRNAs were performed on cDNA templates from uninfected phorbol myristate acetate-differentiated THP-1 cells to confirm the specificity of the designed primers. Each qPCR contained 2× EvaGreen qPCR master mix (ABM), 15 ng of cDNA, and 1 μm of each primer and was analyzed in quantification mode on a DNA Engine Opticon instrument (Bio-Rad). The following cycling conditions were used: 95 °C for 10 min, 40 cycles of 95 °C for 15 s, 52 °C for 15 s, and 60 °C for 30 s with data collection during each cycle. Mock reactions (no reverse transcriptase) were also included with each experiment to confirm the absence of genomic DNA contamination. Ct values were converted to copy numbers using standard curves. Results were analyzed using GraphPad Prism 5.0 software. All values of gene-specific mRNA were internally normalized to cDNA expression levels of the housekeeping gene GAPDH.
TABLE 1.
Oligonucleotides used for quantitative PCR
F indicates forward, and R indicates reverse.
| Oligonucleotides | Sequence (5′ → 3′) |
|---|---|
| Caspase-3 F | TGAGGCGGTTGTAGAAGAGTTT |
| Caspase-3 R | GCTCGCTAACTCCTCACGG |
| GAPDH F | GAAGGTGAAGGTCGGAGTC |
| GAPDH R | GAGGGATCTCGCTCCTGGAAGA |
| GSK3α F | GCTCACCCCTGGACAAAGGTGTT |
| GSK3α R | CGCACAGGCCTCTAGTGGGGA |
Cloning of DNA and Expression of Recombinant Proteins
The list of plasmids and oligonucleotides used for cloning in this study are described in Tables 2 and 3, respectively. GSK3α was obtained as a plasmid (pANT7-GSK3α) from the DNASU Plasmid Repository and PCR-amplified and cloned into the pGEX-6P-3 vector (GE Healthcare). The plasmid pBO1-GSK3α encoding for a His-tagged fusion protein was purchased from GeneCopoeia Inc. The gene-encoding RAB7 was PCR-amplified from cDNA prepared from THP-1 cells and was cloned into pET22b (Millipore Corp.). The M. tuberculosis ptpA gene was cloned into pGEX-6P-3. All plasmid constructs were verified by sequencing (Eurofins MWG Operon). Chemically competent BL21 Escherichia coli cells were transformed with the expression plasmids and expressed according to established protocols. His-tagged recombinant proteins were purified from the soluble fraction by affinity chromatography on nickel-nitrilotriacetic acid polyhistidine tag purification resin (Qiagen) and GST-tagged proteins by affinity chromatography on glutathione-agarose resin (Sigma).
TABLE 2.
Plasmids used for DNA cloning and protein expression
| Plasmids | Characteristics | Resistance Gene | Source |
|---|---|---|---|
| pET22b | Produces C-terminal His6-tagged proteins | Ampicillin | Millipore Corp. |
| pBO1 | Produces N-terminal His6-tagged proteins | Ampicillin | GeneCopoeia Inc. |
| pGEX-6P-3 | Produces N-terminal GST-tagged proteins | Ampicillin | GE Healthcare |
TABLE 3.
Oligonucleotides used for DNA cloning
F indicates forward, and R indicates reverse. The recognition sequence for the restriction site is underlined.
| Oligonucleotides | Sequence (5′ → 3′) | Restriction site |
|---|---|---|
| GSK3α F | TATATAGGATCCATGAGCGGCGGCGGGCCTTCG | BamHI |
| GSK3α R | TATATAGAATTCGGAGGAGTTAGTGAGGGTAGG | EcoRI |
| PtpA F | ATATATGAATTCCGTGTCTGATCCGCTG | EcoRI |
| PtpA R | ATATATCTCGAGTCAACTCGGTCCGTTC | XhoI |
| RAB7 F | TATATAGGATCCATGACCTCTAGGAAGAAAGTG | BamHI |
| RAB7 R | TATATACTCGAGTCAGCAACTGCAGCTTTC | XhoI |
Kinome Analysis by Kinetworks Phospho-site Screen Assay
Kinome analysis was performed as described previously (21). Briefly, THP-1 macrophage-like cells were infected with wild-type M. tuberculosis H37Rv and with the H37Rv strain in which the ptpA gene was deleted (ΔptpA M. tuberculosis) (8), and cellular extracts were harvested 18 h post-infection. The macrophage lysates were prepared for kinome analysis according to the manufacturer's instructions (Kinexus Bioinformatics Corp.). Samples were sent to Kinexus Bioinformatics where the assay was performed. Data were analyzed according to statistical confidence provided by experience in analyzing over 10,000 screens. According to Kinexus Bioinformatics, the significance levels of change are over 25% variability in intensity.
Western Blot Analysis
In vivo Western blot analyses were performed using cellular extracts of infected THP-1 macrophage-like cells harvested 18 and 48 h post-infection as described above. Briefly, 50 μg of THP-1 cellular extracts were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane (Bio-Rad). The blots were probed with affinity-purified rabbit polyclonal anti-phospho-GSK3α (Tyr(P)279) (Invitrogen), anti-GSK3α, or with affinity-purified rabbit polyclonal anti-caspase-3 (Cell Signaling) (final IgG dilution for both antibodies, 1:1000) and incubated overnight at 4 °C. For detection of phosphorylated GSK3α (Tyr279), horseradish peroxidase-conjugated goat anti-rabbit (Sigma) (final IgG dilution, 1:3500) antibody was used as the secondary detection reagent, and the blot was developed by enhanced chemiluminescence (ECL) (Thermo Fisher Scientific). For detection of GSK3α and caspase-3, Alexa Fluor 680 goat anti-rabbit (Invitrogen) antibody was used as the secondary detection reagent (final IgG dilution, 1:10,000), and detection was done using an Odyssey Infrared CLx Imager (LI-COR Biosciences).
For in vitro Western blot analysis, different reactions containing 1–4 μm recombinant GSK3α were incubated with 0.15 mm ATP for 1 h at 37 °C. A fixed concentration of recombinant PtpA (0.04 μm) was added to the different GSK3α reactions, and incubation was continued for 45 min at 37 °C. The resulting samples were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane. The blot was probed with rabbit anti-phospho-GSK3α (Tyr(P)279) IgG, and horseradish peroxidase-conjugated goat anti-rabbit antibody was used as the secondary detection reagent as described previously. The blot was developed by ECL. To ensure identical protein loading of the different samples, Ponceau staining of the blot was performed.
In Vitro Kinase Assay
Three separate subsets of reactions containing 2–4 μm recombinant GSK3α were autophosphorylated in a kinase buffer (50 mm Tris-HCl, 5 mm MgCl2, 2 mm MnCl2, 1 mm DTT, pH 7.5) containing 10 μCi of [γ-32P]ATP (PerkinElmer Life Sciences) for 30 min at 37 °C. After this incubation period, 0.04 μm PtpA was added to the second subset of reactions, and 0.04 μm PtpA and 1.5 mm or 5 μm of the phosphatase inhibitors, Na3VO4 or BVT 948, respectively, to the third subset. Incubation of all three subsets was continued for 15 min at 37 °C. At the end of the incubation period, reactions were stopped with the addition of SDS sample loading buffer and heated at 95 °C for 8 min. The samples were resolved by 12% SDS-PAGE. The gel was silver-stained, dried, and exposed to a screen overnight. The 32P radioactively labeled protein bands were detected by a PhosphorImager SI apparatus (GE Healthcare). Bands corresponding to phosphorylated GSK3α were cut, submerged in scintillation fluid (Beckman Coulter Inc.), and analyzed by scintillation counting using a Beckman Coulter LS 6500 (Beckman Coulter Inc.).
Radiometric Kinase Assay
The kinase assay was performed as described above until the end of the second incubation period. Reactions were spotted onto phosphocellulose paper (GE Healthcare), dried, and washed thoroughly with 1% phosphoric acid six times for 10 min. Radioactivity levels were measured by submerging the phosphocellulose papers in scintillation fluid and analyzed by scintillation counting.
