

Background: Histamine is a major inflammatory mediator.
Results: H1 receptor, STIM1, Orai1, and extracellular calcium are indispensable for histamine-induced intracellular calcium mobilization, NFAT nuclear translocation, and IL-8 production in HUVECs.
Conclusion: STIM1 and Orai1 mediate histamine-evoked calcium entry and NFAT signaling in endothelial cells.
Significance: STIM1 and Orai1 are essential for histamine-induced inflammatory signaling of endothelial cells.
Keywords: Calcium Imaging, Calcium Release-activated Calcium Channel Protein 1 (ORAI1), Interleukin, NFAT Transcription Factor, Stromal Interaction Molecule 1 (STIM1), CRAC Channel, HUVEC, Histamine
Abstract
Histamine is an important immunomodulator involved in allergic reactions and inflammatory responses. In endothelial cells, histamine induces Ca2+ mobilization by releasing Ca2+ from the endoplasmic reticulum and eliciting Ca2+ entry across the plasma membrane. Herein, we show that histamine-evoked Ca2+ entry in human umbilical vein endothelial cells (HUVECs) is sensitive to blockers of Ca2+ release-activated Ca2+ (CRAC) channels. RNA interference against STIM1 or Orai1, the activating subunit and the pore-forming subunit of CRAC channels, respectively, abolishes this histamine-evoked Ca2+ entry. Furthermore, overexpression of dominant-negative CRAC channel subunits inhibits while co-expression of both STIM1 and Orai1 enhances histamine-induced Ca2+ influx. Interestingly, gene silencing of STIM1 or Orai1 also interrupts the activation of calcineurin/nuclear factor of activated T-cells (NFAT) pathway and the production of interleukin 8 triggered by histamine in HUVECs. Collectively, these results suggest a central role of STIM1 and Orai1 in mediating Ca2+ mobilization linked to inflammatory signaling of endothelial cells upon histamine stimulation.
Introduction
Inflammation is a complex biological response to injury and infection; a protective attempt aimed to remove harmful stimuli and to initiate the healing process (1–3). However, when inflammation becomes uncontrolled, it may turn into inflammatory disorders, leading to destruction of healthy tissues (3, 4).
Histamine, a biogenic amine, is one of the major inflammatory mediators that can modulate vascular function (e.g. increased vascular leakage) during inflammation (5–9). Released by mast cells or other leukocytes, histamine induces cytoskeletal reorganization in endothelial cells and intercellular gap formation, leading to endothelial hyperpermeability (9, 10). Histamine also triggers leukocyte extravasation by promoting surface expression of P-selectin (6, 11) and production/secretion of interleukin 8 (IL-8) through calcineurin (12, 13), a Ca2+-dependent phosphatase, in endothelial cells. Activation of calcineurin also contributes to the immune response, signaling by members of the nuclear factor of activated T-cells (NFAT)5 family (14, 15). The effects of histamine are mediated by four G-protein coupled receptors (GPCRs) -H1, H2, H3, and H4 receptors (16, 17). H1 receptors are highly expressed in the endothelium and H1 receptor antagonists suppress histamine-induced endothelial hyperpermeability (9, 18). Histamine binding to H1 receptors activates phospholipase C, which hydrolyzes phosphatidylinositol 4,5-bisphosphate to produce inositol trisphosphate (IP3) and diacylglycerol (19). IP3 in turn triggers Ca2+ release from the endoplasmic reticulum (ER) through IP3 receptors; a key signaling event mediating various physiological and pathophysiological processes of the cell (20–22).
Store-operated Ca2+ entry (SOCE) conducted through the Ca2+ release-activated Ca2+ (CRAC) channels in the plasma membrane (PM) is a key component of the cellular Ca2+ signaling pathway (23–25). Sustained activities of CRAC channels are essential for the activation of NFAT transcription factors in T lymphocytes, which leads to proper immune responses (25, 26). However, it remains unclear whether CRAC channels are necessary for NFAT activation in endothelial cells. Orai1 and STIM1 have been identified and characterized as the pore-forming subunit of CRAC channels and the ER Ca2+ sensor for channel activation, respectively (27–36). Upon Ca2+ release from the ER, STIM1 oligomerizes and subsequently moves to the ER-PM junctions, binding to and activating Orai1 channels for Ca2+ entry (28, 29, 33, 36–41). However, it has not been addressed whether this Ca2+ entry pathway contributes to the Ca2+ mobilization and function of vascular cells in response to histamine.
Recent in vitro studies indicate that both STIM1 and Orai1 are functionally expressed in endothelial cells, mediating SOCE for cell proliferation and migration (42–44). It is also suggested, by animal studies, that STIM1 plays an essential role in coronary endothelial dysfunction associated with diabetes (45), lipopolysaccharide-induced vascular leakage and pulmonary edema (46). However, the role of CRAC channels in the histamine-triggered inflammatory response has not been well examined. In the present study, utilizing both pharmacological and molecular tools specific to CRAC channels, we elucidated the contribution of STIM1 and Orai1 to histamine-evoked intracellular Ca2+ mobilization and downstream cytokine production in endothelial cells.
EXPERIMENTAL PROCEDURES
Chemicals
Histamine, gelatin solution (2%), diphenhydramine hydrochloride, fexofenadine hydrochloride, and GdCl3 were purchased from Sigma. Thapsigargin, 2-aminoethyl diphenylborinate (2-APB), SKF-96365, and BTP2 were purchased from EMD. Cyclosporin A (CsA) was purchased from Alomone Labs.
Molecular Cloning
The generation of pcDNA3/humanSTIM1, eGFP-tagged wild-type (WT) humanOrai1, Orai1-E106A, and Orai1-R91W mutants was described previously (27, 47–49). eGFP-NFATc1 was purchased from Addgene. The AdEasy system was used to create recombinant adenoviruses carrying eGFP-NFATc1 (50, 51). Briefly, a 2.9 kb KpnI-EcoRV eGFP-NFATc1 fragment was subcloned into the pShuttle-CMV vector. The resultant plasmids were linearized and transformed into competent BJ5183 Escherichia coli containing the adenoviral backbone plasmid pAdEasy-1 to generate kanamycin resistant, recombinant adenovirus plasmids, which were then transfected into HEK 293 cells for virus production.
Cells
Human umbilical vein endothelial cells (HUVECs) obtained from Lonza were maintained in EGM-2 medium (Lonza), transfected using Amaxa HUVEC Nucleofector Kit (Lonza), or transduced by the Ad-eGFP-NFATc1 viruses. For [Ca2+]i imaging or confocal imaging, cells were seeded onto glass coverslips pretreated with gelatin.
Single-Cell [Ca2+]i Imaging
Ratiometric [Ca2+]i imaging was performed on an IX-81 microscope (Olympus) based system as described previously (49, 52). HUVECs were incubated with 2 μm Fura-2 AM in the culture medium at 37 °C for 30 min. Transfected cells were identified by the presence of fused or coexpressed eGFP; using Semrock filters (BrightLine single-band multi-exciter filter set, optimized for Fura-2) to minimize contamination of Fura-2 fluorescence by bleed-through of eGFP fluorescence. Solution recipes are summarized in supplemental Table S1. Data were analyzed with Metafluor software (Universal Imaging) and OriginPro 8 software (OriginLab) and are expressed as means ± S.E. (supplemental Table S2).
