

Abstract
The pathophysiological events that lead to renal interstitial fibrogenesis are incompletely understood. Epoxyeicosatrienoic acid (EET), an arachidonic acid metabolite, has anti-inflammatory and profibrinolytic functions. Soluble epoxide hydrolase (sEH) converts EET to less active dihydroxyeicosatrienoic acid. Here, we tested the hypothesis that sEH deficiency would prevent tubulointerstitial fibrosis and inflammation induced by unilateral ureteral obstruction (UUO) in mouse kidneys. The loss of sEH enhanced levels of EET regioisomers and abolished tubulointerstitial fibrosis as demonstrated by reduced collagen deposition and myofibroblast formation at 3 and 10 days after UUO. The inflammatory response was prevented as demonstrated by decreased influx of neutrophil and macrophage, expression of inflammatory cytokines, and chemotactic factors in sEH-deficient UUO kidneys. Pharmacological inhibition of sEH also prevented inflammation and fibrosis after UUO. Next, we delved into the molecular mechanisms piloting the beneficial effects of sEH deficiency in renal fibrosis. UUO upregulated profibrotic factors associated with transforming growth factor (TGF)-β1/Smad3 signaling, oxidative stress, and NF-κB activation, and downregulated antifibrotic factors including peroxisome proliferator-activated receptor (PPAR) isoforms, especially PPARγ, but the loss of sEH prevented these adverse effects in UUO kidneys. Furthermore, administration of PPAR antagonists enhanced myofibroblast formation and activation of Smad3 and NF-κB p65, effects that were prevented by sEH deficiency in UUO kidneys. These data demonstrate that loss of sEH promotes anti-inflammatory and fibroprotective effects in UUO kidneys via activation of PPAR isoforms and downregulation of NF-κB, TGF-β1/Smad3, and inflammatory signaling pathways. Our data suggest the potential use of sEH inhibitors in treating fibrotic diseases.
Keywords: chronic kidney disease, unilateral ureteral obstruction, PPAR isoforms, soluble epoxide hydrolase, tubulointerstitial fibrosis
chronic kidney disease (CKD) is progressive, incurable, and ultimately fatal, either because of the consequences of kidney failure or an alarmingly high level of cardiovascular mortality in the CKD patient population (1). Regardless of the disease etiology, including hypertension and diabetes, tubulointerstitial fibrogenesis is the final common pathway in CKD that leads to disease progression and, ultimately, end-stage renal disease (ESRD). The past decade has seen great strides in our understanding of renal tubulointerstitial fibrosis, particularly as to how inflammation, fibroblast activation, tubular and microvascular injury contribute to fibrogenesis (3). However, pharmacological interventions based on the functions of many identified pathogenic factors have failed, are inadequate, or at best can slow progression to ESRD, and therefore development of effective drugs to treat renal tubulointerstitial fibrosis remains an elusive goal.
The eicosanoid metabolites of the arachidonic acid cascade play important roles in physiology including vasodilatory, anti-inflammatory, and antiapoptotic functions (8, 40). Cytochrome P-450 epoxygenases metabolize arachidonic acid into epoxyeicosatrienoic acid (EET), by catalyzing the epoxidation of the olefinic bonds of arachidonic acid, resulting in the production of four regioisomeric EETs: 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET. Soluble epoxide hydrolase (sEH) enzyme converts the EETs to their corresponding less potent diols termed dihydroxyeicosatrienoic acids (DHETs) and therefore the sEH activity is thought to be a major determinant of EET bioavailability (48). Genetic deficiency of sEH increases EET levels in tissues and plasma, potentiates the effects of EETs, and thus elicits antihypertensive and anti-inflammatory effect (18). Loss of sEH is also shown to decrease glomerular injury and renal inflammation in rodent models of deoxycorticosterone (DOCA)-salt-induced hypertension (33, 34). Inhibiting the sEH enzyme enhances the beneficial cardiovascular properties of EETs (17). Loss of sEH also reduces renal injury in a CKD model of a type 1 diabetes (11); however, a role for sEH in the pathogenesis of renal interstitial fibrosis is not defined. We hypothesized that diminished availability of EETs could be a potential mechanism for the progression of tubulointerstitial fibrosis, and increasing the bioavailability of EETs is therefore a promising therapeutic strategy for CKD. In this study, we tested the premise that genetic deficiency of sEH may prevent fibrogenesis and inflammation in the unilateral ureteral obstruction (UUO) model of renal interstitial fibrogenesis.
MATERIALS AND METHODS
Renal fibrosis model.