Determination of PtpA and GSK3α Dissociation Constant
The interaction between PtpA and GSK3α was measured using a Fusion-α-HT Multimode Microplate Reader (PerkinElmer Life Sciences) and the ALPHAScreen Histidine (Nickel Chelate) Detection Kit (PerkinElmer Life Sciences). Purified GST-tagged recombinant PtpA was biotinylated using the EZ-Link biotinylation kit (Thermo Fisher Scientific) and diluted in the assay buffer (25 mm HEPES, 100 mm NaCl, and 0.1% Tween 20, pH 7.4) in 384-well microplates (PerkinElmer Life Sciences). Purified His-tagged recombinant GSK3α was added to wells containing PtpA. Nickel chelating acceptor beads were further added to the proteins, and the microplate was incubated for 30 min at room temperature. Streptavidin donor beads were then added to the reactions, and incubation was continued for 1 h at room temperature. Kinetics of the reactions was monitored in the ALPHAScreen apparatus by luminescence signals generated from protein-protein interactions (counts/s (cps)). Results obtained were analyzed with GraphPad 5.0 software for dissociation constant determination.
RESULTS
Global Effect of PtpA on Macrophage Proteins
PtpA is secreted into the macrophage cytosol (8). The in vitro dephosphorylation assay, under which recombinant PtpA was incubated with extracts from the host macrophage, resulted in dephosphorylation of multiple host proteins in addition to hVPS33B, a host substrate we have identified previously (Fig. 1 and Table 4) (8). This demonstrates that as an active phosphatase PtpA is capable of interacting with multiple host signaling proteins.
FIGURE 1.
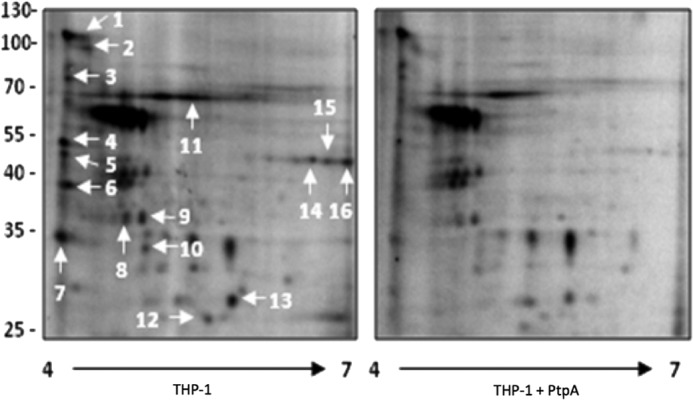
Phosphoproteomic analysis of host macrophage proteins dephosphorylated by PtpA revealed by two-dimensional gel electrophoresis. A cellular extract of differentiated THP-1 cells was incubated with [γ-32P]ATP in a kinase buffer for 30 min at 30 °C. Recombinant PtpA was added to one of the samples for 15 min, and mixtures were electrophoresed onto two-dimensional SDS-polyacrylamide gels using a 4–7 pH gradient for the first dimension and 10% SDS-PAGE for the second dimension. The radiolabeled spot profile was obtained by exposing overnight to a PhosphorImager screen. Sixteen spots demonstrating reduced phosphorylation upon addition of PtpA were identified by mass spectrometry (Table 4).
TABLE 4.
Identification of proteins dephosphorylated by the addition of recombinant PtpA to a phosphorylated THP-1 cellular extract
A cellular extract of differentiated THP-1 cells was incubated with recombinant PtpA. The phosphorylation status of several THP-1 cell proteins was modulated by PtpA. These proteins were identified by mass spectrometry. The numbers shown in this table correspond to spots shown in Fig. 1. Molecule Weight Search (MOWSE) score allows the calculation of the probability of matching N peaks by random chance (34). Accession numbers are shown in parentheses.
| Spot no. | Protein identificationa | MOWSE scoreb | Mr | Coverage |
|---|---|---|---|---|
| pI | % | |||
| 1 | XinB (CAF25191) | 5.8e+16 | 122.1 (5.2) | 15 |
| Phosphoinositide 3-kinase class 3 (NP002638) | 3.17e+15 | 101.5 (6.4) | 12 | |
| 2 | Rabaptin (NP004694) | 3.25e+07 | 99.3 (4.9) | 21 |
| Rabaptin-5 (AAC70781) | 3.25e+07 | 95.6 (4.9) | 19 | |
| Rabaptin-4 (3832516) | 1.63e+07 | 95.5 (4.9) | 19 | |
| VPS39 (AAH15817) | 1.61e+07 | 90.3 (6.6) | 19 | |
| 3 | Exocyst complex component Sec6 (O60645) | 1.31e+02 | 86.8 (5.8) | 12 |
| cGMP-dependent protein kinase 1 (Q13976) | 1.21e+02 | 76.3 (5.7) | 11 | |
| γ-Adducin (Q9UEY8) | 1.56e+02 | 79.1 (5.9) | 7 | |
| 4 | Unknown (AAH04303) | 3.1e+103 | 52.5 (5.9) | 64 |
| Hypothetical protein | 8.36e+97 | 52.4 (5.5) | 51 | |
| 5 | MHC class I antigen Cw*7 (P10321) | 5.31e+01 | 40.7 (5.6) | 7 |
| MHC class I Antigen Cw*1 (P30499) | 2.36e+01 | 40.9 (5.5) | 6 | |
| 6 | Not determined | |||
| 7 | GTPase, IMAP family member 7 (Q8NHV1) | 1.06e+16 | 34.5 (6.1) | 63 |
| 8 | Annexin A13 (P27216) | 1.19e+31 | 35.5 (5.5) | 60 |
| 9 | N-Myc Interactor-STAT Interactor (Q13287) | 3.64e+43 | 35.1 (5.2) | 57 |
| Cdc42 effector protein 4 (Q9H3Q1) | 3.7e+42 | 37.9 (5.1) | 41 | |
| 10 | Not determined | |||
| 11 | Vacuolar protein sorting 33B (AAF91174) | 6.7e+52 | 70.6 (6.3) | 64 |
| 12 | Ras-related protein Rab-7L1 (O14966) | 2.15e+69 | 23.1 (6.7) | 70 |
| 13 | Ras-related protein Rab-28 (Rab-26) (P51157) | 6.5e+48 | 24.9 (5.7) | 44 |
| 14 | Syntaxin 18 (Q9P2W9) | 38.7 (5.4) | 21 | |
| MHC class I antigen Cw*3 (70076) | 1.34e+04 | 40.7 (6.0) | 15 | |
| 15 | Arfaptin-1 (P53367) | 1.86e+01 | 41.7 (6.2) | 10 |
| 16 | MHC class I antigen Cw*17 (Q95604) | 1.63e+01 | 41.2 (6.3) | 19 |
a Accession numbers are shown in parentheses.
b Score is based on peptide frequency.
We have used a specific proteomics approach termed kinome analysis to identify macrophage signaling proteins that might be affected by M. tuberculosis PtpA. This method uses an array of phospho-specific antibodies against defined human signaling proteins and networks (15). To test the effect of PtpA on signal transduction pathways, we monitored and compared the phosphorylation status of a predefined set of signaling proteins from uninfected macrophages, macrophages infected with M. tuberculosis, and macrophages infected with ΔptpA M. tuberculosis. We chose to investigate events occurring 18 h post-infection, since at this time point bacteria are well established in the environment of the host, allowing for the monitoring of macrophage responses to bacteria residing within phagosomes.