RNA Interference (RNAi)
For each of STIM1, Orai1, Orai2, and Orai3, a mixture of four siRNAs was purchased from Dharmacon (47). Control non-targeting siRNA was also obtained from Dharmacon. HUVECs were transfected by siRNAs using Lipofectamine RNAiMAX Transfection Reagent (Life Technologies).
RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from HUVECs using RNeasy Mini Kit (Qiagen) following the manufacturer's protocol. The methods for RT-PCR were the same as described before (47). Quantitative-PCRs were performed using RT2 SYBR Green ROX qPCR Mastermix (Qiagen).
Fractionation and Western Blot
The separation and preparation of cytoplasmic and nuclear extracts from HUVECs were done with the NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific). Samples were resolved by SDS-PAGE and analyzed by standard Western blotting. Immunoblots were incubated with the primary antibodies indicated, including rabbit anti-STIM1 (1:1000) (27), mouse anti-GAPDH (1:10,000, Fitzgerald clone 6C5), rabbit anti-Orai1 (1:500, Alomone Labs), rabbit anti-Orai2 (1:500, Alomone Labs), rabbit anti-Orai3 (1:500, Alomone Labs), mouse anti-HDAC1 (1:500, Upstate clone 2E10), mouse anti-α-tubulin (1:500, ThermoFisher Scientific clone TU-01), and mouse anti-GFP-HRP (1:1000, Miltenyi Biotec).
Confocal Imaging
The methods for cell preparation were described previously (52). Fixed cells were viewed under a confocal laser-scanning microscope (FV300; Olympus) equipped with FluoView software.
Enzyme-linked Immunosorbent Assay (ELISA)
Anti-interleukin 8 ELISA experiments were performed using Human CXCL8/IL-8 Quantikine ELISA Kit (R&D systems) following the manufacturer's protocol.
RESULTS
Histamine Triggers Store Release, Followed by Ca2+ Entry, in HUVECs with Dose-dependent Manner
HUVECs were loaded with Fura-2 AM and imaged under room temperature to evaluate the effect of histamine on intracellular Ca2+ dynamics. The culture medium was replaced by a physiological saline solution containing 2 mm Ca2+ (Ca2, see supplemental Table S1), and histamine was applied at three different final concentrations (1, 10, and 100 μm). As expected, the intracellular Ca2+ ([Ca2+]i) level increased after the addition of histamine (Fig. 1, A–C). At 1 μm histamine, the [Ca2+]i level slowly reached a peak of 97 ± 7 nm in 3 min and remained at a plateau level (88 ± 6 nm, Fig. 1A). In contrast, addition of a vehicle solution did not trigger a detectable intracellular Ca2+ change in HUVECs (data not shown). With 10 μm histamine, the [Ca2+]i level increased to a peak of 223 ± 6 nm in less than 1 min and plateaued for about a minute. Thereafter, the [Ca2+]i level slowly declined to 126 ± 4 nm, measured 4 min after application of histamine (Fig. 1B). Histamine, 100 μm, enhanced the peak response (278 ± 4 nm) but not the sustained level (115 ± 2 nm) of [Ca2+]i (Fig. 1C). To examine whether this histamine-evoked Ca2+ mobilization involved Ca2+ release from the ER, histamine was applied to the cells bathed in a solution without Ca2+ (Ca0, see supplemental Table S1) to eliminate possible Ca2+ entry. Under this condition, the initial [Ca2+]i response was still elicited by histamine in a dose-dependent manner (Fig. 1, D–F). The peak level of [Ca2+]i, with or without extracellular Ca2+, was comparable (Fig. 1G, supplemental Table S2) and the raising phase of [Ca2+]i remained unchanged. However, the sustained [Ca2+]i component was abolished in the absence of extracellular Ca2+ (Fig. 1, D-F and H), indicating that this elevated [Ca2+]i was derived from extracellular sources following intracellular store depletion. Thus, histamine-evoked Ca2+ mobilization in HUVECs is composed of two components; the store release is responsible for the initial peak and the sustained component is mainly derived from Ca2+ influx.
FIGURE 1.

Histamine triggers intracellular calcium mobilization in HUVECs. A–C, representative intracellular free Ca2+ recordings ([Ca2+]i, mean ± S.E.) show cytosolic Ca2+ levels coupled to histamine stimulation. The top bars indicate the type of extracellular solutions (supplemental Table S1) applied to the cells, and the vertical lines on the x axis indicate the time of solution change. Histamine was applied at concentration 1 μm (A, n = 33 cells), 10 μm (B, n = 30 cells), and 100 μm (C, n = 32 cells). D–F, representative [Ca2+]i traces illustrate cellular response to histamine in the absence of extracellular Ca2+ (D, n = 45 cells; E, n = 38 cells; and F, n = 25 cells). G, averaged peak value of [Ca2+]i, with or without extracellular Ca2+, in response to various concentrations of histamine were summarized. H, averaged sustained value of [Ca2+]i were obtained at 4 min after application of histamine.
H1 Receptors Mediate Histamine-induced Mobilization of Intracellular Ca2+
There are four H-subtype cell surface GPCRs that can mediate cellular response to histamine. Because H1 receptors are functionally more dominant in endothelium (9), diphenhydramine (6, 21), and fexofenadine (53, 54), two specific H1 receptor antagonists, were applied to evaluate whether H1 receptors are responsible for the histamine-evoked mobilization of intracellular Ca2+ in HUVECs. As expected, 40 min preincubation with either diphenhydramine (30 μm) or fexofenadine (30 μm) prevented both histamine-triggered store release (Fig. 2, A, D, G, and J) and Ca2+ entry (Fig. 2, B, E, H, and K). In contrast, both diphenhydramine and fexofenadine had no significant effect on the Ca2+ entry induced by thapsigargin (TG, Fig. 2, C, F, I, and L), a sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump inhibitor (55), which passively depletes Ca2+ in the ER store in conjunction with triggering store-operated Ca2+ entry via CRAC channels (Fig. 2C). This observation suggests that the H1 receptor antagonists exert their action upstream from CRAC channels that conduct SOCE. Together, the results from this set of experiments indicate that binding of histamine to H1 receptors causes intracellular Ca2+ release in HUVECs followed by extracellular influx.
FIGURE 2.

Histamine H1 receptor antagonists prevent calcium mobilization elicited by histamine, but not thapsigargin, in HUVECs. A–I, representative [Ca2+]i responses to 10 μm histamine or 2 μm TG were recorded from HUVECs under control (A–C, n = 40, 35, and 51 cells, respectively), treated with either 30 μm diphenhydramine (Dip, D–E, n = 40, 48, and 32 cells, respectively) or 30 μm fexofenadine (FEX, G–I, n = 39, 46, and 39 cells, respectively). J–L, peak and sustained [Ca2+]i values were averaged from control, Dip-treated, and FEX-treated cells for comparison. *: p < 0.01 compared with control.
Histamine-evoked Ca2+ Entry Is Sensitive to CRAC Channel Blockers
To determine whether CRAC channels are involved in conducting the histamine-evoked Ca2+ entry, 10 μm Gd3+ (29), 50 μm 2-APB (56), 20 μm SKF-96365 (57), and 10 μm BTP2 (58) that have been shown to effectively block CRAC channels were independently applied. Each of these agents inhibited not only the TG-evoked Ca2+ entry in HUVECs (Fig. 3, A–F) but also, at the same concentrations, the histamine-evoked Ca2+ entry (Fig. 3, G–L). In another set of experiments, HUVECs were preincubated with 10 μm Gd3+ or 10 μm BTP2 before the application of histamine. Consistently, the sustained component, but not the initial store release, of Ca2+ mobilization by histamine was abolished (Fig. 4, A–D). These results demonstrate that CRAC channels are responsible for conducting histamine-evoked Ca2+ entry. However, the possible contribution of other Ca2+ entry via Ca2+-permeable channels in the PM secondary to the CRAC channel activation cannot be ruled out in this set of studies.