Male C57BL/6J mice aged 8 to 10 wk were purchased from Jackson Laboratories. Male sEH-knockout (KO) mice were previously reported (34). All mouse experiments were performed in accordance with the animal protocols approved by the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. UUO was conducted as previously reported (25). Briefly, the mice were anesthetized with an intraperitoneal injection of a cocktail containing ketamine (200 mg/kg body wt) and xylazine (16 mg/kg body wt). After the left kidney was exposed through the left flank incision, the left ureter was ligated completely near the kidney pelvis using a 5–0 silk tie. Sham-operated mice underwent the same surgical procedure without the ureter ligation. Glomerulonephritis (GN) was developed as previously reported (35). Briefly, mice were immunized with 1 mg of normal sheep IgG (Jackson ImmunoResearch, West Grove, PA) in Freund's complete adjuvant (Santa Cruz Biotechnology, Santa Cruz, CA) and injected intravenously with 100 μl of sheep anti-rat glomerular basement membrane (GBM) serum (Probetex, San Antonio, TX) 10 days later. Control mice underwent the same procedure without the injection of anti-GBM. For the pharmacological inhibition of sEH, trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (tAUCB; 2 or 10 mg/day) (41) or vehicle (10% DMSO in 0.5% methylcellulose) was administered by oral gavage beginning 3 days after UUO. For the pharmacological inhibition of PPAR, a mixture of GW6471 (1 mg·kg body wt−1·day−1; R&D Systems, Minneapolis, MN) against PPARα, GSK0660 (1 mg·kg body wt−1·day−1; R&D Systems) against PPARβ/δ, and T0070907 (1.5 mg·kg body wt−1·day−1; R&D Systems) against PPARγ or vehicle (10% DMSO in 0.9% saline) was intraperitoneally injected on the day of UUO surgery.
Collagen deposition.
Collagen deposition was assessed by both Sirius red staining and hydroxyproline assay as previously described (26). The area of positive Sirius red staining was measured in five randomly chosen high-power (×200) fields per kidney using NIH Image J software.
Histology.
Immunohistochemical staining of the kidneys was performed on paraffin sections as previously described (27). Briefly, 4% paraformaldehyde-fixed kidney sections were rehydrated and labeled with antibodies against α-smooth muscle actin (α-SMA; Sigma, St. Louis, MO), F4/80 (Abcam, Cambridge, MA), or polymorphonuclear neutrophil (PMN; Accurate, Westbury, NY). The sections were then incubated to peroxidase-conjugated secondary antibodies (Vector Laboratories, Burlingame, CA). The quantitative measurement of the immunohistochemical staining was carried out at a suitable magnification (×200; 62,500 μm2). The respective α-SMA- and F4/80-positive areas were measured in five randomly chosen fields per kidney using NIH Image J software. The number of PMN-positive cells in five randomly chosen fields per kidney was directly counted under light microscopy (Leica). Periodic acid-Schiff (PAS)-stained sections were used for tubular injury score as described previously (24). Histological damage of tubular injury was scored by percentage of tubules that displayed tubular necrosis, cast formation, and tubular dilation as follows: 0 = normal, 1 = <10%, 2 = 10 to 25%, 3 = 26 to 50%, 4 = 51 to 75%, and 5 = >75%. Ten randomly chosen high-power (×200 magnification) fields per kidney were used for the counting.
Western blot.
We performed electrophoresis of protein extracts using tris-glycine buffer systems and subsequent blotting as previously described (24). Membranes were incubated with antibodies against α-SMA (Sigma), fibronectin (Cedarlane, Hornby, Ontario, Canada), phosphorylated Smad3 (p-Smad3), phosphorylated NF-κB p65 (p-p65), poly(ADP-ribose) polymerase 1 (PARP1), caspase-3 (Cell Signaling, Beverly, MA), poly ADP-ribose (PAR; BD Pharmingen, San Jose, CA), intercellular adhesion molecule-1 (ICAM-1; Santa Cruz Biotechnology), TNF-α (Abcam), or sEH (Cayman). Peroxidase-conjugated secondary antibodies (Vector Laboratories) were applied, and a chemiluminescence reagent (PerkinElmer, Boston, MA) was used to detect proteins. Anti-β-actin antibody (Sigma) was used for loading controls on stripped membranes. The bands were quantified using Lab Works analysis software (Ultra-Violet Products, Cambridge, UK).
ELISA.