As illustrated in Fig. 2, cellular extracts of uninfected and M. tuberculosis-infected macrophages were subjected to simultaneous screens (Fig. 2, A–C). Changes in phosphorylation were measured based on the intensity of 38 predefined phosphoproteins (Table 5) shown as bands in each gel. The phosphorylation levels of the three different treatments were compared in terms of the relative fold change in phosphorylation. The fold change was calculated by comparing the accumulated signal of proteins obtained over a given scan time (normalized counts/min) from uninfected macrophages and macrophages treated with M. tuberculosis with the accumulated signal of proteins from macrophages treated with ΔptpA M. tuberculosis (control; accumulated signal set as 1) (Table 5). Because of the high sensitivity of the assay in determining the phosphorylation state of phosphoproteins, a change in phosphorylation greater than 25% between treated cells is considered significant according to Kinexus Bioinformatics Corp., which provides the screening kit. A change in phosphorylation of less than this percentage may be due to experimental variation.
FIGURE 2.

Global kinome analysis of THP-1 cells infected with M. tuberculosis and ΔptpA M. tuberculosis. Simultaneous detection of selected host proteins and their activation status using a multiple immunoblotting technique are shown. Accurate intensity values for each protein are the accumulated signal obtained over a given scan time for each blot. These are shown as numerical values in Table 5. The three gels represent uninfected THP-1 cells (A), THP-1 cells infected with M. tuberculosis (B), and THP-1 cells infected with ΔptpA M. tuberculosis (C). Each lane was probed with one or more antibodies. The highlighted proteins are host signaling proteins showing a phosphorylation change greater than 25%. Antibodies against the phosphorylated proteins were as follows: lane 1, molecular size standard; lane 2, NR1 (Ser896); lane 3, PKR1 (Thr451); lane 4, STAT5A (Tyr694); lane 5, PKCα (Ser657) and SRC (Tyr418); lane 6, JNK (Thr183 + Tyr185) and RSK1/3 (Thr359 + Ser363/Thr356 + Ser360); lane 7, MEK3/6 (Ser189/Ser207) and PKCα/β2 (Thr638 + Thr641); lane 8, ERK1 (Thr202 + Tyr204), ERK2 (Thr185 + Tyr187), S6Kα P70 (Thr389), and S6Kα P85 (Thr389); lane 9, PKCϵ (Ser729) and SMAD1/5/9 (Ser463 + Ser363/Ser463 + Ser465/Ser465 + Ser467); lane 10, STAT3 (Ser727); lane 11, JUN (Ser73); lane 12, RAF1 (Ser259), STAT1α (Tyr701), and STAT1β (Tyr701); lane 13, PKBα (Akt1) (Thr308) and PKCδ (Thr507); lane 14, PKBα (Akt1) (Ser473); lane 15, GSK3α (Ser21), GSK3β (Ser9), and MSK1 (Ser376); lane 16, adducin α (Ser726), adducin γ (Ser693), CDK1/2 (Tyr15), and SRC (Tyr529); lane 17, GSK3α (Tyr279) and GSK3β (Tyr216); lane 18, p38α MAPK (Thr180 + Tyr182) and RB (Ser780); lane 19, NPM (Ser4), and MEK1/2 (Ser218 + Ser222); and lane 20, CREB1 (Ser133) and RB (Ser807 + Ser811).
TABLE 5.
Kinome analysis of host signaling proteins affected by PtpA
A total of 38 phospho-specific antibodies targeting key host signaling proteins were used. The trace quantity of each protein band was measured by the area under its intensity profile curve and corrected for the individual scan times (recorded time before saturation occurs). Values for the control samples were set to 1 or 0. A value of 0 indicates that no immunoreactive signal was detected for this protein. An immunoreactive signal was detected for only 17 proteins. Values for uninfected THP-1 cells and cells infected with M. tuberculosis show the fold change relative to their respective control samples (ΔptpA M. tuberculosis).
| Protein full Name | Abbreviation | Epitopes | Control ΔptpA M. tuberculosis | Fold change |
|
|---|---|---|---|---|---|
| Uninfected | M. tuberculosis | ||||
| Adducin α (ADD1) | α-Adducin | Ser726 | 0 | ||
| Adducin γ (ADD3) | γ-Adducin | Ser693 | 0 | ||
| B23 (Nucleophosmin, Numatrin, nucleolar protein NO38) | B23 (NPM) | Ser4 | 0 | ||
| Cyclin-dependent protein kinase 1/2 | CDK1/2 | Tyr15 | 1 | 2.00 | NDa |
| cAMP-response element-binding protein 1 | CREB1 | Ser133 | 1 | 0.70 | 0.84 |
| Extracellular regulated protein kinase 1 (p44 MAPK) | ERK1 | Thr202 + Tyr204 | 0 | ||
| Extracellular regulated protein kinase 2 (p42 MAPK) | ERK2 | Thr185 + Tyr187 | 0 | ||
| Glycogen synthase kinase 3α | GSK3 | Ser21 | 0 | ||
| Glycogen synthase kinase 3α | GSK3α | Tyr279 | 1 | 0.65 | 0.33 |
| Glycogen synthase kinase 3β | GSK3β | Ser9 | 0 | ||
| Glycogen synthase kinase 3β | GSK3β | Tyr216 | 1 | 0.93 | 0.61 |
| Jun N-terminal protein kinase (stress-activated protein kinase (SAPK)) 1/2/3 | JNK | Thr183 + Tyr185 | 1 | ND | 1.15 |
| Jun proto-oncogene-encoded AP1 transcription factor S73 | JUN | Ser73 | 0 | ||
| MAPK/ERK protein kinase 1/2 (MKK1/2) | MEK1/2 (MAP2K1/2) | Ser217 + Ser221 | 0 | ||
| MAPK protein kinase 3/6 (MKK3/6) | MEK3/6 (MAP2K3/6) | Ser189/Ser207 | 0 | ||
| MAPK protein kinase 6 (MKK6) | MEK6 (MAP2K6) | Ser207 | 0 | ||
| Mitogen- and stress-activated protein kinase 1 | MSK1 | Ser376 | 1 | 0.57 | 0.67 |
| N-Methyl-d-aspartate (NMDA) glutamate receptor 1 subunit ζ | NR1 | Ser896 | 1 | 0.85 | 0.90 |
| Mitogen-activated protein kinase p38α | P38α MAPK | Thr180 + Tyr182 | 0 | ||
| Protein kinase Bα (Akt1) | PKBα (Akt1) | Thr308 | 0 | ||
| Protein kinase Bα (Akt1) | PKBα (Akt1) | Ser473 | 0 | ||
| Protein kinase Cα | PKCα | Ser657 | 1 | 1.50 | 1.25 |
| Protein kinase Cα/β2 | PKCα/β2 | Thr638/Thr641 | 1 | 0.94 | 0.62 |
| Protein kinase Cδ | PKCδ | Thr507 | 1 | 0.81 | 0.51 |
| Protein kinase Cϵ | PKCϵ | Ser729 | 0 | ||
| Double-stranded RNA-dependent protein kinase | PKR1 | Thr451 | 1 | 1.31 | 1.54 |
| Raf1 proto-oncogene-encoded protein kinase | RAF1 | Ser259 | 1 | 0.88 | 0.47 |
| Retinoblastoma-associated protein | RB | Ser780 | 1 | 0.45 | 0.78 |
| Retinoblastoma-associated protein | RB | Ser807 + Ser811 | 0 | ||
| Ribosomal S6 protein kinase 1/3 | RSK1/3 | Thr359 + Ser363/ Thr356 + Ser360 | 0 | ||
| p85 ribosomal protein S6 kinase 2 | S6K2 p85 | Thr412 | 1 | 1.16 | 0.88 |
| p70 ribosomal protein S6 kinase α | S6Kα p70 | Thr389 | 1 | 1.03 | 0.85 |
| SMA- and mothers against decapentaplegic homologs 1/5/9 | SMAD1/5/9 | Ser463 + Ser465/Ser463 + Ser465/Ser465 + Ser467 | 0 | ||
| Src proto-oncogene-encoded protein kinase | SRC | Tyr418 | 0 | ||
| Src proto-oncogene-encoded protein kinase | SRC | Tyr529 | 1 | 0.89 | 1.57 |
| Signal transducer and activator of transcription 1α | STAT1α | Tyr701 | 1 | 0.56 | 0.86 |
| Signal transducer and activator of transcription 1β | STAT1β | Tyr701 | 1 | 1.04 | 0.90 |
| Signal transducer and activator of transcription 3 | STAT3 | Ser727 | 1 | 0.79 | 0.86 |
| Signal transducer and activator of transcription 5 | STAT5 | Tyr694 | 0 | ||
a ND means not determined.