FIGURE 3.

CRAC channel blockers inhibit histamine-evoked calcium entry. A–E, representative [Ca2+]i traces from HUVECs show store-operated Ca2+ entry following store depletion by 2 μm TG (A, n = 40 cells), in a manner sensitive to CRAC channel blockers: 10 μm Gd3+ (B, n = 27 cells), 50 μm 2-APB (C, n = 40 cells), 20 μm SKF-96365 (D, n = 22 cells), or 10 μm BTP2 (E, n = 33 cells). F, averaged [Ca2+]i values were obtained at two-minute after application of vehicle control (Ctl) or CRAC channel blockers. G–K, representative [Ca2+]i traces from HUVECs show histamine-evoked [Ca2+]i mobilization (G, n = 37 cells), in a manner sensitive to various CRAC channel blockers (H–K, n = 34, 39, 47, and 33 cells, respectively). L, averaged [Ca2+]i values were obtained at two-minute after application of vehicle control (Ctl) or various CRAC channel blockers (i.e. 4 min after application of 100 μm histamine). *: p < 0.01 compared with control.
FIGURE 4.
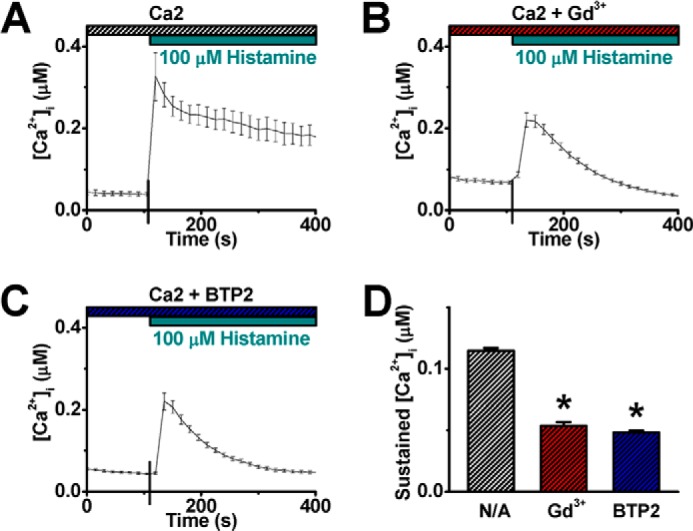
CRAC channel blockers eliminate the sustained component of histamine-evoked calcium mobilization. A–C, representative [Ca2+]i traces show histamine-evoked [Ca2+]i mobilization from control HUVECs (A, n = 10 cells) or cells treated with 10 μm Gd3+ (B, n = 33 cells) or 10 μm BTP2 (C, n = 30 cells) before the application of 100 μm histamine. D, averaged value of sustained [Ca2+]i was obtained at 4 min after histamine application. *: p < 0.01 compared with control.
Inhibiting STIM1 Expression by RNA Interference Eliminates Histamine-evoked Ca2+ Entry
It was reported that CRAC channels were formed by STIM1 and Orai1 in endothelial cells (42, 43). To examine the role of STIM1 in histamine-evoked Ca2+ entry, HUVECs were transfected with siRNA against STIM1 to disrupt the endogenous expression of STIM1. Cells treated with non-targeting siRNA were tested in parallel. While non-targeting siRNA did not significantly alter TG-induced Ca2+ influx, compared with untreated cells (Fig. 5, A, B, and G), STIM1-siRNA treated cells showed very little SOCE upon TG stimulation (Fig. 5C), indicating that STIM1 is necessary for CRAC channel functioning in HUVECs. Consistently, RNAi against STIM1 also prevented HUVECs from responding to histamine for intracellular Ca2+ mobilization (Fig. 5, D–F and H). The effect of RNAi on STIM1 expression was confirmed by RT-PCR (supplemental Fig. S1, A and B) and Western blot (Fig. 5I).
FIGURE 5.

Silencing of the STIM1 gene abolishes both thapsigargin- and histamine-evoked calcium entries in HUVECs. A–C, representative [Ca2+]i recordings show TG-triggered SOCE in HUVECs treated with vehicle control (A, n = 25 cells), non-targeting siRNA (B, n = 28 cells), or siRNA against STIM1 (C, n = 25 cells). D–F, representative [Ca2+]i responses to histamine from cells treated with vehicle control (D, n = 41 cells), non-targeting siRNA (E, n = 31 cells), or STIM1-siRNA (F, n = 51 cells). G–H, averaged peak (G) and sustained (H) values of [Ca2+]i for TG-evoked SOCE and histamine-induced Ca2+ entry, respectively, were compared among the treatments above. *: p < 0.01 compared with control. I, Western blot validation of siRNA-mediated gene silencing of STIM1 in HUVECs is shown. The protein levels of GAPDH from individual samples were examined to verify the application of equal amount of total cell lysates for Western blot assays. MW: molecular weight; Ctl: control; NT: non-targeting.
Gene Silencing of Orai1, but Not Orai2 or Orai3, Hinders Ca2+ Influx Triggered by Histamine
The involvement of three Orai channels (36) in histamine-evoked Ca2+ entry was also tested in HUVECs using RNAi approach. In cells transfected with siRNA specifically targeting Orai1, TG and histamine failed to stimulate Ca2+ influx (Fig. 6, A, B, E, and F). In contrast, knocking-down Orai2 or Orai3 did not reduce Ca2+ influx in response to TG or histamine (Fig. 6, C, D, G, and H). The specificity and efficacy of all the siRNA species on HUVECs were verified by RT-PCR (supplemental Fig. S1, C–F). Several anti-Orai2 and anti-Orai3 antibodies were tested, but none of them could detect endogenous Orai2 and Orai3 proteins in HUVECs, respectively (Fig. 6I and supplemental Fig. S2). In contrast, these proteins were detected in the cells transfected with human Orai2 or Orai3 plasmids (Fig. 6I and supplemental Fig. S2), indicating that Orai2 and Orai3 are not expressed or the expression levels are not detectable in HUVECs. Notably, endogenous Orai1 proteins were detected and the expression was effectively knocked down by Orai1 RNAi (Fig. 6I). Our results indicate that Orai1 and STIM1, the pore-forming and activating subunits, respectively, form the main type of endothelial CRAC channels, downstream of H1 receptors, mediating Ca2+ mobilization upon histamine stimulation.
FIGURE 6.
