

Background: Inhibition of bacterial nitric oxide synthase (NOS) improves the efficacy of antibiotics.
Results: Development and characterization of a novel bacterial NOS chimera supports NO production in the presence of NADPH and a flavodoxin reductase.
Conclusion: Structure activity relationship between NOS inhibitors and bacterial NOS can now be evaluated.
Significance: Assays can be adapted for high-throughput screening to identify new bacterial NOS inhibitors.
Keywords: Crystallography, Electron Transfer, Enzyme Inhibitor, Isothermal Titration Calorimetry (ITC), Nitric Oxide
Abstract
Production of nitric oxide (NO) by nitric oxide synthase (NOS) requires electrons to reduce the heme iron for substrate oxidation. Both FAD and FMN flavin groups mediate the transfer of NADPH derived electrons to NOS. Unlike mammalian NOS that contain both FAD and FMN binding domains within a single polypeptide chain, bacterial NOS is only composed of an oxygenase domain and must rely on separate redox partners for electron transfer and subsequent activity. Here, we report on the native redox partners for Bacillus subtilis NOS (bsNOS) and a novel chimera that promotes bsNOS activity. By identifying and characterizing native redox partners, we were also able to establish a robust enzyme assay for measuring bsNOS activity and inhibition. This assay was used to evaluate a series of established NOS inhibitors. Using the new assay for screening small molecules led to the identification of several potent inhibitors for which bsNOS-inhibitor crystal structures were determined. In addition to characterizing potent bsNOS inhibitors, substrate binding was also analyzed using isothermal titration calorimetry giving the first detailed thermodynamic analysis of substrate binding to NOS.
Introduction
Both mammals and select bacterial species oxidize l-Arg to l-citrulline and NO using nitric oxide synthase (NOS,2 (mNOS, mammalian NOS; bNOS, bacterial NOS) as the enzyme catalyst. NO production by NOS requires electrons to be transferred from NADPH to the heme active site via redox partners. Between the three multidomain mNOS isoforms, inducible NOS, endothelial NOS, and neuronal NOS, electron transfer relies on calmodulin-dependent interdomain protein-protein interactions (1) between the heme containing oxygenase domain and the NADPH/FAD/FMN-containing reductase domain. In sharp contrast, bNOS from Staphyloccocus aureus, Bacillus anthracis, and Bacillus subtilis are only composed of an oxygenase domain and thus must rely on other redox partners (2).
Across species, the biological function of NO is quite variable. In mammals, NO function ranges from neural communication to immune function and blood pressure homeostasis (3). Recently, significant efforts have been made toward the development and characterization of isoform specific neuronal NOS inhibitors to modulate the pathological effects of NO overproduction by neuronal NOS (4). Much of this work has relied on characterizing the inhibitor structure-activity relationship for structure-based drug design (5). In Gram-positive organisms, NO function is diverse and varied across species (6). Specifically, bNOS-generated NO renders the highly drug-resistant pathogen S. aureus less sensitive to antibiotics (7). Previously, we identified and structurally characterized several nonspecific bNOS inhibitors that improved the efficacy of an antimicrobial (8).
Initial kinetic analyses of bNOS relied on the mNOS reductase domain for electron transfer (9) or peroxide-dependent oxidation of N-omega-hydroxy-l-arginine (NOHA) (10) to circumvent the requirement for native redox partners. In these experiments, NO production was either indirectly quantified by the Griess reaction because NO is oxidized to nitrite/nitrate (11) or monitored by stop-flow analysis (9, 12). Later identification of native flavodoxins that could support bNOS activity led to the development of a three-component reaction composed an E. coli-derived ferredoxin reductase, B. subtilis-derived flavodoxin YkuN (or YkuP), and Bacillus subtilis NOS (bsNOS) (13). NO production was also measured using the three-component system by Griess reaction and electron transfer between YkuN and bsNOS was measured using stop-flow spectrophotometry (13). Although bsNOS activity could be measured in a reconstituted system, the relevance of using a heterologous ferredoxin reductase was unknown. More importantly, despite the advances made thus far to characterize NO production by a bNOS, a simple and rigorous assay for purposes of screening inhibitors against a bNOS has yet to be established.
To characterize and rapidly evaluate bNOS inhibitors for structure-based drug design, it was essential to first develop a robust enzyme assay. To carry this out, we first identified flavoproteins from B. subtilis that would support bsNOS activity. Because the flavodoxin YkuN was previously established as a redox partner for bsNOS (13), we hypothesized that electron transfer rates to YkuN would be improved using a B. subtilis-derived reductase. In part because a blast search of the previously utilized E. coli ferredoxin/flavodoxin reductase (FLDR), for bsNOS activity, revealed no sequence homology to the B. subtilis proteome. This was not surprising because unlike the Gram-positive B. subtilis bacterium that have evolved the ability to produce NO, Gram-negative E. coli does not contain a bNOS and thus has not evolved redox partners to support bNOS activity. Therefore, identification of a native redox system that promotes electron transfer from NAD(P)H to bsNOS would likely improve the flux of electrons to bsNOS for activity measurements.
Fortunately, a ferredoxin reductase from B. subtilis, YumC, has been previously identified and characterized (14, 15). YumC shares high sequence homology to thioredoxin reductase but lacks the di-cysteine motif (CXXC) essential for thioredoxin function (14). Interestingly, YumC also shares high sequence homology to another B. subtilis protein YcgT, a putative ferredoxin reductase, that is induced under nitrosative stress (16–18) and regulated by the ferric uptake regulator (19). Unlike YumC, characterization of YcgT as a ferredoxin reductase has yet to be reported. Because YumC has previously been characterized as a redox partner to iron-sulfur ferredoxins (20), we elected to evaluate YumC as a potential redox partner to the flavodoxin YkuN.
To evaluate whether YumC and YkuN would support bsNOS activity, we report here several enzyme assays that confirm YumC to function as a native redox partner involved in bsNOS activity. Because substrate hydroxylation by bsNOS requires electron transfer and electron transfer between proteins is a distant dependent event (21), we anticipated that we could further improve bsNOS activity by engineering a bsNOS-YkuN chimera (bBiDomain), using a linker similar in length to mNOS. To this end, we found the chimera bBiDomain to support NO production at conditions in which the three-component reaction produced negligible amounts of NO. We also have used this new assay to screen a series of potential bNOS inhibitors and have solved the crystal structure of a select few.