As seen in Fig. 2, and detailed in Table 5, out of 38 tested signaling proteins, an accumulated signal was detected for 17 macrophage phosphoproteins. Among these, several displayed significant changes in phosphorylation between macrophages infected with M. tuberculosis and the control ΔptpA mutant. These include the following: protein kinase Cα (PKCα; fold change of 1.25 compared with the control); double-stranded RNA-dependent protein kinase (PKR1; 1.54); protein kinase C α/β2 (PKCα/β2; 0.62); Raf1 proto-oncogene-encoded protein kinase (RAF1; 0.47); protein kinase Cδ (PKCδ; 0.51); mitogen- and stress-activated protein kinase 1 (MSK1; 0.67); Src proto-oncogene-encoded protein kinase (SRC; 1.57); glycogen synthase kinase 3α (GSK3α; 0.33); and glycogen synthase kinase 3β (GSK3β; 0.61). Among these, SRC kinase was the only protein previously shown to be associated with M. tuberculosis infection (22).
Interestingly, the serine/threonine protein kinase GSK3α was identified as the protein dephosphorylated the most by M. tuberculosis. GSK3α Tyr279 showed a 67% decrease in phosphorylation when macrophages infected with M. tuberculosis were compared with macrophages infected with the ΔptpA mutant strain. Because of its status as a key player in the regulation of cell fate in both pro- and anti-apoptotic processes (23, 24), GSK3α was selected for further analysis.
PtpA Does Not Influence GSK3α Transcription Levels
To rule out the possibility that the GSK3α dephosphorylation observed in the kinome analysis (Fig. 2) was caused by reduced levels of expression due to the effect of PtpA on GSK3α transcription levels, we examined levels of GSK3α transcripts by qPCR. RNA from uninfected THP-1 cells and from THP-1 cells infected with M. tuberculosis and ΔptpA M. tuberculosis was harvested 18 h post-infection corresponding to the time point at which lysates were harvested for the kinome analysis. As seen in Fig. 3, qPCR profiling revealed a general modest increase in GSK3α transcript levels in cells infected with both M. tuberculosis and the ΔptpA mutant (Fig. 3, A and B) without significant difference between the two. Therefore, we concluded that PtpA does not have an impact on GSK3α expression levels and that the dephosphorylation observed in the kinome analysis is not due to the effect of PtpA on GSK3α transcription level but rather on bona fide dephosphorylation of GSK3α by PtpA.
FIGURE 3.
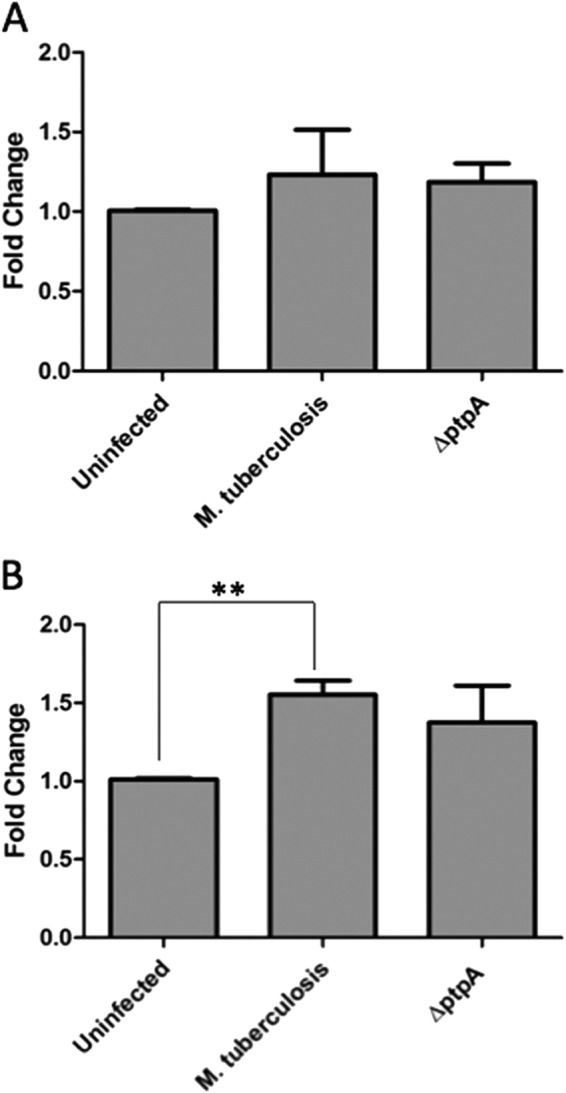
Transcriptional levels of GSK3α post-infection. Quantitative PCR analysis comparing mRNA levels of GSK3α from different infection conditions. RNA from uninfected and treated THP-1 cells (treated with M. tuberculosis and ΔptpA M. tuberculosis) was extracted 4 h (A) and 18 h (B) after infection and reverse-transcribed. Data observed show the expression levels of GSK3α. Transcript abundance was determined relative to the housekeeping gene GAPDH. Data shown are the means ± S.D. of three independent experiments. The difference in GSK3α transcript levels between M. tuberculosis-infected and ΔptpA M. tuberculosis-infected cells was not significant (p value of 0.8868 for A and 0.5193 for B). **, p < 0.001. Significant difference compared by Student's t test.
PtpA Dephosphorylates GSK3α under in Vivo and in Vitro Growth Conditions
To examine the dephosphorylation of GSK3α by PtpA, we conducted a Western blot assay in which we tested cellular extracts of macrophages infected with M. tuberculosis and the ΔptpA mutant (Fig. 4A). The phosphorylation level was monitored using the same anti-phospho-GSK3α (Tyr(P)279) antibody used in the kinome analysis (Fig. 2). As seen in Fig. 4A, GSK3α phosphorylation levels were found to be higher in extracts obtained from macrophages infected with the ΔptpA mutant compared with macrophages extracts obtained from infection by the parental M. tuberculosis strain, confirming our kinome analysis screening. We used an anti-GSK3α antibody to confirm that total protein levels of GSK3α did not change between samples.
FIGURE 4.
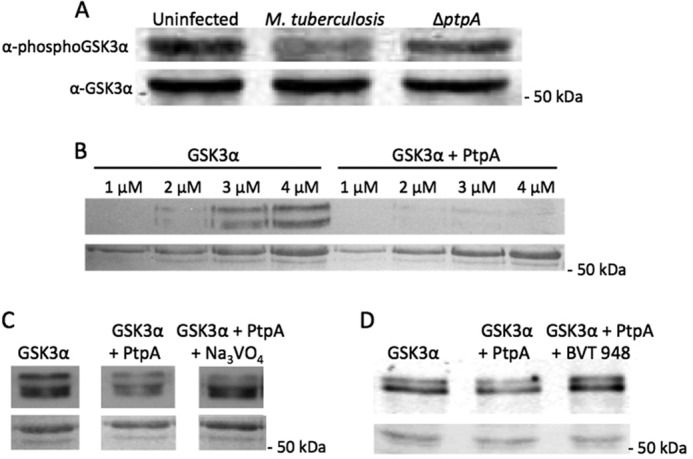
Western blot analyses of PtpA dephosphorylation of GSK3α in vivo and in vitro. A, THP-1 cells were infected with M. tuberculosis or ΔptpA M. tuberculosis. Cellular extracts were harvested 18 h post-infection, and 50 μg of it was used for Western blotting in which the anti-phospho-GSK3α (Tyr(P)279) antibody was utilized. The bottom panel represents the membrane probed with anti-GSK3α. The molecular mass of GSK3α is 50.981 kDa. B, different concentrations of GSK3α (1–4 μm) with and without PtpA (0.04 μm) were incubated and developed by enhanced chemiluminescence. The bottom panel represents the Ponceau-stained membrane showing equal loading of samples. C and D, fixed concentration of GSK3α (3 μm) with and without PtpA (0.04 μm) was incubated with the tyrosine phosphatase inhibitor Na3VO4 (1.5 mm) or BVT 948 (5 μm) and developed by enhanced chemiluminescence. The bottom panels represent the Ponceau-stained membranes showing equal loading of samples.