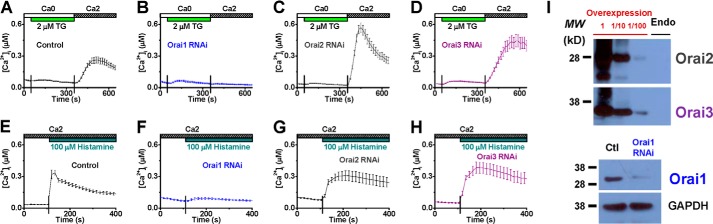
RNA interference against Orai1, but not Orai2 or Orai3, diminishes intracellular calcium mobilization triggered by thapsigargin or histamine in HUVECs. A–H, averaged cytosolic Ca2+ concentrations were obtained from HUVECs treated with vehicle control (A and E), siRNA targeting Orai1 (B and F), Orai2 (C and G), or Orai3 (D and H) in response to TG (A–D, n = 27, 12, 29, and 25 cells, respectively) or histamine (E–H, n = 23, 22, 23, and 25 cells, respectively). I, representative Western blot results show the lack of endogenous Orai2 and Orai3 protein signals from HUVECs (top) and the reduction of the Orai1 proteins in HUVECs treated with Orai1-siRNA (bottom). Cells transfected with human Orai2 or Orai3 plasmids were used as positive controls, and the corresponding cell lysates were loaded with (1:10 and 1:100) or without dilutions. GAPDH protein levels were examined to verify the equal loading of Western blot assays. MW: molecular weight; Endo: endogenous.
Overexpression of Dominant-negative Orai1 Mutants Suppresses Histamine-evoked Ca2+ Entry
Dominant-negative Orai1 mutants were employed to suppress the activity of endogenous Orai1 channels. The Orai1-E106A mutant (33, 34, 48) and Orai1-R91W mutant (30, 49, 59), with altered selectivity filter and inner gate of the channel pore, respectively, have been shown to disrupt channel function. The corresponding eGFP-fused cDNA constructs were delivered into HUVECs via nucleofection. Cells expressing either eGFP-Orai1-E106A or eGFP-Orai1-R91W led to diminished Ca2+ influx in response to TG (Fig. 7, A–D) as well as histamine (Fig. 7, E–H), emphasizing the indispensable role of Orai1 in histamine-evoked Ca2+ influx.
FIGURE 7.

Overexpression of dominant-negative Orai1 mutants suppresses calcium entry evoked by thapsigargin or histamine in HUVECs. A–C, representative [Ca2+]i recordings illustrate TG-triggered SOCE in HUVECs transfected with eGFP (A, control, n = 6 cells), eGFP-Orai1-E106A (B, n = 14 cells), or eGFP-Orai1-R91W (C, n = 13 cells). D, peak [Ca2+]i values were averaged for comparison. E–G, representative [Ca2+]i traces show histamine-evoked Ca2+ mobilization in cells transfected with eGFP (E, n = 5 cells), eGFP-Orai1-E106A (F, n = 11 cells), or eGFP-Orai1-R91W (G, n = 11 cells). H, sustained [Ca2+]i values, obtained at 4 min after histamine application, were averaged for comparison. *: p < 0.01 compared with control.
Histamine-evoked Ca2+ Entry Is Amplified by Overexpression of Both STIM1 and Orai1
The role of STIM1 and Orai1 in mediating histamine-evoked Ca2+ entry was further tested by overexpressing both channel molecules, STIM1 and eGFP-tagged Orai1, in HUVECs. Control cells were transfected with eGFP alone. As expected, co-expression of STIM1 and Orai1 significantly increased Ca2+ influx induced by TG or histamine (Fig. 8).
FIGURE 8.
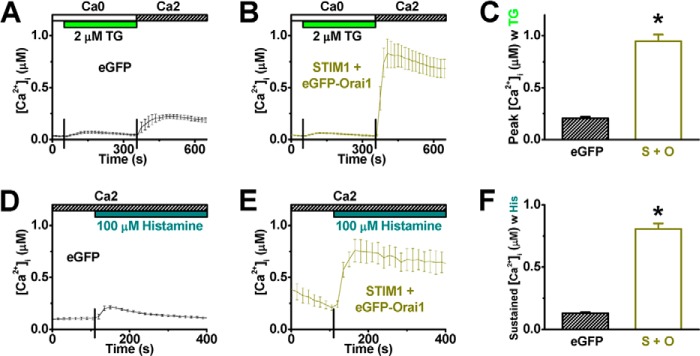
Co-expression of STIM1 and Orai1 amplifies calcium entry evoked by thapsigargin or histamine in HUVECs. A and B, TG-induced Ca2+ entry was traced from cells transfected with eGFP only (A, negative control, n = 9 cells) or STIM1 + eGFP-Orai1 (B, n = 11 cells). C, averaged peak [Ca2+]i values from cells transfected with eGFP or STIM1 + eGFP-Orai1 (S + O) were shown. D and E, mobilization of cytosolic Ca2+ in response to histamine was also recorded from cells transfected with eGFP only (D, n = 9 cells) or STIM1 + eGFP-Orai1 (E, n = 3 cells). F, sustained [Ca2+]i values were obtained at 4 min after histamine application. *: p < 0.01 compared with control.
Histamine-evoked Ca2+ Entry Causes Nuclear Import of NFAT
Members of the NFAT family are Ca2+-sensitive transcription factors (14, 60). After dephosphorylation by calcineurin, NFAT proteins translocate from the cytosol to the nucleus where they bind to promoter regions to turn on expression of numerous genes. However, the role of CRAC channels in mediating NFAT signaling in endothelial cells remains unclear, especially in relation to the inflammatory agent histamine. To address this issue, the subcellular localization of NFATc1 was examined by confocal imaging of HUVECs transfected with eGFP-NFATc1. In control cells, the exogenous NFATc1 molecules appeared to be distributed in the cytosol (Fig. 9, A and I), which exhibited a low level of Ca2+ (Fig. 1). In contrast, in cells with 100 μm histamine for 15–30 min in the presence of 2 mm extracellular Ca2+, eGFP-NFATc1 translocated to the cell nuclei (Fig. 9, B and J). This nuclear translocation process was blocked by H1 receptor antagonist (Fig. 9C). These results suggest a pivotal role of Ca2+ elevation, via extracellular Ca2+ entry (Fig. 9D), in mediating the NFAT nuclear translocation following H1 receptor activation by histamine. Moreover, the nuclear translocation of eGFP-NFATc1 is sensitive to both cyclosporin A (Fig. 9E), a calcineurin inhibitor (61), and Gd3+ (Fig. 9F), a CRAC channel blocker. These data support the idea that Ca2+ entry through CRAC channels activates calcineurin for NFATc1 dephosphorylation and subsequently leads to the nuclear import. Knocking-down STIM1 or Orai1 in HUVECs also prevented histamine-induced NFAT nuclear translocation (Fig. 9, G-H and K-L), further supporting a role of CRAC channel in regulating gene expression through the calcineurin-NFAT pathway. In parallel, fractionation experiments were performed to demonstrate nuclear import of NFAT transcription factors. An adenovirus vector carrying eGFP-NFATc1 was initially created to identify exogenous NFAT expression in HUVECs. Confocal imaging results confirmed a histamine-induced nuclear import of eGFP-NFATc1 in HUVECs transduced by the Ad-eGFP-NFATc1 viruses (data not shown). Next, the cytoplasmic and nuclear components were extracted from total HUVEC lysates and HDAC1 and alpha-tubulin were stained as nuclear and cytoplasmic markers, respectively. In consistence with the confocal imaging data, histamine (100 μm) elicited nuclear translocation of eGFP-NFATc1 in HUVECs with extracellular Ca2+. The increased eGFP-NFATc1 level in nuclear fraction was inhibited or abolished by pretreating the cells with cyclosporin A, Gd3+, STIM1-siRNA, or Orai1-siRNA (Fig. 10).