EXPERIMENTAL PROCEDURES
Cloning of yumC Gene
The yumC gene was PCR-amplified from chromosomal DNA of B. subtilis (ATCC 23857) using primers YumC-NdeI-F (5′-cgccatatgcgagaggatacaaaggtttatgatattacaattatag-3′) and YumC-XhoI-R (5′-cggctcgagttatttattttcaaaaagacttgttgagtgaagagg-3′). The PCR product was digested with restriction enzymes NdeI and XhoI, gel-purified, and ligated into pET28a (Novagen) using the NdeI and XhoI restriction sites to generate pJH085.
Cloning of ykuN
The B. subitilis YkuN protein sequence was obtained from GenbankTM (22). The DNA sequence was then codon-optimized for E. coli expression, synthesized, and ligated into pET21a vector (Novagen) using the NdeI and XhoI restriction sites by Genewiz to generate pJH060.
FLDR
The E. coli ferredoxin NADP+ reductase encoded plasmid was kindly provided by Dr Michael Waterman (University of Southern California) and subcloned into pET28a (Novagen) using the NdeI and XhoI restriction sites to generate overexpression plasmid pJH063.
Cloning of bBiDomain
A linker encoding the (GGGS)5 sequence, synthesized by Genewiz, was ligated into the BamH1 and SalI restriction sites of pET28a (Novagen) to generate the primary shuttle vector pJH045. DNA encoding bsNOS from (8) was PCR-amplified and ligated into the shuttle vector pJH045 using NdeI and BamH1 restriction sites to generate the secondary shuttle vector pJH067. YkuN DNA was also PCR amplified and ligated into pJH067 using the SalI and XhoI restriction sites to produce pJH070.
Expression and Purification
YumC containing expression plasmid pJH085 was transformed into chemically competent BL21 DE3 (Invitrogen) E. coli and expressed as reported previously (14). YumC-containing cell pellets were resuspended and lysed by microfluidics in buffer containing 50 mm KPi (pH 7.4), 100 mm NaCl, 5 μm FAD, and 10 mm imidazole. Cell lysate was loaded onto a nickel-nitrilotriacetic acid agarose (Qiagen) column and washed with buffer containing 50 mm KPi (pH 7.4), 200 mm NaCl, 5 μm FAD, and 18 mm imidazole. YumC was then eluted off the nickel-nitrilotriacetic acid column using 50 mm KPi (pH 7.4), 10 mm NaCl, 5 μm FAD, and 200 mm imidazole. The eluted YumC was then diluted 10-fold into 50 mm KPi (pH 7.4) and loaded onto a Q Sepharose column that was pre-equilibrated in 50 mm KPi (pH 7.4). The Q Sepharose column was washed with buffer containing 50 mm KPi (pH 7.4) and 10 mm NaCl. YumC was eluted with buffer contaiing 50 mm KPi (pH 7.4) and 200 mm NaCl. Protein was concentrated and further purified over an Superdex 75 column, using an AKTA FPLC (GE Life Sciences), in buffer containing 50 mm KPi (pH 7.4), 5% (v/v) glycerol, and 5 μm FAD. YumC purity was checked by SDS-PAGE gel analysis and concentration was calculated using the BCA assay kit (Pierce).
Expression of YkuN using plasmid pJH060 was followed as reported previously (23). Cells containing YkuN were purified as described for YumC except purification buffers contained 5 μm FMN instead of FAD.
FLDR expression plasmid pJH063 was transformed into chemically competent BL21 DE3 (Invitrogen) E. coli and plated on kanamycin containing LB agar plates. Starter cultures from individual colonies were grown for 8 h and used to inoculate 1 liter of LB media at 37 °C and shaken overnight at 200 rpm. The following morning cultures were induced at 25 °C with 1 mm isopropyl 1-thio-β-d-galactopyranoside and 3 μm riboflavin, and shaking was lowered to 80 RPM. Bacterial cultures were harvested 24 h post-induction. FLDR was purified as described for YkuN.
Expression and purification of bsNOS followed as reported previously (8). Expression of bBiDomain also followed the previous bsNOS expression protocol (8) except 3 μm riboflavin and 1 mm 5-aminolevulinic acid (Biosynth International, Inc.) were added at the time of induction to improve cofactor biosynthesis. The bBiDomain and bsNOS concentrations were determined from the 444-nm ferrous-CO complex absorption using a molar extinction coefficient of 76 mm−1 cm−1 (24).
Cytochrome c Reduction
Horse heart cytochrome c reduction assays were performed at room temperature and cytochrome c reduction was monitored at 550 nm using a Cary 3E UV-visible spectrophotometer. The assays were carried out in 25 mm KPi (pH 7.4), 1 μm FMN, 1 μm FAD, 40 μm horse heart cytochrome c (Affymetrix USB), 5 nm YkuN, and varying amounts of either FLDR or YumC. Individual reactions were initiated with 100 μm NADPH and the steady-state oxidation of cytochrome c reduction was calculated as described previously using Δϵ550 = 21 mm−1 cm−1 (25).
NO-mediated Nitrite Production by a Three-component Reaction
Reactions were carried out at 35 °C in a buffered solution composed of 25 mm KPi (pH 7.4), 100 mm NaCl, 10 units/ml SOD, 10 units/ml catalase, 5 μm FMN, 5 μm FAD, 50 μm H4B, and 200 μm NOHA. To the buffered solution, bsNOS, YkuN, and YumC were also added. Protein concentrations evaluated for NO-mediated nitrite production using a three-component reaction included 0.1 μm bsNOS, 0.1 μm YkuN, and 1 μm YumC. In addition, the three-component reaction was also evaluated with 1 μm bsNOS, 5 μm YkuN, and 5 μm YumC. Reactions were initiated by addition of NADPH at a final concentration of 400 μm and quenched by thermal denaturation at 100 °C for 10 min. Excess NADPH was consumed by adding 10 units/ml lactate dehydrogenase and 150 mm sodium pyruvate. Reactions were aliquoted to a 96-well microplate and mixed in a 1:1 ratio with Griess reagents as described previously (26) with the exception that nitrate was not reduced to nitrite. Nitrite concentrations were determined from a standard curve of nitrite solutions based on absorbance at 548 nm.