To determine whether GSK3α is a direct substrate of PtpA, we used two separate approaches as follows: biochemical assays to monitor catalysis, and a protein-protein interaction analysis to determine interaction between the two proteins. Western blot analysis of recombinant GSK3α to which PtpA was added was performed, and the result demonstrated that Tyr279 is dephosphorylated by PtpA in vitro (Fig. 4B). To assess the veracity of the dephosphorylating effect of PtpA on GSK3α, two Western blot analyses were performed in which the tyrosine phosphatase inhibitors sodium orthovanadate (Na3VO4) and BVT 948 were added to a GSK3α reaction containing PtpA. Although these are nonspecific protein-tyrosine phosphatase inhibitors, their inhibitory effect on PtpA is noticeable bringing the GSK3α Tyr279 phosphorylation level closer to its basal level (Fig. 4, C and D).
A more sensitive radioactive assay monitoring GSK3α kinase activity revealed that its autophosphorylation levels were significantly reduced in the presence of recombinant PtpA (Fig. 5A). This phenomenon was ameliorated upon addition of the tyrosine phosphatase inhibitor Na3VO4 and completely reversed by the addition of BVT 948 (Fig. 5, B and C). The extent of [γ-32P]ATP incorporation into GSK3α confirms that GSK3α is a self-phosphorylating autokinase dephosphorylated by PtpA.
FIGURE 5.
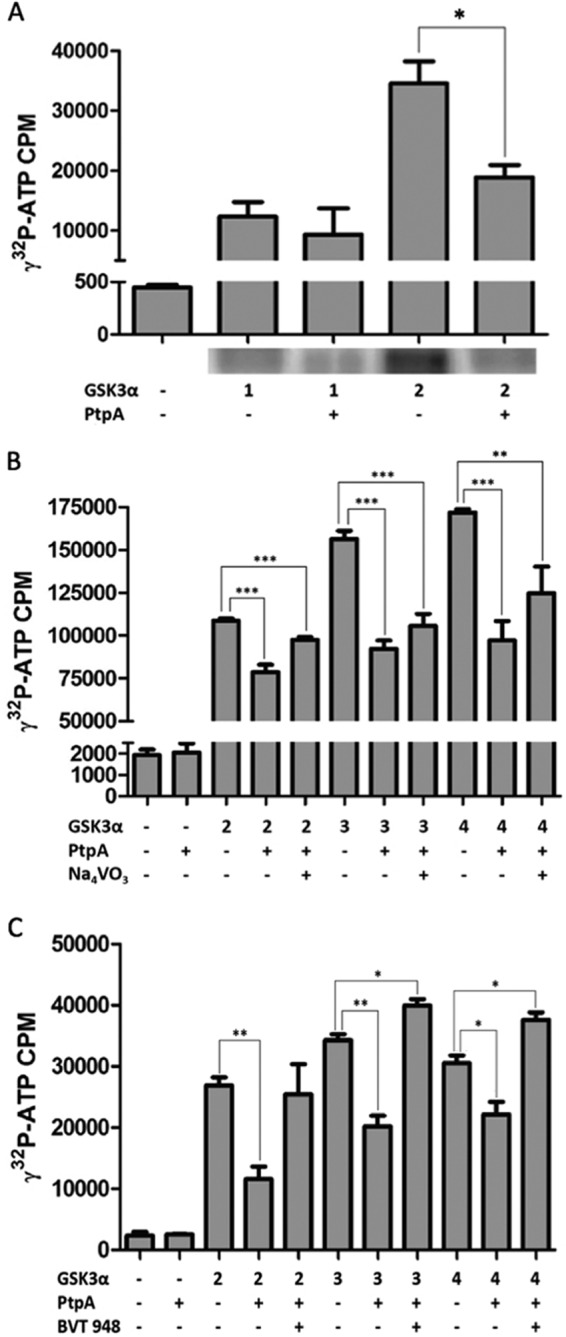
Kinase assays of PtpA dephosphorylation of GSK3α in vitro. A, dephosphorylation of GSK3α by PtpA was tested in an in vitro kinase assay. GSK3α (1 and 2 μm) was autophosphorylated in a kinase buffer containing 10 μCi of [γ-32P]ATP with or without PtpA (0.04 μm) and was resolved onto a 12% SDS gel and exposed to a PhosphorImager screen for radiolabeled band localization. After drying the gel, bands corresponding to phosphorylated GSK3α were cut from the gel, and the radioactive incorporation was measured by a scintillation counter. This graph represents the radioactivity of the dried gel. Results are expressed as ± S.D. of three independent experiments. The p value of 2 μm GSK3α with PtpA is 0.0214. B and C, inhibiting effect of PtpA on GSK3α's activity was tested by radiometric analysis. GSK3α (2–4 μm) was autophosphorylated in a reaction containing kinase buffer and 10 μCi of [γ-32P]ATP. PtpA (0.04 μm) and Na3VO4 (1.5 mm) or PtpA (0.04 μm) and BVT 948 (5 μm) were added to the sample mixtures and were spotted onto phosphocellulose paper. Radioactivity levels were measured by a scintillation apparatus. *, p < 0.05; **, p < 0.001; ***, p < 0.0001. Significant difference is compared by Student's t test.
To assess whether PtpA binds to GSK3α, ALPHAScreen (amplified luminescent proximity homogeneous assay), which is used to monitor protein-protein interactions, was performed. GSK3α was immobilized to beads by His tag, whereas GST-PtpA was immobilized by biotinylation according to the manufacturer's protocol (PerkinElmer Life Sciences). The results show direct and dose-dependent interaction between PtpA and GSK3α (Fig. 6). A hyperbolic curve fitting the 1:1 Langmuir binding model and a dissociation constant (Kd) of 4.023 × 10−9 m indicate a high level of affinity between the two proteins. PtpA binding to its known host substrate, hVPS33B, has similar strength with an assessed Kd value of 2.1 × 10−9 m (8).
FIGURE 6.
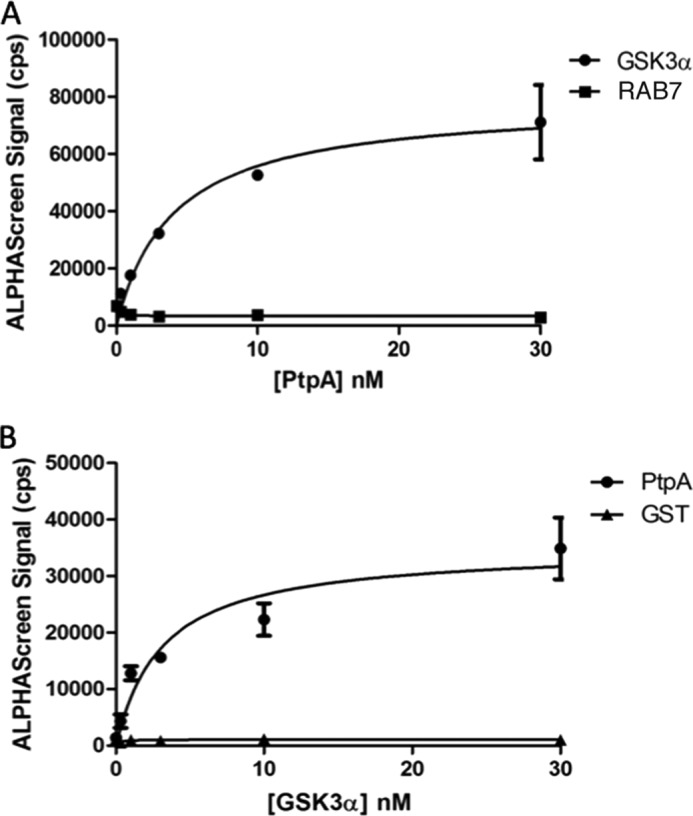
PtpA interacts with GSK3α in vitro. Protein-protein interaction between GSK3α and PtpA was determined using ALPHAScreen technology. A, Kd value was calculated using biotinylated PtpA and increasing concentrations of His-tagged GSK3α. His-tagged RAB7 protein served as the negative control. Curve fitting yielded a Kd of 4.023 × 10−9 m. B, reciprocal experiment with increasing concentrations of PtpA yielded a Kd of 3.125 × 10−9 m. GST was used as negative control.