FIGURE 9.

CRAC channel-mediated calcium entry is essential for NFAT nuclear translocation in HUVECs upon histamine stimulation. A, representative confocal image of HUVECs transfected with eGFP-NFATc1 (green) is shown. The eGFP-NFATc1 molecules were mainly distributed in the cytosol under the control condition (in the Ca2 solution, see supplemental Table S1, without histamine). B, after 30-min stimulation with 100 μm histamine in the Ca2 solution, the majority of eGFP-NFATc1 molecules were translocated to the nuclei. C, cells were treated with 30 μm fexofenadine for 30 min before histamine stimulation. D, cells were stimulated with 100 μm histamine in the Ca0 solution (see supplemental Table S1). E and F, cells were treated with calcineurin inhibitor, cyclosporin A (CsA, 1 μm, E), or CRAC channel blocker, Gd3+ (10 μm, F), for 30 min before histamine stimulation. G and H, cells were treated with siRNA targeting STIM1 (G) or Orai1 (H) before histamine stimulation. I–K, enlarged areas marked by the squares in panels A, B, and G are displayed. Scale bars, 50 μm. L, quantitative results on NFAT nuclear translocation from 40 to 140 cells for each set of experiments are shown. *: p < 0.01 compared with column B. Ctl: control.
FIGURE 10.
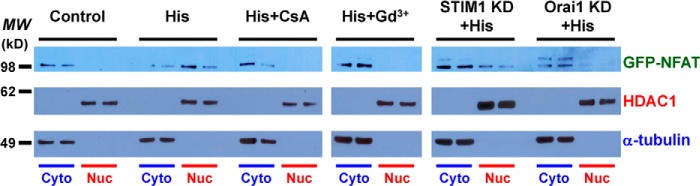
Confirmation of STIM1/Orai1-mediated nuclear import of NFAT by fractionation assays. Representative Western blot data following fractionation assays indicate a complete separation of cytoplasmic (Cyto) and nuclear (Nuc) contents from HUVECs expressing eGFP-NFATc1. The presence of eGFP-NFATc1 in the cytoplasmic and/or nuclear extracts under six different conditions was assessed by anti-GFP blotting. MW: molecular weight; CsA: cyclosporin A; KD: knocking-down; HDAC1: a nuclear marker; α-tubulin: a cytoplasmic marker.
STIM1 and Orai1 Are Indispensable for Histamine-promoted Interleukin 8 Expression
Interleukin 8, a chemokine produced by macrophages and endothelial cells, is essential for leukocyte chemoattraction and phagocytosis during inflammation (62, 63). The production of IL-8 in endothelial cells has been suggested to be enhanced by histamine in a calcineurin-dependent manner (12, 13). To determine whether functional CRAC channels are central to the regulation of IL-8 expression, the IL-8 mRNA level was measured by RT-PCR. Incubating HUVECs with 100 μm histamine for one hour significantly up-regulated IL-8 mRNA in a manner sensitive to cyclosporin A (1 μm, Fig. 11A). More importantly, the histamine-promoted IL-8 expression was also abolished by RNAi against Orai1 or STIM1 (Fig. 11A). Moreover, 24-hour treatment of HUVECs with 100 μm histamine led to a more than 3-fold increase in IL-8 production/secretion from these cells (Fig. 11B). This response was blocked by treating the cells with cyclosporin A (1 μm) or siRNA against Orai1 or STIM1 (Fig. 11B), indicating the close link between calcineurin-dependent signaling and CRAC channel activation for IL-8 synthesis/release in HUVECs upon histamine stimulation.
FIGURE 11.
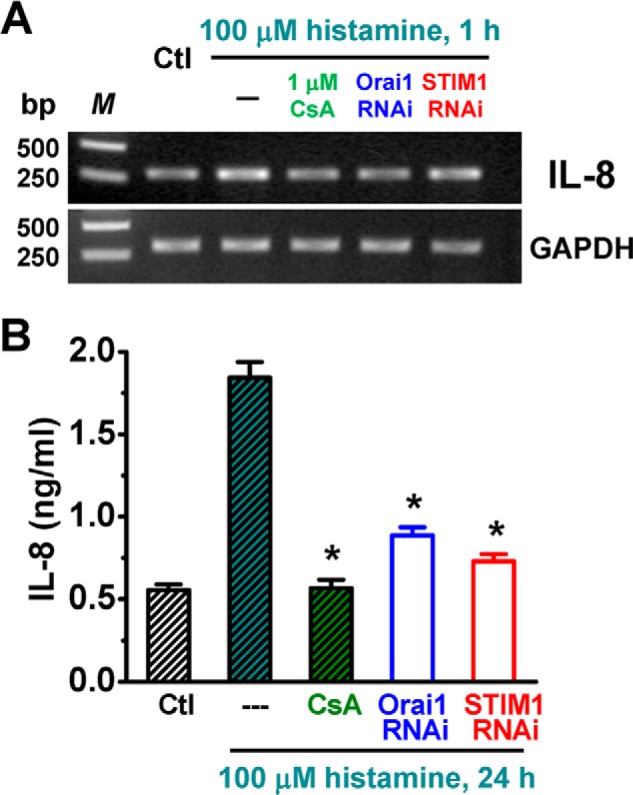
CRAC channels are critical for histamine-evoked interleukin 8 production and secretion in HUVECs. A, IL-8 expression was measured by RT-PCR. Cells were treated with 1 μm cyclosporin A, siRNA against STIM1 or Orai1 before vehicle (Ctl) or histamine (100 μm) administration. GAPDH expression was referenced as the loading control. B, secretion of IL-8 was measured by ELISA. Cells were treated with or without histamine (100 μm) for 24 h. *: p < 0.01 compared with cells treated with histamine only.
DISCUSSION
Inflammation is the bodies primary immune response to injury and infection; however, unregulated inflammation would cause damage to healthy tissues (2–4). Inflammatory disorders are considered to be one of the major pathogenic factors that cause a wide variety of human diseases, including but not limited to asthma, atherosclerosis, autoimmune diseases, inflammatory bowel diseases, reperfusion injury, and rheumatoid arthritis (2, 4, 64, 65).
Histamine is an important inflammatory mediator that has been commonly targeted to alleviate allergies and inflammatory responses (66, 67). In this study, we have demonstrated that the CRAC channel, formed by STIM1 and Orai1, mediates the histamine-evoked Ca2+ entry and the downstream NFAT nuclear translocation, as well as cytokine production, in HUVECs. These results suggest a unique role for CRAC channels in endothelium and its potential contribution to the development of inflammatory response.
Accumulating evidence on the Ca2+ entry through store-operated channels (e.g. CRAC channels) following depletion of the intracellular Ca2+-store suggests a ubiquitous and essential homeostatic signaling mechanism in immune cells (23, 25, 26, 36). Using pharmacological tools in combination with ratiometric Ca2+ imaging approach, we have elucidated that the intracellular Ca2+ mobilization in response to histamine involves H1 receptor-dependent store release followed by a sustained extracellular Ca2+ influx through CRAC channels in vascular endothelial cells (Figs. 1–4). This observation indicates that histamine-initiated vascular response in inflammation is partially due to the activation of CRAC channels.