NO-mediated Nitrite Production by bBiDomain
Reactions were carried out using the buffered solution reported for the three-component reaction. The bBiDomain reactions included 100 nm bBiDomain and either YumC or FLDR at 1 μm; except for reactions in which YumC concentrations were varied. For inhibition assays, inhibitors were added at 30 μm. Similar to the three-component reaction, bBiDomain reactions were initiated with NADPH at a final concentration of 400 μm, quenched by thermal denaturation, and analyzed by Griess reaction.
IC50 Determination
Reactions were run analogous to bBiDomain activity assays except inhibitors were included at varying concentrations. Nitrite concentration was measured 4 min after NADPH addition by the Griess reaction. Percent activity was calculated based on the fraction of nitrite detected at a specific inhibitor concentration as compared with no inhibitor. To calculate the IC50, the data were fit to a curve using SigmaPlot (version 10.0, Systat Software, Inc.). IC50 was calculated from the line of best fit.
Imidazole Displacement
Conversion of a low spin imidazole-bound species to a high spin species was monitored by the imidazole displacement assay as described previously for bsNOS (8, 27). The same conditions for bsNOS were also used for bBiDomain. Additional imidazole displacement assays using bsNOS and YkuN were composed of 2 and 20 μm protein, respectively. An apparent spectral binding constant (KS,app) was calculated from a non-linear regression analysis using SigmaPlot (version 10.0, Systat Software, Inc.) and fit to Equation 1.
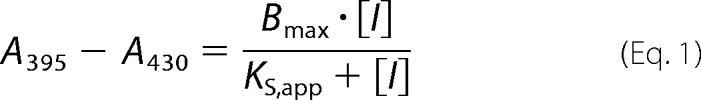 |
Assuming the KD of imidazole to be 384 μm (12), the spectral binding constant KS was calculated as described previously (27).
Crystallization and Structure Determination
Inhibitor bound bsNOS crystals were prepared and cryoprotected as previously described (8). X-ray data were collected at beamlines ALS 8.2.1 and SSRL 7-1. Data were indexed and integrated using MOSFLM (28) and scaled using Aimless (29). Protein and ligand were modeled in COOT (30) and refined with REFMAC (31).
Isothermal Titration Calorimetry
A VP-ITC titration calorimeter instrument (GE Healthcare) was used to measure l-Arg binding to bsNOS. l-Arg was prepared in 50 mm KPi (pH 7.4), and protein samples were exchanged into 50 mm KPi (pH 7.4) using Superdex 75 (GE Life Sciences). All samples were degassed under vacuum at −30 mm Hg for ∼20 min. The sample cell was filled with 118.14 μm bsNOS, and the injection syringe contained 3.0 mm l-Arg. All experiments were performed in triplicate at 25 °C using 30 injections at a reference power of 10 μcal/sec, stirring speed of 307 rpm and a 240-s time delay between injections. The first injection volume was set at 3 μl, and the remaining 29 injections were set to 5 μl. Heat measured for each injection was integrated, and the data were fit to a one-to-one binding mode using Origin (version 7.0, OriginLab Corp.).
RESULTS AND DISCUSSION
Cytochrome c Reduction by YkuN
Previous structural and biochemical analyses revealed YumC functions as a FAD-containing reductase (14, 15). To compare the rates of electron transfer from either YumC or FLDR to the flavodoxin YkuN, we monitored cytochrome c reduction by YkuN because cytochrome c is easily reduced by FMN containing flavodoxins (32). As shown in Fig. 1A, the rate of cytochrome c reduction is dependent on the concentration of either YumC or FLDR with YumC reaching saturation at ≈600 nm, whereas FLDR is unable to saturate YkuN. At saturation levels of YumC (≈600 nm), the rate of cytochrome c reduction is ∼20-fold faster than FLDR. This suggests that electron flux through YkuN is significantly improved when YumC is used as the reductase as compared with FLDR. The improved rates of electron transfer by YumC over FLDR also suggest that both YumC and YkuN function as native B. subtilis redox partners. This is also the first time YumC has been observed to reduce a flavodoxin. Because YumC now is established as a probable redox partner for multiple proteins in B. subtilis and is encoded by an essential gene (33), it is likely to play a necessary yet promiscuous role as a ferredoxin/flavodoxin reductase in supporting critical cellular redox processes.
FIGURE 1.
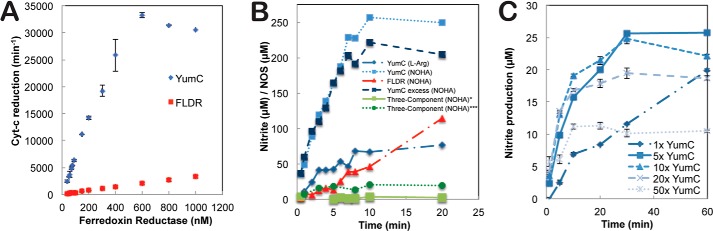
Catalytic activity of redox partners involved in bsNOS/bBiDomain production of NO. A, YumC more efficiently transfers electrons from NADPH to YkuN then FLDR based on cytochrome c reductase activity. B, time-dependent production of NO, as measured by nitrite, is improved using a combination of bBiDomain and YumC. The substrate used, either NOHA or l-Arg is indicated for each trial. Reactions YumC (l-Arg), YumC (NOHA), and FLDR (NOHA) were carried out with 0.1 μm bBiDomain and 1 μm YumC or FLDR as indicated. The three component reaction (single asterisk) was composed of 0.1 μm bsNOS, 0.1 μm YkuN, and 1 μm YumC. Separately, the three-component reaction (triple asterisks) was carried out with 1 μm bsNOS, 5 μm YkuN, and 5 μm YumC. C, stoichiometric ratio of YumC to bBiDomain influences activity over time. Error bars represent S.E.
bsNOS and bBiDomain Activity
Based on previous experiments, bsNOS-mediated NO production requires YkuN and a flavodoxin reductase (13). We confirmed that the three-component reaction utilizing YumC produces measurable quantities of NO oxidized nitrite over time (Fig. 1B, indicated by triple asterisks) in the presence of substrate NOHA. To improve the production of bsNOS generated NO, we chose to generate a catalytically self-sufficient system by fusing the N-terminal end of YkuN to the C-terminal end of bsNOS. This is similar to P450BM3 where the FMN/FAD reductase is fused to the C-terminal end of the P450 (34). The rate of the P450BM3 fatty acid hydroxylation using the individual modules is only 5% that of holo-P450BM3 (35), thus illustrating that fusion of redox partners could well increase activity. Assuming the site of electron transfer to the NOS oxygenase domain is conserved across species, we selected a linker length of 25 amino acids based on a sequence alignment between mammalian NOS redox partners (Fig. 2). The linker was composed of a GGGS motif and was chosen to promote flexibility and prevent proteolysis during expression and purification.