PtpA Interferes with Host Macrophage Apoptosis Early during Infection
GSK3α plays a key role in the control of cell fate (23, 24). Previous studies have shown that phosphorylation of GSK3α Tyr279 is essential for the full activity of the enzyme (19) and that apoptotic stimuli increase its tyrosine phosphorylation activity (20). To check whether dephosphorylation of GSK3α Tyr279 by PtpA affects apoptosis, measurements of transcriptional and translational expression levels of the apoptotic executioner, caspase-3, were taken (25).
To investigate caspase-3 transcription levels, comparative qPCR levels of caspase-3 were performed on cellular extracts from either uninfected macrophages or infected with M. tuberculosis, the ΔptpA mutant, or with the complemented ΔptpA mutant (ΔptpA::ptpA). As seen in Fig. 7, A and B, we observed a significant difference in the expression levels of caspase-3 between cells infected with M. tuberculosis and ones infected with the ΔptpA mutant. Eighteen hours post-infection, caspase-3 transcription levels were 2-fold higher in the ΔptpA mutant-infected macrophages compared with wild-type M. tuberculosis-infected macrophages (Fig. 7A), indicating that suppression of caspase-3 expression by M. tuberculosis is PtpA-dependent early during infection. Caspase-3 expression levels were even lower in macrophages infected with M. tuberculosis than in uninfected macrophages (Fig. 7A). Interestingly, the inhibition of caspase-3 expression is overturned between 18 and 48 h post-infection where the caspase-3 transcript in M. tuberculosis-infected macrophages increases, surpassing its levels in the ΔptpA mutant-infected macrophages (Fig. 7B). This turn of events indicates that PtpA interference with the apoptotic pathway is transient, and macrophages are capable of initiating apoptosis regardless of the presence of PtpA (Fig. 7B).
FIGURE 7.
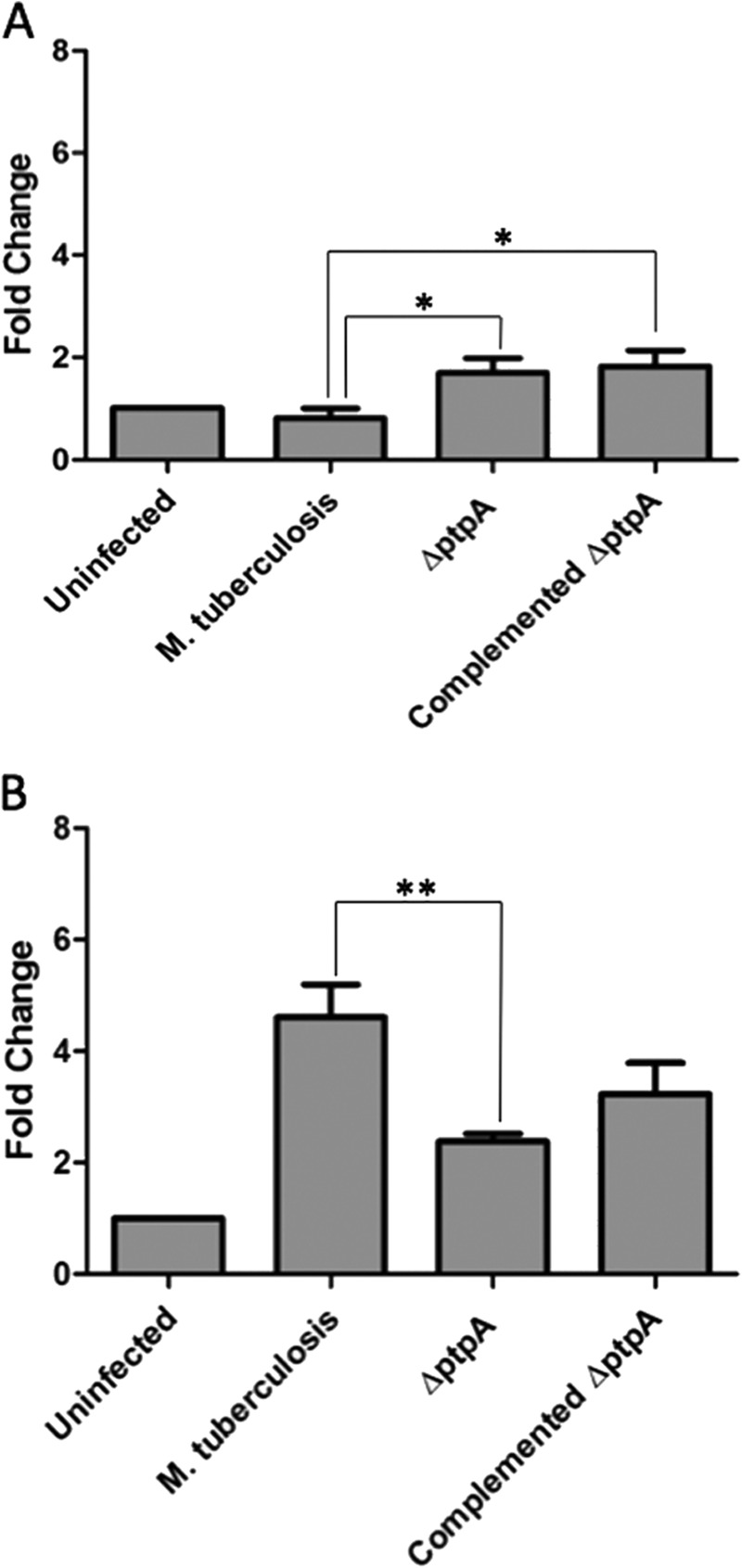
PtpA reduces transcriptional levels of caspase-3 early in infection. Quantitative PCR analysis comparing mRNA levels of caspase-3 from different infection conditions. RNA from uninfected and treated THP-1 cells (treated with M. tuberculosis, ΔptpA M. tuberculosis, and ΔptpA::ptpA M. tuberculosis) was extracted 18 h (A) and 48 h (B) after infection and reverse-transcribed. Data observed show the expression levels of caspase-3. Transcript abundance was determined relative to the housekeeping gene GAPDH. Data shown are the means ± S.D. of three independent experiments. The differences in transcript levels in cells infected with M. tuberculosis and ΔptpA M. tuberculosis 18 and 48 h post-infection were significant (p value of 0.0179 for A and 0.0097 for B). *, p < 0.05; **, p < 0.001. Significant difference compared by Student's t test.
M. tuberculosis Blocks Proteolytic Cleavage of Inactive Caspase-3 into Active Caspase-3
To test whether PtpA's dephosphorylation of GSK3α results in modulation of caspase-3 activity, we monitored caspase-3 proteolytic degradation using anti-caspase-3 antibody. As seen in Fig. 8, M. tuberculosis infection inhibits the cleavage of inactive caspase-3 (31.6 kDa) into its active forms (17/19 kDa). Cellular extracts from uninfected cells and from cells infected with the ΔptpA mutant show both active and inactive caspase-3, whereas those from M. tuberculosis-infected macrophages show only inactive caspase-3. Macrophages infected with the ΔptpA::ptpA strain show only limited activation of caspase-3, suggesting that the observed effect of the complemented strain is not optimal and in agreement with other complementation phenotypes we have observed (8, 11).