In the current study, for the first time, we applied molecular tools at the gene level to determine the contribution of CRAC channels in mediating the histamine-elicited biological response in HUVECs. The human genome contains two STIM (STIM1 and STIM2) and three Orai (Orai1, Orai2, and Orai3) genes (36). Genetic defects in both STIM1 and Orai1 have been shown to cause immunodeficiency and autoimmunity in human patients (30, 68–70). Our results from the RNAi study demonstrated that STIM1 is indispensable for histamine-evoked Ca2+ entry in HUVECs (Fig. 5). It has been suggested that STIM2 may be functionally different from STIM1, that it is probably involved in the regulation of basal cytosolic Ca2+ level (71). Although it was not tested in the present study, the role of STIM2 in modulating intracellular Ca2+ level during histamine stimulation deserves future investigation. It is worth noting that the biophysical properties of ionic currents mediated by Orai2 and Orai3 channels have been shown to be very close to that of Orai1 channels (47, 72–74). However, our RNAi data indicate that Orai1, but not Orai2 or Orai3, is critical in mediating the histamine-evoked Ca2+ entry (Fig. 6). The necessity of CRAC/Orai1 channels to conduct Ca2+ influx in response to histamine was further confirmed by the reversed Ca2+ phenotypes following overexpression of wild-type versus dominant-negative Orai1 mutants (Figs. 7 and 8). The designation that specific Orai channels are linked to the activation of corresponding receptors might be important for the selective activation of a biological function in association with Ca2+ mobilization in the biological system.
Leukocytes are critical for the initiation and maintenance of inflammation (65). Leukocyte extravasation is a process of cell migration from the blood to the injured tissue across the vascular wall, including the steps of chemoattraction, rolling adhesion, tight adhesion, and transmigration (75, 76). It has been indicated that leukocyte recruitment can be interrupted when any of these steps is suppressed. IL-8 is a chemoattractant that directs neutrophils and other granulocytes to migrate toward the site of injury or infection, and anti-IL-8 therapy has been initiated for the treatment of inflammatory disorders (62, 63, 77). IL-8 production in endothelial cells has been reported to be up-regulated through the calcineurin/NFAT pathway upon histamine stimulation (12, 13). Our results suggest that extracellular Ca2+ entry, via Orai1/STIM1 activation, is essential for the nuclear translocation of NFAT and IL-8 synthesis in response to histamine (Figs. 9, 10, and 11). Thus, the CRAC channel may be a useful target for the control of leukocyte extravasation by modulating IL-8 synthesis in the endothelium.
In summary, the current study elucidates the fundamental role of STIM1 and Orai1 in the signaling cascade initiated by histamine (Fig. 12). This is a critical step in the development of the inflammatory pathologies associated with numerous disease processes. Understanding the functional mechanisms of CRAC channels in inflammation could lead to the development of CRAC channel blockers as potential therapeutic tools for treating inflammatory disorders.
FIGURE 12.
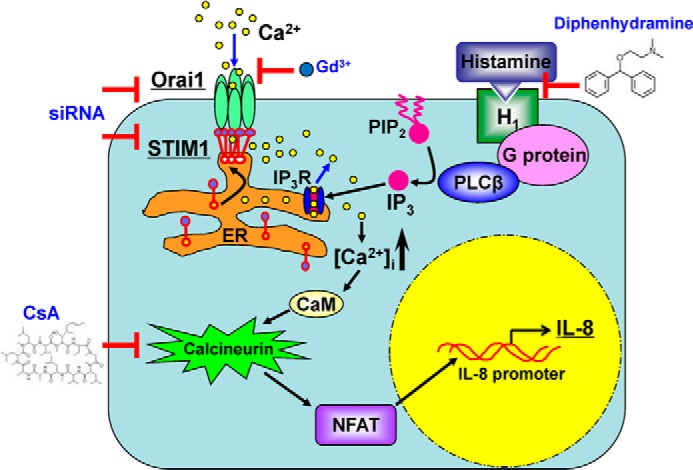
Schematic model for the CRAC channel signaling in endothelial cells upon histamine stimulation. The abbreviations used in this figure: PLC: phospholipase C; PIP2: phosphatidylinositol 4,5-bisphosphate; IP3R: inositol trisphosphate receptor; CaM: calmodulin; CsA: cyclosporin A.
Supplementary Material
Acknowledgments
We thank A. Webb for technical support with confocal imaging. Confocal microscopy was performed at the Integrated Microscopy and Imaging Laboratory, Texas A&M Health Science Center.
This work was supported by National Institutes of Health Grant R01 HL096552 (to D. C. Z.), and the LLS 3013-12 and National Institutes of Health Grant R01 GM112003 (to Y. Z.), the AHA NCRP 13SDG17200006 (to S. L. Z.), Kruse Family Centennial Chair Fund, Scott & White Memorial Hospital, and the 2013 TAMU-NSFC Program M1303600 (to L. K.), the 2012 Texas A&M-Weizmann Program (to S. L. Z. and X. P.), and the 2014 TAMU-CONACYT Program (to S. L. Z. and C. M. G.).

This article contains supplemental Tables S1 and S2 and Figs. S1 and S2.
- NFAT
- nuclear factor of activated T-cells
- STIM1
- stromal interaction molecule 1
- HUVEC
- human umbilical vein endothelial cell
- CRAC channel
- Ca2+ release-activated Ca2+ channel
- GPCR
- G-protein coupled receptor
- IP3
- inositol trisphosphate
- ER
- endoplasmic reticulum
- SOCE
- store-operated Ca2+ entry
- PM
- plasma membrane
- 2-APB
- 2-aminoethyl diphenylborinate
- CsA
- cyclosporin A
- RNAi
- RNA interference
- RT-PCR
- reverse transcription polymerase chain reaction
- TG
- thapsigargin
- SERCA
- sarco/endoplasmic reticulum Ca2+-ATPase
- eGFP
- enhanced green fluorescent protein.