FIGURE 2.

The chimera bBidomain contains a (GGGS)5 linker. A, partial protein sequence alignment of human inducible NOS (iNOS), human endothelial NOS (eNOS), rat neuronal NOS (nNOS) and bBiDomain. Sequences were manually aligned based on crystal structure comparison of oxygenase domain using the homologous residues indicated by (single asterisk) and the FMN-binding domains are underlined. bBidomain oxygenase and FMN domain sequences are highlighted red and yellow, respectively. B, model for electron transfer from NADPH to bBiDomain.
Similar to the three-component reaction, time-dependent bBiDomain activity was evaluated using NADPH-dependent nitrite formation from either NOHA or l-Arg (Fig. 1B). Although l-Arg supported NO production, as measured by nitrite, the nitrite detected is less then half the amount produced by NOHA after 20 min. The bBiDomain dramatically improved bsNOS activity over the three-component system and is ∼8-fold more active, as shown in Fig. 1B. In addition, adding excess amounts of YkuN did not significantly alter bBiDomain activity (Fig. 1B). This is expected because electron transfer from YumC to either free YkuN or the YkuN within bBidomain is not limiting, and intramolecular electron transfer from YkuN to bsNOS in bBiDomain is much more efficient than with the free proteins. Electron transfer as a function of bBiDomain activity was also improved using YumC over FLDR as the ferredoxin reductase (Fig. 1B). Thus, the combination of YumC and bBiDomain significantly improved NOS-like activity and further corroborated the cytochrome c assay results (Fig. 1A) to provide additional support that YumC functions as a likely redox partner for YkuN to support bsNOS activity.
Interestingly, the ratio of YumC to bBidomain was critical for measuring bBiDomain activity (Fig. 1C). Even though a 200-fold excess of YumC was required to saturate YkuN, as observed by cytochrome c activity (Fig. 1A), excess YumC clearly lowers the production of NO, as measured by nitrite concentration. This might be expected if the site for electron donation on YkuN from YumC and to the heme active site of bBiDomain is the same. At high concentrations of YumC, the amount of YumC-YkuN complex would increase thus limiting electron transfer to the bBiDomain active site.
NO Production and Inhibition of bBiDomain
To test the new assay for the ability to quickly and simply estimate inhibitor potency, we first tested the well known NOS inhibitor N-omega-nitro-l-arginine (l-NNA). The reactions remained linear for the first 4 min, and both reactions reached completion after ∼10 min (Fig. 3A). We next screened several commercial NOS inhibitors and previously identified bsNOS inhibitors 1 and 2 that we have shown to exhibit antimicrobial properties (Fig. 3B) (8). These assays were carried out for 4 min prior to nitrite concentration being determined because we know from Fig. 3A that the reaction is still within the linear phase and thus is a direct measure of activity and inhibitor potency. Although all inhibitors reduced NO production, both 7-NI and 2 stand out as the most potent. This is consistent with our previous findings where 2 was found to be superior to l-NNA in bacterial growth inhibition assays (8). Moreover, these results show that a single time point Griess determination of nitrite using bBiDomain can provide a high throughput method for screening bNOS inhibitors.
FIGURE 3.
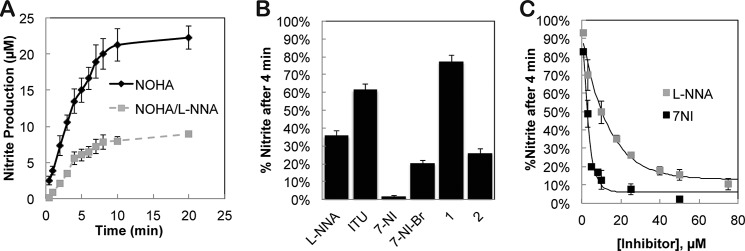
Time-dependent bBiDomain activity and inhibition as measured by Griess reaction. A, nitrite production remains linear in the first 4 min with bBiDomain and YumC at 100 nm and 1 μm, respectively. B, percent nitrite detected relative to no inhibitor after a 4-min incubation at 35 °C with 30 μm inhibitor. C, % nitrite detected as a function of inhibitor concentration for l-NNA and 7-NI. Error bars represent the S.E. of three separate experiments for inhibitor concentration measured.
Similarly, we also demonstrated the single-time point measurements to be a useful approach for determining inhibitor IC50 by measuring the fraction of nitrite produced by bBiDomain as a function of inhibitor concentration (Fig. 3C). IC50 values for both l-NNA and 7-NI were evaluated and determined to be 10.2 and 2.7 μm, respectively. Interestingly, these data compare well with the previously determined mammalian NOS IC50 values using separate and distinct enzyme assays (Table 1). Therefore, by utilizing a series of single time points as a function of inhibitor concentration, we can determine accurate IC50 values with bBiDomain and YumC.
TABLE 1.
Comparison of NOS IC50 determined for l-NNA and 7-NI
Imidazole Displacement
To further evaluate ligand binding to the bsNOS active site, we also measured the spectral binding constant, KS, for each ligand (Table 2). All substrates/inhibitors evaluated were found to function as type I ligands in both bsNOS and bBiDomain and shift the heme iron from low spin to high spin. In the presence of 10 molar excess YkuN or by using bBiDomain to evaluate binding, we consistently observed substrate/inhibitor binding in the low μm range, based on KS. These data demonstrate that YkuN does not influence substrate/inhibitor binding to the heme active site. Interestingly, based on the KS values for bsNOS, 2 binds ∼4-fold better than 1, whereas based on the enzyme assays, 2 is a ∼13-fold better inhibitor than 1. This pattern of KS values giving less dramatic differences than the enzyme assay is consistent with the other inhibitors tested, and we consider this reasonable agreement between the two methods given the complexity of the components required for the enzyme assay and that nitrite was measured at only a single time point.
TABLE 2.