FIGURE 8.
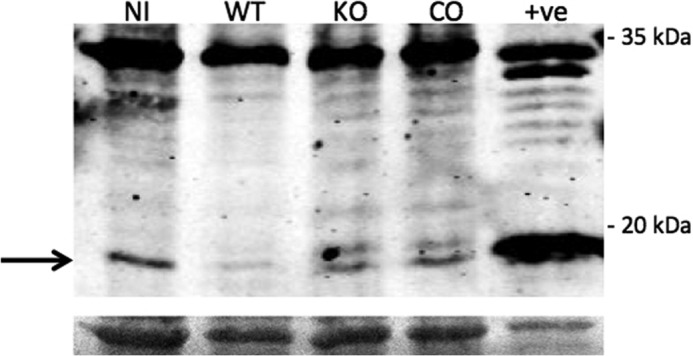
M. tuberculosis blocks activation of caspase-3 in vivo. THP-1 cells were uninfected (NI), infected with M. tuberculosis (WT), the ΔptpA mutant (KO), or the complement ΔptpA mutant (CO) and cellular extracts were harvested 48 h post-infection. A total of 50 μg of cellular extracts was used for Western blotting in which the anti-caspase-3 antibody was utilized. The molecular mass of inactive caspase-3 is 31.608 and 17/19 kDa for active caspase-3. The cellular extract of RAW 264.7 cells treated with 5 μm staurosporine for 5 h was used as the positive control (+ve). The bottom panel represents the Ponceau-stained membrane showing equal loading of samples.
DISCUSSION
M. tuberculosis pathogenicity relies upon its ability to sense changes in the environment and respond to host defense assaults. It does so by actively interfering with macrophage physiological pathways (26). One specific strategy utilizes the secreted phosphatase, PtpA, to block both phagosome maturation and acidification (8, 11), which are the two key processes required for digestion of invading microorganisms and initiation of an adaptive immune response (3).
In a previous study, we showed that the global kinome of macrophages changes significantly upon mycobacterial infection (21). Following the rationale that some of these changes are dependent on the signaling protein PtpA of M. tuberculosis, this study was designed to comparatively monitor PtpA's contribution to the kinome status of key human signal transduction proteins during M. tuberculosis infection. We have now shown that PtpA modulates global phosphorylation patterns of macrophage proteins and that these modulations can impact the host cell fate.
In our previous kinome analysis, we compared the effect of infecting macrophages with live or dead Mycobacterium bovis BCG on phosphoprotein levels (21). Notably, GSK3β was among the most phosphorylated proteins upon M. bovis BCG infection (21). Interestingly, the phosphorylation patterns of M. bovis BCG and M. tuberculosis kinome analyses show some contradicting results exemplified by GSK3α and GSK3β. GSK3α and GSK3β were hyperphosphorylated on Tyr279/216 in cells infected with live M. bovis BCG with a fold change of 1.29 and 1.57, respectively, compared with the uninfected control cells (21). Kinome analysis of M. tuberculosis infection compared with the uninfected cells shows that GSK3α and GSK3β were dephosphorylated and had a fold change of 0.51 and 0.66, respectively. This discrepancy can be attributed to the genotypic differences of the two strains as M. bovis BCG is an avirulent vaccine strain (27). It is also well documented that macrophages respond differently to M. bovis BCG than they do to M. tuberculosis (28). The function of PtpA in M. bovis BCG is still under investigation. Potential reduction or the absence of secreted PtpA in this vaccine strain could explain the hyperphosphorylation occurring in macrophages harboring this nonvirulent strain.
It is worth noting that, in comparison with uninfected cells, the relative fold change of the GSK3 isoforms in macrophages infected with ΔptpA M. tuberculosis resembles that of the isoforms in macrophages infected with live M. bovis BCG. In fact, an increase in phosphorylation for GSK3α (fold change of 1.54) and for GSK3β (1.07) is observed in macrophages infected with ΔptpA M. tuberculosis when compared with the phosphorylation status of these isoforms in uninfected macrophages (Table 5). These data are similar to the results obtained from our previous kinome analysis when comparing the phosphorylation status of the isoforms in M. bovis BCG-infected cells versus uninfected cells (21). It appears that the attenuation caused by the ΔptpA mutation (8) makes it behave more like the avirulent M. bovis BCG strain.
GSK3α and GSK3β play an essential role in the regulation of the apoptotic pathway that functions as a host defense mechanism in mycobacterial infection (29). Studies have shown that phosphorylation of GSK3 Tyr279/216 is critical for the full activation of the kinases (19) and the induction of apoptosis (20). We showed that infection of macrophages with an attenuated mycobacterial strain primes macrophages for apoptosis via increased phosphorylation of Tyr279/216 and activation of the isoforms (21). Results from both kinome analyses suggest that the apoptotic pathway is turned on in macrophages infected with the attenuated strains, i.e. in M. bovis BCG and ΔptpA M. tuberculosis, via phosphorylation of GSK3α and GSK3β Tyr279/216. Alternatively, our study demonstrates that the virulent strain M. tuberculosis H37Rv inactivates GSK3α and GSK3β by dephosphorylation of Tyr279/216 that promotes survival of the host cell (Table 5).
Several in vitro studies have shown that the apoptosis rate is increased in macrophages infected with mycobacteria (30, 31). However, virulent strains of mycobacteria seem to induce less apoptosis compared with avirulent or attenuated strains (32, 33), reinforcing the idea that M. tuberculosis has developed strategies to block apoptosis to promote host cell survival. The ability of M. tuberculosis to block apoptosis is of great importance for the pathogen as death of the host cell removes its supportive growth environment (33). In agreement with this, our experiments have shown that GSK3α dephosphorylation on Tyr279 can be interpreted as an anti-apoptotic signal targeted by this pathogen.
Transcriptional levels of caspase-3, a protease that plays a critical role in the execution phase of apoptosis of the host, were suppressed in M. tuberculosis-infected cells (Fig. 7A), indicating that M. tuberculosis blocks early expression of caspase-3 to prevent apoptosis. Moreover, a significant difference in transcript levels exists between cells infected with M. tuberculosis and those infected with ΔptpA M. tuberculosis (Fig. 7A) confirming that this suppression is PtpA-dependent. This phenomenon might not necessarily be a direct effect of PtpA but rather a result of ΔptpA mutant attenuation.
Dephosphorylation of the host GSK3α by PtpA leads to prevention of host cell apoptosis during early stages of infection. The anti-apoptotic role of PtpA fades at later stages of infection but does not necessarily signify resumption of host macrophage apoptosis. To the contrary, we found that activation of caspase-3 by proteolytic cleavage in M. tuberculosis-infected macrophages is blocked 48 h post-infection despite nonengagement of PtpA. As shown in Fig. 8, inactive caspase-3 is expressed in all four treatments but is only cleaved to active caspase-3 in two treatments as follows: in uninfected cells and in cells infected with ΔptpA M. tuberculosis. However, macrophages infected with M. tuberculosis and the complemented mutant strain show no cleavage and limited cleavage of caspase-3, respectively.
To summarize, our study presents for the first time evidence that M. tuberculosis modulates host macrophage apoptosis using PtpA dephosphorylation of GSK3α early in infection. This provides novel insight into the pathogenicity of M. tuberculosis within macrophages and a better mechanistic understanding of how it is able to circumvent the macrophage killing machinery.
Acknowledgments
We thank Stefan Szary for help with graphical illustrations, Jeffrey Helm for proofreading of our manuscript, and Louise Creagh for technical assistance. We also thank Kinexus Bioinformatics Corp. for their multiphosphoprotein analysis and the British Columbia Centre for Disease Control for the use of the containment level 3 facility.