REFERENCES
- 1. Mueller K. (2013) Inflammation. Inflammation's yin-yang. Introduction. Science 339, 155. [DOI] [PubMed] [Google Scholar]
- 2. Buckley C. D., Gilroy D. W., Serhan C. N., Stockinger B., Tak P. P. (2013) The resolution of inflammation. Nature Reviews. Immunology 13, 59–66 [DOI] [PubMed] [Google Scholar]
- 3. Wallach D., Kang T. B., Kovalenko A. (2014) Concepts of tissue injury and cell death in inflammation: a historical perspective. Nature Reviews. Immunology 14, 51–59 [DOI] [PubMed] [Google Scholar]
- 4. Tabas I., Glass C. K. (2013) Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science 339, 166–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yuan Y., Granger H. J., Zawieja D. C., DeFily D. V., Chilian W. M. (1993) Histamine increases venular permeability via a phospholipase C-NO synthase-guanylate cyclase cascade. Am. J. Physiol. 264, H1734–H1739 [DOI] [PubMed] [Google Scholar]
- 6. Asako H., Kurose I., Wolf R., DeFrees S., Zheng Z. L., Phillips M. L., Paulson J. C., Granger D. N. (1994) Role of H1 receptors and P-selectin in histamine-induced leukocyte rolling and adhesion in postcapillary venules. J. Clin. Investig. 93, 1508–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kurose I., Pothoulakis C., LaMont J. T., Anderson D. C., Paulson J. C., Miyasaka M., Wolf R., Granger D. N. (1994) Clostridium difficile toxin A-induced microvascular dysfunction. Role of histamine. J. Clin. Investig. 94, 1919–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu M. H., Yuan S. Y., Granger H. J. (2005) The protein kinase MEK1/2 mediate vascular endothelial growth factor- and histamine-induced hyperpermeability in porcine coronary venules. J. Physiol. 563, 95–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumar P., Shen Q., Pivetti C. D., Lee E. S., Wu M. H., Yuan S. Y. (2009) Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Exp. Rev. Mol. Med. 11, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ehringer W. D., Edwards M. J., Miller F. N. (1996) Mechanisms of alpha-thrombin, histamine, and bradykinin induced endothelial permeability. J. Cell. Physiol. 167, 562–569 [DOI] [PubMed] [Google Scholar]
- 11. Hattori R., Hamilton K. K., Fugate R. D., McEver R. P., Sims P. J. (1989) Stimulated secretion of endothelial von Willebrand factor is accompanied by rapid redistribution to the cell surface of the intracellular granule membrane protein GMP-140. J. Biol. Chem. 264, 7768–7771 [PubMed] [Google Scholar]
- 12. Jeannin P., Delneste Y., Gosset P., Molet S., Lassalle P., Hamid Q., Tsicopoulos A., Tonnel A. B. (1994) Histamine induces interleukin-8 secretion by endothelial cells. Blood 84, 2229–2233 [PubMed] [Google Scholar]
- 13. Boss V., Wang X., Koppelman L. F., Xu K., Murphy T. J. (1998) Histamine induces nuclear factor of activated T cell-mediated transcription and cyclosporin A-sensitive interleukin-8 mRNA expression in human umbilical vein endothelial cells. Mol. Pharmacol. 54, 264–272 [DOI] [PubMed] [Google Scholar]
- 14. Müller M. R., Rao A. (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nature Reviews. Immunology 10, 645–656 [DOI] [PubMed] [Google Scholar]
- 15. Pan M. G., Xiong Y., Chen F. (2013) NFAT gene family in inflammation and cancer. Curr. Mol. Med. 13, 543–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Walter M., Stark H. (2012) Histamine receptor subtypes: a century of rational drug design. Frontiers Biosciences 4, 461–488 [DOI] [PubMed] [Google Scholar]
- 17. Seifert R., Strasser A., Schneider E. H., Neumann D., Dove S., Buschauer A. (2013) Molecular and cellular analysis of human histamine receptor subtypes. Trends Pharmacol. Sci. 34, 33–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Srinivas S. P., Satpathy M., Guo Y., Anandan V. (2006) Histamine-induced phosphorylation of the regulatory light chain of myosin II disrupts the barrier integrity of corneal endothelial cells. Investig. Ophthalmol. Visual Sci. 47, 4011–4018 [DOI] [PubMed] [Google Scholar]
- 19. Hegyesi H., Darvas Z., Thurmond R., Falus A. (2005) Histamine Receptors and Signaling in Biophysical Aspects of Transmembrane Signaling (Damjanovich S., ed), pp. 265–291, Springer; Berlin Heidelberg [Google Scholar]
- 20. Jacob R. (1990) Agonist-stimulated divalent cation entry into single cultured human umbilical vein endothelial cells. J. Physiol. 421, 55–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jow F., Numann R. (2000) Histamine increases [Ca(2+)](in) and activates Ca-K and nonselective cation currents in cultured human capillary endothelial cells. J. Membr. Biol. 173, 107–116 [DOI] [PubMed] [Google Scholar]
- 22. Berridge M. J., Bootman M. D., Roderick H. L. (2003) Calcium signalling: dynamics, homeostasis and remodelling. Nature Reviews. Molecular Cell Biology 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 23. Parekh A. B., Putney J. W., Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810 [DOI] [PubMed] [Google Scholar]
- 24. Clapham D. E. (2007) Calcium signaling. Cell 131, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 25. Parekh A. B. (2010) Store-operated CRAC channels: function in health and disease. Nature Reviews. Drug Discovery 9, 399–410 [DOI] [PubMed] [Google Scholar]
- 26. Feske S. (2007) Calcium signalling in lymphocyte activation and disease. Nature Reviews. Immunology 7, 690–702 [DOI] [PubMed] [Google Scholar]
- 27. Roos J., DiGregorio P. J., Yeromin A. V., Ohlsen K., Lioudyno M., Zhang S., Safrina O., Kozak J. A., Wagner S. L., Cahalan M. D., Veliçelebi G., Stauderman K. A. (2005) STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169, 435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liou J., Kim M. L., Heo W. D., Jones J. T., Myers J. W., Ferrell J. E., Jr., Meyer T. (2005) STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15, 1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang S. L., Yu Y., Roos J., Kozak J. A., Deerinck T. J., Ellisman M. H., Stauderman K. A., Cahalan M. D. (2005) STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature 437, 902–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S. H., Tanasa B., Hogan P. G., Lewis R. S., Daly M., Rao A. (2006) A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441, 179–185 [DOI] [PubMed] [Google Scholar]
- 31. Zhang S. L., Yeromin A. V., Zhang X. H., Yu Y., Safrina O., Penna A., Roos J., Stauderman K. A., Cahalan M. D. (2006) Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. U. S. A. 103, 9357–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vig M., Peinelt C., Beck A., Koomoa D. L., Rabah D., Koblan-Huberson M., Kraft S., Turner H., Fleig A., Penner R., Kinet J. P. (2006) CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science 312, 1220–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yeromin A. V., Zhang S. L., Jiang W., Yu Y., Safrina O., Cahalan M. D. (2006) Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature 443, 226–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P. G. (2006) Orai1 is an essential pore subunit of the CRAC channel. Nature 443, 230–233 [DOI] [PubMed] [Google Scholar]
- 35. Vig M., Beck A., Billingsley J. M., Lis A., Parvez S., Peinelt C., Koomoa D. L., Soboloff J., Gill D. L., Fleig A., Kinet J. P., Penner R. (2006) CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 16, 2073–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hogan P. G., Lewis R. S., Rao A. (2010) Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 28, 491–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wu M. M., Buchanan J., Luik R. M., Lewis R. S. (2006) Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J. Cell Biol. 174, 803–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luik R. M., Wang B., Prakriya M., Wu M. M., Lewis R. S. (2008) Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature 454, 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Park C. Y., Hoover P. J., Mullins F. M., Bachhawat P., Covington E. D., Raunser S., Walz T., Garcia K. C., Dolmetsch R. E., Lewis R. S. (2009) STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136, 876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yuan J. P., Zeng W., Dorwart M. R., Choi Y. J., Worley P. F., Muallem S. (2009) SOAR and the polybasic STIM1 domains gate and regulate Orai channels. Nature Cell Biology 11, 337–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muik M., Fahrner M., Derler I., Schindl R., Bergsmann