The ratios of EETs to DHETs in the kidneys were assessed using EET/DHET ELISA immunoassay kit (Detroit R&D, Detroit, MI). The levels of transforming growth factor (TGF)-β1, KC, macrophage inflammatory protein-2 (MIP-2), and monocyte chemotactic protein-1 (MCP-1) in the kidneys were measured using multiplex immunoassay (Millipore, Bedford, MA). The activities of peroxisome proliferator-activated receptor (PPAR) isoforms in the kidneys were measured using PPARα, β/δ, γ transcription factor assay kit (Cayman). The level of lipid hydroperoxide in the kidneys was measured using lipid hydroperoxide assay kit (Cayman).
Oxylipin profiling.
Oxylipin concentrations in kidney tissues were measured by liquid chromatography/tandem MS analysis as previously described (47).
Statistical analyses.
ANOVA was used to compare data among groups. Differences between two groups were assessed by two-tailed unpaired Student's t-test. P values <0.05 were considered statistically significant.
RESULTS
Loss of sEH prevents EETs to DHETs conversion in UUO kidneys.
To assess the role of sEH during renal interstitial fibrogenesis in vivo, we used a mouse model of obstructive nephropathy. UUO significantly increased the expression of sEH protein in wild-type (WT) mice at 3 and 10 days postinjury compared with that in sham-operated mice (Fig. 1A). No expression of sEH was observed in sEH-deficient mice at any of the time points studied. The ratio of EETs to DHETs was assessed by immunoassay. The ratio of both 11,12-EET/DHET and 14,15-EET/DHET decreased in WT compared with that in sEH-KO mice (Ephx2−/−) kidneys at 10 days after UUO. Levels of sEH-independent oxylipin metabolites showed no significant difference between WT and sEH-KO mice (Supplementary Table S1; the online version of this article contains supplemental data). These data indicate that the increased sEH activity reduced the ratio of EETs to DHETs in WT UUO kidneys consistent with its increased expression levels (Fig. 1B).
Fig. 1.
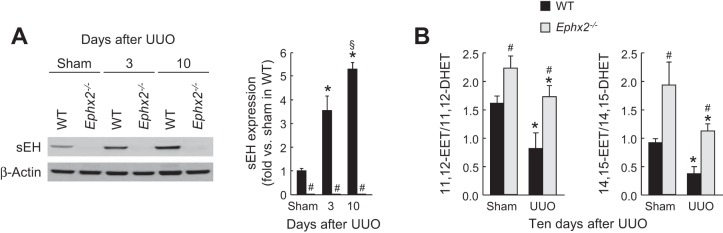
Unilateral ureteral obstruction (UUO) increases soluble epoxide hydrolase (sEH) expression and activity. A: expression of kidney sEH in wild-type (WT) or sEH-knockout (KO) mice at 3 or 10 days after sham operation or UUO using Western blot. The bands were quantified using Lab Works analysis software. B: sEH activity presented by ratio of epoxyeicosatrienoic acid (EET) to dihydroxyeicosatrienoic acid (DHET) in the kidneys using EET/DHET ELISA kit (Detroit R&D, Detroit, MI). Error bars represent SD (n = 5). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Loss of sEH prevents renal fibrosis in UUO and GN models.
To determine whether sEH activation contributes to interstitial fibrosis induced by UUO, Collagen deposition was assessed by Sirius red staining and hydroxyproline measurement. During UUO, WT kidneys showed a time-dependent increase of collagen deposition as demonstrated by increased Sirius red-positive area (Fig. 2, A and B) and hydroxyproline level (Fig. 2C), whereas the loss of sEH markedly reduced the collagen deposition (Fig. 2, A–C). Tubulointerstitial expression of α-SMA, a marker of myofibroblast formation, was induced at 3 and 10 days in WT UUO-induced kidneys but was significantly diminished by the loss of sEH at both time points (Fig. 2, A, D, and E). Three and 10 days after UUO, fibronectin expression level in whole kidneys was also diminished by the loss of sEH (Fig. 2E). We also tested whether loss of sEH prevented renal interstitial fibrosis during GN, another model of renal fibrosis. During GN, collagen deposition in WT kidneys was time-dependently increased as demonstrated by Sirius red-positive area (Fig. 3, A and B) and hydroxyproline level (Fig. 3C), whereas the loss of sEH significantly reduced the collagen deposition at 10 days of GN (Fig. 3, A–C). These data suggest that the loss of sEH has anti-fibrotic effect in different experimental models of CKD.
Fig. 2.