Chemical name and calculated KS for substrate/inhibitor to active site of bsNOS, bsNOS in the presence of a 10 molar excess YkuN and bBiDomain
Error represents the error associated with the fit curve used to calculate the KS. Values not determined (n.d.) are indicated.
| Compound | Chemical name | bsNOS | bsNOS + 10x YkuN | bBiDomain |
|---|---|---|---|---|
| KS, μm | KS, μm | KS, μm | ||
| l-Arg | (S)-2-Amino-5-guanidinopentanoic acid | 0.8 ± 0.1 (8) | 6.5 ± 0.2 | 16 ± 2 |
| NOHA | N-Omega-hydroxy-l-arginine | 3.2 ± 0.5 | 2.2 ± 0.1 | 13 ± 5 |
| l-NNA | N-Omega-nitro-l-arginine | 1.3 ± 0.1 (8) | 4.9 ± 0.9 | 24 ± 7 |
| l-NAME | N-Omega-nitro-l-arginine methyl ester | 66 ± 46 | n.d. | n.d. |
| ITU | Ethylisothiourea | 9.3 ± 1.2 | n.d. | n.d. |
| 7-NI | 7-Nitroindazole | 3.5 ± 0.4 | 6.8 ± 0.8 | 27 ± 4 |
| 7-NI-Br | 3-Bromo-7-nitroindazole | 12 ± 1 | n.d. | n.d. |
| 1 | 6-({[(3R,5S)-5-{[(6-Amino-4-methylpyridin-2-yl)methoxy]methyl}pyrrolidin-3-yl]oxy}methyl)-4-methylpyridin-2-amine | 4.4 ± 0.1 (8) | n.d. | n.d. |
| 2 | 6,6′-{[(2S,3S)-2-Aminobutane-1,3-diyl]bis(oxymethanediyl)}bis(4-methylpyridin-2-amine) | 1.1 ± 0.1 (8) | n.d. | n.d. |
Ligand-bound Crystal Structures
Because both nitroindazoles and l-NNA are good bBiDomain inhibitors and structures of these inhibitors bound to a bacterial NOS have not been reported previously, we solved the crystal structures of these inhibitors bound to bsNOS (Table 3 and Fig. 4). Even though 7-NI-Br shares no structural similarity to l-Arg, it is still able to bind at the active site and induce a spin shift (Table 2). The 7-NI-Br-bsNOS crystal structure was refined to 2.15-Å resolution with clear electron density for the entire molecule (Fig. 4A). Similar to previous mNOS crystal structures (Fig. 4B), 7-NI-Br stacks parallel within a van der Waal's contact of the bsNOS heme group to form a hydrogen bond with the O-carbonyl of Trp-238 and NH of Met-240 (Fig. 4A). Also similar to mNOS, the active site Glu-243 must assume a different rotomeric position. Unlike mNOS (40) the reorientation of Glu-243 toward the pterin binding site does not induce a significant conformational change in heme propionate A. Regardless, the bsNOS pterin site is distorted due to the reorientation of Glu-243 and a pterin molecule is not observed in the crystal structure. In mNOS, the pterin is also displaced but a second 7-NI-Br molecule binds in the pterin site. We most likely do not observe a second 7-NI-Br in bsNOS because the pterin site in bsNOS is much larger and weakly binds pterins, in the 10–20 μm range (36), as compared with mNOS, which binds pterins in the nanomolar range (37).
TABLE 3.
Data collection, processing, and refinement statistics of the NOS ligand-bound structures
Values in parentheses are for the highest resolution shell. PDB, Protein Data Bank.
| PDB code | bsNOS-l-NNA (4UQR) | bsNOS-7NI-Br (4UQS) |
|---|---|---|
| Data collection | ||
| Wavelength (Å) | 1.127 | 0.918 |
| Space group | P21212 | P21212 |
| Cell dimension | a = 80.6, b = 94.8, and c = 62.9 Å; α = 90, β = 90, and γ = 90° | a = 80.4, b = 94.5, c = 63.2 Å; α = 90, β = 90, and γ = 90° |
| Unique reflections | 50,723 | 26,866 |
| Resolution (Å) | 61.41-1.72 (1.75-1.72) | 49.70-2.15 (2.21-2.15) |
| Rmergea | 0.064 (0.555) | 0.196 (0.720) |
| Rp.i.m.b | 0.045 (.509) | .080 (.429) |
| CC½c | 0.998 (0.812) | 0.992 (0.827) |
| I/σI | 12.5 (1.9) | 7.4 (1.3) |
| Completeness (%) | 97.9 (94.2) | 99.8 (99.5) |
| Multiplicity | 4.4 (3.6) | 5.7 (3.5) |
| Refinement | ||
| Resolution (Å) | 50.00-1.72 | 50.00-2.15 |
| No. of reflections | 42,876 | 24,931 |
| Rworkd | 0.168 | 0.229 |
| Rfreee | 0.199 | 0.283 |
| No. of atoms | 3402 | 3144 |
| Macromolecule | 2949 | 2941 |
| Ligand | 123 | 64 |
| Water | 329 | 138 |
| B-factors | ||
| Macromolecule | 28.5 | 53.2 |
| Ligand/ion | 30.9 | 44.4 |
| Solvent | 34.9 | 50.5 |
| r.m.s.d.f | ||
| Bond lengths (Å) | 0.012 | 0.019 |
| Bond angles | 1.77° | 2.05° |
a Rmerge = ΣhklΣi|Ii(hkl) − 〈I(hkl)〉|/ΣhklΣiIi(hkl), where 〈I(hkl)〉 is the mean value for its unique reflection; summations are over all reflections.
b Rp.i.m. = Σhkl[1/(N − 1)]½Σi|Ii(hkl) − 〈Ii(hkl)〉|/ΣhklΣiIi(hkl).
c CC½ was calculated from Ref. 48.
d Rwork = Σhkl‖Fobs| − Fcalc‖/Σhkl|Fobs|.
e Rfree = Rwork calculated using 5% of the reflection data chosen randomly and not included in the refinement.
f r.m.s.d., root mean square deviation.
FIGURE 4.

Inhibitor binding models of 7-NI-Br and l-NNA complexed to NOS active sites. Molecules bound near the active site are colored yellow, and the active site residues are colored white, with the heme shown in stick form and colored pink. A, the 2Fo − Fc map of 7-NI-Br bound to bsNOS active site and contoured to 1.5σ induces a conformational change in Glu-243. Also observed in the crystal structure is a PEG molecule and chloride ion. B, 7-NI-Br bound to endothelial NOS (eNOS; Protein Data Bank code 8NSE) reveals a near identical binding mode as observed in bsNOS except a second molecule of 7-Ni-Br forms a stacking interaction with Trp-449 to occupy the H4B co-substrate binding site. C, l-NNA binds to a network of active site hydrogen bonds and the 2Fo − Fc map of l-NNA is contoured at 2.0σ along with H4B bound to pterin site and binds in a similar orientation to endothelial NOS (D; eNOS; Protein Data Bank code 1D0C).