J., Frischauf I., Groschner K., Romanin C. (2009) A Cytosolic Homomerization and a Modulatory Domain within STIM1 C Terminus Determine Coupling to ORAI1 Channels. J. Biol. Chem. 284, 8421–8426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Abdullaev I. F., Bisaillon J. M., Potier M., Gonzalez J. C., Motiani R. K., Trebak M. (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circulation Research 103, 1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li J., Cubbon R. M., Wilson L. A., Amer M. S., McKeown L., Hou B., Majeed Y., Tumova S., Seymour V. A., Taylor H., Stacey M., O'Regan D., Foster R., Porter K. E., Kearney M. T., Beech D. J. (2011) Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circulation Research 108, 1190–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsai F. C., Seki A., Yang H. W., Hayer A., Carrasco S., Malmersjö S., Meyer T. (2014) A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nature Cell Biology 16, 133–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Estrada I. A., Donthamsetty R., Debski P., Zhou M. H., Zhang S. L., Yuan J. X., Han W., Makino A. (2012) STIM1 restores coronary endothelial function in type 1 diabetic mice. Circulation Res. 111, 1166–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gandhirajan R. K., Meng S., Chandramoorthy H. C., Mallilankaraman K., Mancarella S., Gao H., Razmpour R., Yang X. F., Houser S. R., Chen J., Koch W. J., Wang H., Soboloff J., Gill D. L., Madesh M. (2013) Blockade of NOX2 and STIM1 signaling limits lipopolysaccharide-induced vascular inflammation. J. Clin. Investig. 123, 887–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang S. L., Kozak J. A., Jiang W., Yeromin A. V., Chen J., Yu Y., Penna A., Shen W., Chi V., Cahalan M. D. (2008) Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J. Biol. Chem. 283, 17662–17671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lioudyno M. I., Kozak J. A., Penna A., Safrina O., Zhang S. L., Sen D., Roos J., Stauderman K. A., Cahalan M. D. (2008) Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc. Natl. Acad. Sci.U. S. A. 105, 2011–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang S. L., Yeromin A. V., Hu J., Amcheslavsky A., Zheng H., Cahalan M. D. (2011) Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc. Natl. Acad. Sci.U. S. A. 108, 17838–17843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Melkoumian Z. K., Peng X., Gan B., Wu X., Guan J. L. (2005) Mechanism of cell cycle regulation by FIP200 in human breast cancer cells. Cancer Research 65, 6676–6684 [DOI] [PubMed] [Google Scholar]
- 51. Luo J., Deng Z. L., Luo X., Tang N., Song W. X., Chen J., Sharff K. A., Luu H. H., Haydon R. C., Kinzler K. W., Vogelstein B., He T. C. (2007) A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nature Protocols 2, 1236–1247 [DOI] [PubMed] [Google Scholar]
- 52. Zheng H., Zhou M. H., Hu C., Kuo E., Peng X., Hu J., Kuo L., Zhang S. L. (2013) Differential roles of the C and N termini of Orai1 protein in interacting with stromal interaction molecule 1 (STIM1) for Ca2+ release-activated Ca2+ (CRAC) channel activation. J. Biol. Chem. 288, 11263–11272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abdelaziz M. M., Devalia J. L., Khair O. A., Bayram H., Prior A. J., Davies R. J. (1998) Effect of fexofenadine on eosinophil-induced changes in epithelial permeability and cytokine release from nasal epithelial cells of patients with seasonal allergic rhinitis. J. Allergy Clinical Immunol. 101, 410–420 [DOI] [PubMed] [Google Scholar]
- 54. Triggiani M., Gentile M., Secondo A., Granata F., Oriente A., Taglialatela M., Annunziato L., Marone G. (2001) Histamine induces exocytosis and IL-6 production from human lung macrophages through interaction with H1 receptors. J. Immunol. 166, 4083–4091 [DOI] [PubMed] [Google Scholar]
- 55. Zweifach A., Lewis R. S. (1993) Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc. Natl. Acad. Sci.U. S. A. 90, 6295–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prakriya M., Lewis R. S. (2001) Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J. Physiol. 536, 3–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chung S. C., McDonald T. V., Gardner P. (1994) Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br. J. Pharmacol. 113, 861–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zitt C., Strauss B., Schwarz E. C., Spaeth N., Rast G., Hatzelmann A., Hoth M. (2004) Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J. Biol. Chem. 279, 12427–12437 [DOI] [PubMed] [Google Scholar]
- 59. Hou X., Pedi L., Diver M. M., Long S. B. (2012) Crystal structure of the calcium release-activated calcium channel Orai. Science 338, 1308–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Crabtree G. R., Olson E. N. (2002) NFAT signaling: choreographing the social lives of cells. Cell 109, S67–S79 [DOI] [PubMed] [Google Scholar]
- 61. Fruman D. A., Klee C. B., Bierer B. E., Burakoff S. J. (1992) Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc. Natl. Acad. Sci.U. S. A. 89, 3686–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zeilhofer H. U., Schorr W. (2000) Role of interleukin-8 in neutrophil signaling. Curr. Opin. Hematol. 7, 178–182 [DOI] [PubMed] [Google Scholar]
- 63. Mukaida N. (2000) Interleukin-8: an expanding universe beyond neutrophil chemotaxis and activation. Int. J. Hematol. 72, 391–398 [PubMed] [Google Scholar]
- 64. Goel S., Duda D. G., Xu L., Munn L. L., Boucher Y., Fukumura D., Jain R. K. (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiological Reviews 91, 1071–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Swirski F. K., Nahrendorf M. (2013) Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 339, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bachert C. (1998) Histamine–a major role in allergy? Clin. Exp. Allergy 28, 15–19 [DOI] [PubMed] [Google Scholar]
- 67. Thurmond R. L., Gelfand E. W., Dunford P. J. (2008) The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nature Reviews. Drug Discovery 7, 41–53 [DOI] [PubMed] [Google Scholar]
- 68. Picard C., McCarl C. A., Papolos A., Khalil S., Lüthy K., Hivroz C., LeDeist F., Rieux-Laucat F., Rechavi G., Rao A., Fischer A., Feske S. (2009) STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N.E. J. Med. 360, 1971–1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. McCarl C. A., Picard C., Khalil S., Kawasaki T., Röther J., Papolos A., Kutok J., Hivroz C., Ledeist F., Plogmann K., Ehl S., Notheis G., Albert M. H., Belohradsky B. H., Kirschner J., Rao A., Fischer A., Feske S. (2009) ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J. Allergy Clinical Immunology 124, 1311–1318.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C., Jouanguy E., Feske S., Abel L., Casanova J. L. (2010) Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 207, 2307–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Brandman O., Liou J., Park W. S., Meyer T. (2007) STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell 131, 1327–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Soboloff J., Spassova M. A., Tang X. D., Hewavitharana T., Xu W., Gill D. L. (2006) Orai1 and STIM reconstitute store-operated calcium channel function. J. Biol. Chem. 281, 20661–20665 [DOI] [PubMed] [Google Scholar]
- 73. Mercer J. C., Dehaven W. I., Smyth J. T., Wedel B., Boyles R. R., Bird G. S., Putney J. W., Jr. (2006) Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 281, 24979–24990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lis A., Peinelt C., Beck A., Parvez S., Monteilh-Zoller M., Fleig A., Penner R. (2007) CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr. Biol. 17, 794–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Vestweber D. (2012) Novel insights into leukocyte extravasation. Curr. Opin. Hematol. 19, 212–217 [DOI] [PubMed] [Google Scholar]
- 76. Vestweber D. (2012) Relevance of endothelial junctions in leukocyte extravasation and vascular permeability. Ann. NY Acad. Sci. 1257, 184–192 [DOI] [PubMed] [Google Scholar]
- 77. Skov L., Beurskens F. J., Zachariae C. O., Reitamo S., Teeling J., Satijn D., Knudsen K. M., Boot E. P., Hudson D., Baadsgaard O., Parren P. W., van de Winkel J. G. (2008) IL-8 as antibody therapeutic target in inflammatory diseases: reduction of clinical activity in palmoplantar pustulosis. J. Immunol. 181, 669–679 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.