Loss of sEH prevents interstitial fibrogenesis induced by UUO. A: collagen deposition detected by Sirius red stain and α-smooth muscle actin (α-SMA) expression stained with α-SMA monoclonal antibody on kidney sections in WT or sEH-KO mice at 10 days after sham operation or UUO. Arrow heads indicate glomeruli. Scale bars = 50 μm. To demonstrate the specific staining of α-SMA in the myofibroblasts in the interstitial area, representative high magnification of the α-SMA staining regions from UUO-induced kidneys is shown in the bottom panels. B: percent of Sirius red-positive area on kidney sections. C: collagen content represented by hydroxyproline level in the kidneys. D: percent of α-SMA-positive area on the kidney sections. E: profibrotic expression of α-SMA and fibronectin in the kidneys evaluated by Western blot analyses. The bands were quantified using Lab Works analysis software. Error bars represent SD (n = 5). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Fig. 3.
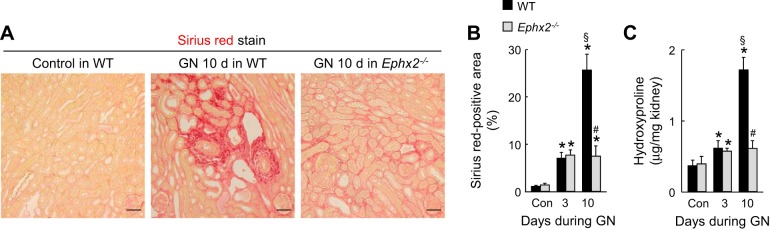
Loss of sEH prevents interstitial fibrosis induced by glomerulonephritis (GN). A: collagen deposition detected by Sirius red stain and α-SMA expression stained with α-SMA monoclonal antibody on kidney sections in WT or sEH-KO mice at 10 days after nonadministration or administration of anti-glomerular basement membrane (GBM) serum. Scale bars = 50 μm. B: percent of Sirius red-positive area on kidney sections. C: collagen content represented by hydroxyproline level in the kidneys. Error bars represent SD (n = 4). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Loss of sEH prevents TGF-β expression and its downstream signaling in UUO kidneys.
Since myofibroblast differentiation and activation by TGF-β signaling are critical events in renal fibrosis (6), we tested whether the TGF-β1 level is affected by sEH upregulation in WT UUO kidneys. The TGF-β1 level and its downstream signaling mediator p-Smad3 are significantly reduced in sEH-KO kidneys at 3 and 10 days after UUO, compared with those in WT kidneys (Fig. 4, A and B). These data suggest that sEH upregulation contributes to tubulointerstitial fibrogenesis via TGF-β1/Smad3 signaling.
Fig. 4.
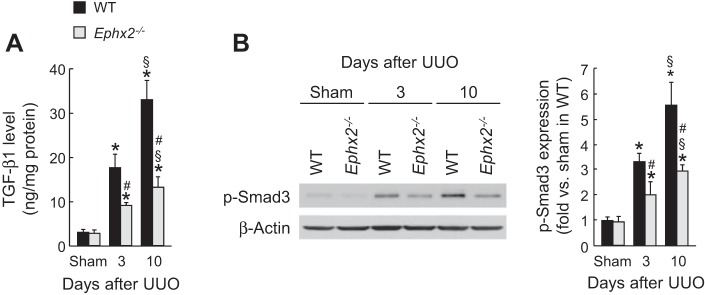
Loss of sEH reduces transforming growth factor (TGF)-β signaling during UUO. A: kidney TGF-β1 level in WT or sEH-KO mice after sham operation or UUO was measured by multiplex immunoassay. B: Smad3 phosphorylation (p-Smad3) in the kidneys was analyzed by Western blot. The bands were quantified using Lab Works analysis software. Error bars represent SD (n = 5). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Loss of sEH diminishes infliltration of neutrophils and macrophages after UUO.
To determine whether sEH is implicated in renal inflammation in obstructive nephropathy, we next examined leukocyte influx and proinflammatory response in UUO kidneys. Three and 10 days after UUO, a prominent influx of both PMN-positive neutrophil and F4/80-positive macrophage occurred in WT kidneys, but the loss of sEH significantly prevents inflammatory cell infiltration in UUO kidneys (Fig. 5).
Fig. 5.