We also solved and refined the l-NNA-bound bsNOS crystal structure to 1.72 Å (Fig. 4C and Table 3). The binding mode of l-NNA is similar to l-Arg and analogous l-NNA mNOS structures for which bsNOS utilizes Glu-243 and the backbone atoms of both Met-240 and Trp-238 to H-bond the guanodinium group. Unlike l-Arg, the nitro group of l-NNA introduces new H-bonded interactions, as observed in mNOS structures (Fig. 4D) with Gly-237 and Trp-238 backbone atoms H bonding to the oxygen atoms of the nitro group (38). The additional H-bond interactions result in a lower KD, as compared with l-Arg, in mNOS systems (38). The same pattern for l-NNA KD likely holds true in bsNOS due to the high sequence similarity at the NOS active site across species and the observation that inhibitor binding modes are near identical.
l-Arg Binding Affinity
Further characterization of substrate binding to NOS active site was evaluated using isothermal titration calorimetry, as shown in Fig. 5, and represents the first thermodynamic analysis completed for l-Arg binding to a NOS protein. Accurate determination of l-Arg KD was necessary for future inhibition studies that evaluate Ki using the Cheng-Prusoff equation (39), as is commonly done for mNOS (40). The KD of l-Arg to bsNOS was determined to be 5.6 ± 0.1 μm and is in close agreement with the S. aureus NOS KD of 4.2 ± 0.2 μm determined by laser flash photolysis (41). Compared with neuronal NOS, which has an l-Arg Km of 8.4 ± 2.7 μm (42), the bsNOS KD is also within the same low μm range of mNOS. This is not surprising because the mNOS and bsNOS active sites are nearly identical (6). Moreover, l-Arg binding was also determined to be exothermic with a ΔH of −7.73 ± 0.02 kcal/mol at 25 °C. This thermodynamic result and additional characterization of l-Arg binding as enthalpically favored provides a better understanding for the thermodynamic considerations of future inhibitor based drug design.
FIGURE 5.
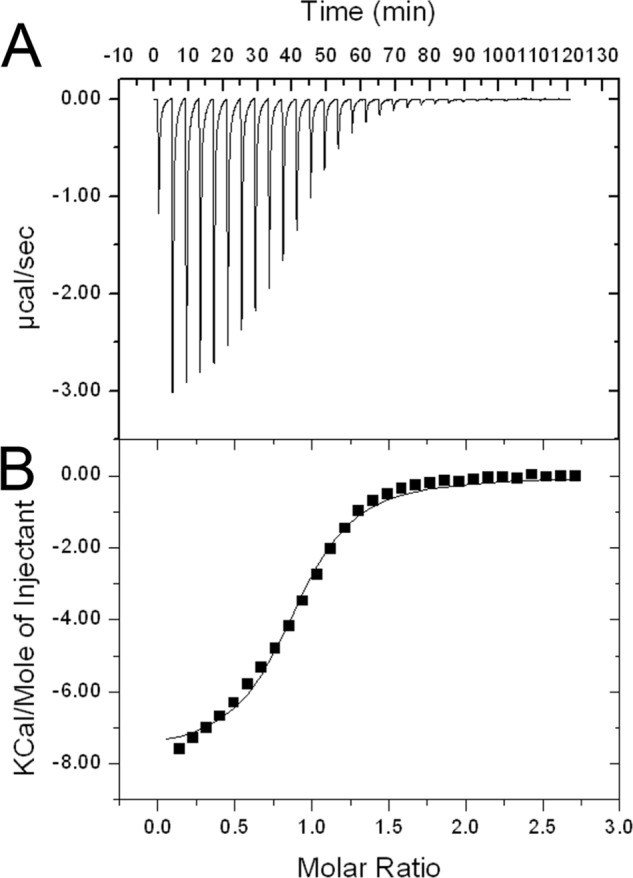
Isothermal titration calorimetry data of l-Arg titrated into bsNOS in 50 mm KPi, pH 7.4, at 25 °C. A, raw isothermal titration calorimetry data plotted as heat generated versus time for one of three trials. B, integrated and concentration-normalized heat evolved for one of three trials at each injection. A one-site model was used to generate the thermodynamic values for l-Arg binding KD (or 1/KA) = 5.6 ± 0.1 μm, ΔH = −7.73 ± 0.02 kcal/mol and TΔS = −0.047 ± 0.002 kcal/mol based on the average and S.E.M. of three separate trials.
Conclusion
Between a series of recent gene knock-out experiments and subsequent biological assays, it is clear that bNOS functions in pathogenic bacteria to provide a protective barrier against reactive oxygen species and a variety of antibiotics (7, 8, 43, 44). Therefore, by inhibiting the enzymatic function of bNOS, it is plausible that we can improve the efficacy of antibiotic treatments targeting pathogenic bacteria such as S. aureus and B. anthracis. To first identify potential inhibitors of bNOS, it was essential to develop a protein-based system in which ligand inhibition could be rapidly evaluated in vitro. Although previous studies have reported bNOS activity and rigorously characterized bNOS substrate catalysis (9, 10, 12, 13), this is the first study to utilize native redox partners for a bNOS and evaluate bNOS inhibition. Ultimately, this led us to the identification of YumC as a likely native redox partner for YkuN and the development of a novel chimera, bBiDomain, for activity analysis. In addition, this is also the first study to characterize the thermodynamics of substrate binding to a NOS protein and illustrates that detailed thermodynamic information can be obtained for ligand-bNOS interactions. Although the calorimetric approach will not be useful for high throughput screening, a careful thermodynamic analysis of a select few tight binding inhibitors can provide important insights into the inhibitor design effort.
Acknowledgments
This work is indebted to the many thought-provoking conversations with Drs. Huiying Li, Irina Sevrioukova, and Yarrow Madrona as well as Jerry Richo for initial help with protein expression and purification. We also thank the beamline staff at Stanford Synchrotron Radiation Laboratory and Advanced Light Source for assistance during the remote x-ray diffraction data collections.