This work was supported by Canadian Institutes of Health Research Operating Grant MOP-106622 (to Y. A.-G.).
- qPCR
- quantitative PCR.
REFERENCES
- 1. Dolin P. J., Raviglione M. C., Kochi A. (1994) Global tuberculosis incidence and mortality during 1990–2000. Bull. World Health Organ. 72, 213–220 [PMC free article] [PubMed] [Google Scholar]
- 2. Hestvik A. L., Hmama Z., Av-Gay Y. (2005) Mycobacterial manipulation of the host cell. FEMS Microbiol. Rev. 29, 1041–1050 [DOI] [PubMed] [Google Scholar]
- 3. Armstrong J. A., Hart P. D. (1971) Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 134, 713–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sturgill-Koszycki S., Schaible U. E., Russell D. G. (1996) Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. EMBO J. 15, 6960–6968 [PMC free article] [PubMed] [Google Scholar]
- 5. Clemens D. L., Horwitz M. A. (1995) Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J. Exp. Med. 181, 257–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hoflack B., Kornfeld S. (1985) Lysosomal enzyme binding to mouse P388D1 macrophage membranes lacking the 215-kDa mannose 6-phosphate receptor: evidence for the existence of a second mannose 6-phosphate receptor. Proc. Natl. Acad. Sci. U.S.A. 82, 4428–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xu S., Cooper A., Sturgill-Koszycki S., van Heyningen T., Chatterjee D., Orme I., Allen P., Russell D. G. (1994) Intracellular trafficking in Mycobacterium tuberculosis and Mycobacterium avium-infected macrophages. J. Immunol. 153, 2568–2578 [PubMed] [Google Scholar]
- 8. Bach H., Papavinasasundaram K. G., Wong D., Hmama Z., Av-Gay Y. (2008) Mycobacterium tuberculosis virulence is mediated by PtpA dephosphorylation of human vacuolar protein sorting 33B. Cell Host Microbe 3, 316–322 [DOI] [PubMed] [Google Scholar]
- 9. Bach H., Sun J., Hmama Z., Av-Gay Y. (2006) Mycobacterium avium subsp. paratuberculosis PtpA is an endogenous tyrosine phosphatase secreted during infection. Infect. Immun. 74, 6540–6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banta L. M., Robinson J. S., Klionsky D. J., Emr S. D. (1988) Organelle assembly in yeast: characterization of yeast mutants defective in vacuolar biogenesis and protein sorting. J. Cell Biol. 107, 1369–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wong D., Bach H., Sun J., Hmama Z., Av-Gay Y. (2011) Mycobacterium tuberculosis protein tyrosine phosphatase A disrupts phagosome acidification by exclusion of host vacuolar H+-ATPase. Proc. Natl. Acad. Sci. U.S.A. 108, 19371–19376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hackam D. J., Rotstein O. D., Zhang W. J., Demaurex N., Woodside M., Tsai O., Grinstein S. (1997) Regulation of phagosomal acidification. Differential targeting of Na+/H+ exchangers, Na+/K+-ATPases, and vacuolar-type H+-ATPases. J. Biol. Chem. 272, 29810–29820 [DOI] [PubMed] [Google Scholar]
- 13. Mukherjee S., Ghosh R. N., Maxfield F. R. (1997) Endocytosis. Physiol. Rev. 77, 759–803 [DOI] [PubMed] [Google Scholar]
- 14. Wong D., Chao J. D., Av-Gay Y. (2013) Mycobacterium tuberculosis-secreted phosphatases: from pathogenesis to targets for TB drug development. Trends Microbiol. 21, 100–109 [DOI] [PubMed] [Google Scholar]
- 15. Pelech S., Zhang H. (2002) Plasticity of the kinomes in monkey and rat tissues. Sci. STKE 2002, 162, pe50. [DOI] [PubMed] [Google Scholar]
- 16. Embi N., Rylatt D. B., Cohen P. (1980) Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527 [PubMed] [Google Scholar]
- 17. Woodgett J. R. (1990) Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 9, 2431–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plyte S. E., Hughes K., Nikolakaki E., Pulverer B. J., Woodgett J. R. (1992) Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim. Biophys. Acta 1114, 147–162 [DOI] [PubMed] [Google Scholar]
- 19. Hughes K., Nikolakaki E., Plyte S. E., Totty N. F., Woodgett J. R. (1993) Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 12, 803–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bhat R. V., Shanley J., Correll M. P., Fieles W. E., Keith R. A., Scott C. W., Lee C. M. (2000) Regulation and localization of tyrosine 216 phosphorylation of glycogen synthase kinase-3β in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U.S.A. 97, 11074–11079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hestvik A. L., Hmama Z., Av-Gay Y. (2003) Kinome analysis of host response to mycobacterial infection: a novel technique in proteomics. Infect. Immun. 71, 5514–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karim A. F., Chandra P., Chopra A., Siddiqui Z., Bhaskar A., Singh A., Kumar D. (2011) Express path analysis identifies a tyrosine kinase Src-centric network regulating divergent host responses to Mycobacterium tuberculosis infection. J. Biol. Chem. 286, 40307–40319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cross D. A., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378, 785–789 [DOI] [PubMed] [Google Scholar]
- 24. Hoeflich K. P., Luo J., Rubie E. A., Tsao M. S., Jin O., Woodgett J. R. (2000) Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature 406, 86–90 [DOI] [PubMed] [Google Scholar]
- 25. Wang Z. B., Liu Y. Q., Cui Y. F. (2005) Pathways to caspase activation. Cell Biol. Int. 29, 489–496 [DOI] [PubMed] [Google Scholar]
- 26. Poirier V., Av-Gay Y. (2012) Mycobacterium tuberculosis modulators of the macrophage's cellular events. Microbes Infect. 14, 1211–1219 [DOI] [PubMed] [Google Scholar]
- 27. Behr M. A., Wilson M. A., Gill W. P., Salamon H., Schoolnik G. K., Rane S., Small P. M. (1999) Comparative genomics of BCG vaccines by whole genome DNA microarray. Science 284, 1520–1523 [DOI] [PubMed] [Google Scholar]
- 28. Yadav M., Schorey J. S. (2006) The β-glucan receptor dectin-1 functions together with TLR2 to mediate macrophage activation by mycobacteria. Blood 108, 3168–3175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oddo M., Renno T., Attinger A., Bakker T., MacDonald H. R., Meylan P. R. (1998) Fas ligand-induced apoptosis of infected human macrophages reduces the viability of intracellular Mycobacterium tuberculosis. J. Immunol. 160, 5448–5454 [PubMed] [Google Scholar]
- 30. Fratazzi C., Arbeit R. D., Carini C., Remold H. G. (1997) Programmed cell death of Mycobacterium avium serovar 4-infected human macrophages prevents the mycobacteria from spreading and induces mycobacterial growth inhibition by freshly added, uninfected macrophages. J. Immunol. 158, 4320–4327 [PubMed] [Google Scholar]
- 31. Riendeau C. J., Kornfeld H. (2003) THP-1 cell apoptosis in response to mycobacterial infection. Infect. Immun. 71, 254–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Balcewicz-Sablinska M. K., Keane J., Kornfeld H., Remold H. G. (1998) Pathogenic Mycobacterium tuberculosis evades apoptosis of host macrophages by release of TNF-R2, resulting in inactivation of TNF-α. J. Immunol. 161, 2636–2641 [PubMed] [Google Scholar]
- 33. Keane J., Remold H. G., Kornfeld H. (2000) Virulent Mycobacterium tuberculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol. 164, 2016–2020 [DOI] [PubMed] [Google Scholar]
- 34. Pappin D. J., Hojrup P., Bleasby A. J. (1993) Rapid identification of proteins by peptide-mass fingerprinting. Curr. Biol. 3, 327–332 [DOI] [PubMed] [Google Scholar]