This work was supported, in whole or in part, by National Institutes of Health Grant GM57353 (to T. L. P.).
The atomic coordinates and structure factors (codes 4UQR and 4UQS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NOS
- nitric oxide synthase(s)
- bNOS
- bacterial nitric oxide synthase(s)
- bsNOS
- Bacillus subtilis nitric oxide synthase(s)
- FLDR
- E. coli flavodoxin/ferredoxin reductase
- NOHA
- N-omega-hydroxy-l-arginine
- l-NNA
- N-omega-nitro-l-arginine
- 7-NI
- 7-nitroindazole
- 7-NI-Br
- 3-bromo-7-nitroindazole.
REFERENCES
- 1. White K. A., Marletta M. A. (1992) Nitric-oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry 31, 6627–6631 [DOI] [PubMed] [Google Scholar]
- 2. Adak S., Bilwes A. M., Panda K., Hosfield D., Aulak K. S., McDonald J. F., Tainer J. A., Getzoff E. D., Crane B. R., Stuehr D. J. (2002) Cloning, expression, and characterization of a nitric oxide synthase protein from Deinococcus radiodurans. Proc. Natl. Acad. Sci. U.S.A. 99, 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Moncada S., Palmer R. M., Higgs E. A. (1991) Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol. Rev. 43, 109–142 [PubMed] [Google Scholar]
- 4. Silverman R. B. (2009) Design of selective neuronal nitric oxide synthase inhibitors for the prevention and treatment of neurodegenerative diseases. Acc. Chem. Res. 42, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Poulos T. L., Li H. (2013) Structural basis for isoform-selective inhibition in nitric oxide synthase. Acc. Chem. Res. 46, 390–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sudhamsu J., Crane B. R. (2009) Bacterial nitric oxide synthases: what are they good for? Trends Microbiol. 17, 212–218 [DOI] [PubMed] [Google Scholar]
- 7. Gusarov I., Shatalin K., Starodubtseva M., Nudler E. (2009) Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325, 1380–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holden J. K., Li H., Jing Q., Kang S., Richo J., Silverman R. B., Poulos T. L. (2013) Structural and biological studies on bacterial nitric oxide synthase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 110, 18127–18131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adak S., Aulak K. S., Stuehr D. J. (2002) Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis. J. Biol. Chem. 277, 16167–16171 [DOI] [PubMed] [Google Scholar]
- 10. Bird L. E., Ren J., Zhang J., Foxwell N., Hawkins A. R., Charles I. G., Stammers D. K. (2002) Crystal structure of SANOS, a bacterial nitric oxide synthase oxygenase protein from Staphylococcus aureus. Structure 10, 1687–1696 [DOI] [PubMed] [Google Scholar]
- 11. Pufahl R. A., Wishnok J. S., Marletta M. A. (1995) Hydrogen peroxide-supported oxidation of NG-hydroxy-l-arginine by nitric oxide synthase. Biochemistry 34, 1930–1941 [DOI] [PubMed] [Google Scholar]
- 12. Wang Z. Q., Wei C. C., Sharma M., Pant K., Crane B. R., Stuehr D. J. (2004) A conserved Val to Ile switch near the heme pocket of animal and bacterial nitric-oxide synthases helps determine their distinct catalytic profiles. J. Biol. Chem. 279, 19018–19025 [DOI] [PubMed] [Google Scholar]
- 13. Wang Z. Q., Lawson R. J., Buddha M. R., Wei C. C., Crane B. R., Munro A. W., Stuehr D. J. (2007) Bacterial flavodoxins support nitric oxide production by Bacillus subtilis nitric-oxide synthase. J. Biol. Chem. 282, 2196–2202 [DOI] [PubMed] [Google Scholar]
- 14. Seo D., Kamino K., Inoue K., Sakurai H. (2004) Purification and characterization of ferredoxin-NADP+ reductase encoded by Bacillus subtilis yumC. Arch. Microbiol. 182, 80–89 [DOI] [PubMed] [Google Scholar]
- 15. Komori H., Seo D., Sakurai T., Higuchi Y. (2010) Crystal structure analysis of Bacillus subtilis ferredoxin-NADP(+) oxidoreductase and the structural basis for its substrate selectivity. Protein Sci. 19, 2279–2290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Moore C. M., Nakano M. M., Wang T., Ye R. W., Helmann J. D. (2004) Response of Bacillus subtilis to nitric oxide and the nitrosating agent sodium nitroprusside. J. Bacteriol. 186, 4655–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakano M. M. (2002) Induction of ResDE-dependent gene expression in Bacillus subtilis in response to nitric oxide and nitrosative stress. J. Bacteriol. 184, 1783–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reents H., Münch R., Dammeyer T., Jahn D., Härtig E. (2006) The Fnr regulon of Bacillus subtilis. J. Bacteriol. 188, 1103–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ollinger J., Song K. B., Antelmann H., Hecker M., Helmann J. D. (2006) Role of the Fur regulon in iron transport in Bacillus subtilis. J. Bacteriol. 188, 3664–3673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seo D., Okabe S., Yanase M., Kataoka K., Sakurai T. (2009) Studies of interaction of homo-dimeric ferredoxin-NAD(P)+ oxidoreductases of Bacillus subtilis and Rhodopseudomonas palustris, that are closely related to thioredoxin reductases in amino acid sequence, with ferredoxins and pyridine nucleotide coenzymes. Biochim. Biophys. Acta 1794, 594–601 [DOI] [PubMed] [Google Scholar]
- 21. Gray H. B., Winkler J. R. (2003) Electron tunneling through proteins. Q. Rev. Biophys. 36, 341–372 [DOI] [PubMed] [Google Scholar]
- 22. Kunst F., Ogasawara N., Moszer I., Albertini A. M., Alloni G., Azevedo V., Bertero M. G., Bessières P., Bolotin A., Borchert S., Borriss R., Boursier L., Brans A., Braun M., Brignell S. C., Bron S., Brouillet S., Bruschi C. V., Caldwell B., Capuano V., Carter N. M., Choi S. K., Cordani J. J., Connerton I. F., Cummings N. J., Daniel R. A., Denziot F., Devine K. M., Düsterhöft A., Ehrlich S. D., Emmerson P. T., Entian K. D., Errington J., Fabret C., Ferrari E., Foulger D., Fritz C., Fujita M., Fujita Y., Fuma S., Galizzi A., Galleron N., Ghim S. Y., Glaser P., Goffeau A., Golightly E. J., Grandi G., Guiseppi G., Guy B. J., Haga K., Haiech J. (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256 [DOI] [PubMed] [Google Scholar]
- 23. Lawson R. J., von Wachenfeldt C., Haq I., Perkins J., Munro A. W. (2004) Expression and characterization of the two flavodoxin proteins of Bacillus subtilis, YkuN and YkuP: biophysical properties and interactions with cytochrome P450 BioI. Biochemistry 43, 12390–12409 [DOI] [PubMed] [Google Scholar]
- 24. Stuehr D. J., Ikeda-Saito M. (1992) Spectral characterization of brain and macrophage nitric oxide synthases: cytochrome P-450-like hemeproteins that contain a flavin semiquinone radical. J. Biol. Chem. 267, 20547–20550 [PubMed] [Google Scholar]
- 25. Martásek P., Miller R. T., Roman L. J., Shea T., Masters B. S. (1999) Assay of isoforms of Escherichia coli-expressed nitric oxide synthase. Methods Enzymol. 301, 70–78 [DOI] [PubMed] [Google Scholar]
- 26. Miranda K. M., Espey M. G., Wink D. A. (2001) A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 5, 62–71 [DOI] [PubMed] [Google Scholar]
- 27. Roman L. J., Sheta E. A., Martasek P., Gross S. S., Liu Q., Masters B. S. (1995) High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 8428–8432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. W. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Cryst. D67, 271–281. 10.1107/S0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 30. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 32. Simondsen R. P., Weber P. C., Salemme F. R., Tollin G. (1982) Transient kinetics of electron transfer reactions of flavodoxin: ionic strength dependence of semiquinone oxidation by cytochrome c, ferricyanide, and ferric ethylenediaminetetraacetic acid and computer modeling of reaction complexes. Biochemistry 21, 6366–6375 [DOI] [PubMed] [Google Scholar]
- 33. Kobayashi K., Ehrlich S. D., Albertini A., Amati G., Andersen K. K., Arnaud M., Asai K., Ashikaga S., Aymerich S., Bessieres P., Boland F., Brignell S. C., Bron S., Bunai K., Chapuis J., Christiansen L. C., Danchin A., Débarbouille M., Dervyn E., Deuerling E., Devine K., Devine S. K., Dreesen O., Errington J., Fillinger S., Foster S. J., Fujita Y., Galizzi A., Gardan R., Eschevins C., Fukushima T., Haga K., Harwood C. R., Hecker M., Hosoya D., Hullo M. F., Kakeshita H., Karamata D., Kasahara Y., Kawamura F., Koga K., Koski P., Kuwana R., Imamura D., Ishimaru M., Ishikawa S., Ishio I., Le Coq D., Masson A., Mauël C., Meima R. (2003) Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 100, 4678–4683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Narhi L. O., Fulco A. J. (1986) Characterization of a catalytically self-sufficient 119,000-dalton cytochrome P-450 monooxygenase induced by barbiturates in Bacillus megaterium. J. Biol. Chem. 261, 7160–7169 [PubMed] [Google Scholar]
- 35. Sevrioukova I., Truan G., Peterson J. A. (1997) Reconstitution of the fatty acid hydroxylase activity of cytochrome P450BM-3 utilizing its functional domains. Arch. Biochem. Biophys. 340, 231–238 [DOI] [PubMed] [Google Scholar]
- 36. Crane B. R., Sudhamsu J., Patel B. A. (2010) Bacterial nitric oxide synthases. Annu. Rev. Biochem. 79, 445–470 [DOI] [PubMed] [Google Scholar]
- 37. Tejero J., Stuehr D. (2013) Tetrahydrobiopterin in nitric oxide synthase. IUBMB Life 65, 358–365 [DOI] [PubMed] [Google Scholar]
- 38. Raman C. S., Li H., Martásek P., Southan G., Masters B. S., Poulos T. L. (2001) Crystal structure of nitric oxide synthase bound to nitro indazole reveals a novel inactivation mechanism. Biochemistry 40, 13448–13455 [DOI] [PubMed] [Google Scholar]
- 39. Cheng Y., Prusoff W. H. (1973) Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 22, 3099–3108 [DOI] [PubMed] [Google Scholar]
- 40. Xue F., Fang J., Delker S. L., Li H., Martásek P., Roman L. J., Poulos T. L., Silverman R. B. (2011) Symmetric double-headed aminopyridines, a novel strategy for potent and membrane-permeable inhibitors of neuronal nitric oxide synthase. J. Med. Chem. 54, 2039–2048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chartier F. J., Couture M. (2007) Interactions between substrates and the haem-bound nitric oxide of ferric and ferrous bacterial nitric oxide synthases. Biochem. J. 401, 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Knowles R. G., Palacios M., Palmer R. M., Moncada S. (1990) Kinetic characteristics of nitric oxide synthase from rat brain. Biochem. J. 269, 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van Sorge N. M., Beasley F. C., Gusarov I., Gonzalez D. J., von Köckritz-Blickwede M., Anik S., Borkowski A. W., Dorrestein P. C., Nudler E., Nizet V. (2013) Methicillin-resistant Staphylococcus aureus bacterial nitric-oxide synthase affects antibiotic sensitivity and skin abscess development. J. Biol. Chem. 288, 6417–6426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gusarov I., Nudler E. (2005) NO-mediated cytoprotection: instant adaptation to oxidative stress in bacteria. Proc. Natl. Acad. Sci. U.S.A. 102, 13855–13860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alderton W. K., Cooper C. E., Knowles R. G. (2001) Nitric oxide synthases: structure, function and inhibition. Biochem. J. 357, 593–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Boer R., Ulrich W. R., Klein T., Mirau B., Haas S., Baur I. (2000) The inhibitory potency and selectivity of arginine substrate site nitric-oxide synthase inhibitors is solely determined by their affinity toward the different isoenzymes. Mol. Pharmacol. 58, 1026–1034 [PubMed] [Google Scholar]
- 47. Wolff D. J., Gribin B. J. (1994) The inhibition of the constitutive and inducible nitric oxide synthase isoforms by indazole agents. Arch. Biochem. Biophys. 311, 300–306 [DOI] [PubMed] [Google Scholar]
- 48. Karplus P. A., Diederichs K. (2012) Linking crystallographic model and data quality. Science 336, 1030–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]