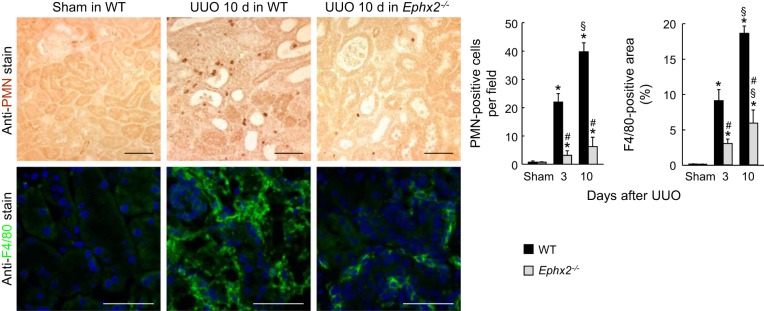
Loss of sEH prevents infiltration of neutrophil and macrophage during UUO. The number of polymorphonuclear neutrophil (PMN)-positive neutrophils and the percent of F4/80-positive area on kidney sections in WT or sEH-KO mice after sham operation or UUO was analyzed using immunohistochemistry. The number of neutrophils or macrophages was counted. Error bars represent SD (n = 5). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Loss of sEH diminishes expression of inflammatory cytokines and chemotactic factors after UUO.
To determine whether sEH alters the inflammatory milieu in the UUO-induced kidney, we assessed the levels of various inflammatory molecules. Loss of sEH decreased levels of keratinocyte-derived chemokine (KC), MIP-2, and MCP-1 proteins, potent leukocyte chemotactic factors upregulated by UUO (Fig. 6A). Since these chemotactic factors can be transactivated by NF-κB transcription factor (38, 43, 45), we assessed NF-κB activation by quantifying p-p65 in UUO kidneys. The level of p-p65 was significantly reduced in sEH-KO kidneys during UUO compared with that in WT kidneys, respectively (Fig. 6B). Expression levels of NF-κB target gene proteins, TNF-α and ICAM-1, were increased in WT kidneys, but were prevented by the loss of sEH during UUO (Fig. 6B). These data suggest that sEH upregulation exacerbates renal inflammation at least partly through an NF-κB-dependent signaling pathway during interstitial fibrogenesis.
Fig. 6.

Loss of sEH reduces inflammatory gene expression during UUO. A: levels of chemokine KC, macrophage inflammatory protein-2 (MIP-2), and monocyte chemotactic protein-1 (MCP-1) in the kidneys in WT or sEH-KO mice after sham operation or UUO using multiplex immunoassay. B: protein expression of p-p65, TNF-α, and intercellular adhesion molecule-1 (ICAM-1) in the kidneys using Western blot. The bands were quantified using Lab Works analysis software. Error bars represent SD (n = 5). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Pharmacological inhibition of sEH post-UUO attenuates increase of fibrotic and inflammatory protein expression.
To determine whether treatment with tAUCB, a pharmacological inhibitor of sEH, during UUO attenuates renal fibrosis, we measured fibrotic protein expression. Treatment with tAUCB beginning 3 days post-UUO significantly reduced α-SMA and fibronectin expression compared with vehicle-treated animals (Fig. 7A). We also assessed the expression of inflammatory proteins in kidneys with or without treatment with tAUCB during UUO. UUO-induced expression of TNF-α and ICAM-1 was significantly diminished by pharmacological inhibition of sEH at 10 days after the injury (Fig. 7B). These data suggest that sEH inhibitors may be used as a prophylactic agent to attenuate progression of fibrosis in CKD.
Fig. 7.

Pharmacological inhibition of sEH attenuates fibrotic and inflammatory protein expressions induced by UUO. A: profibrotic expression of α-SMA and fibronectin in sham or UUO kidneys of mice treated with or without trans-4-[4-(3-adamantan-1-yl-ureido)-cyclohexyloxy]-benzoic acid (tAUCB), a sEH inhibitor, evaluated by Western blot analyses. B: proinflammatory expression of TNF-α and ICAM-1 in the kidneys evaluated by Western blot analyses. The bands were quantified using Lab Works analysis software. Error bars represent SD (n = 4). *P < 0.05 vs. sham. †P < 0.05 vs. vehicle.
Loss of sEH diminishes cell death and oxidative stress after UUO.
Since inflammation and fibrogenesis can be initiated by cell death (37) and vice versa, we assessed tubular injury and cell death in UUO-subjected kidneys with or without the sEH deficiency. Tubular injury score based on PAS-stained kidney sections was increased at 3 and 10 days after UUO, but the loss of sEH significantly prevented the tubular injury in UUO kidneys (Fig. 8, A and B). The loss of sEH also reduced UUO-induced tubular cell death including both necrosis as demonstrated by PARP1-dependent increased PAR formation and PARP1 expression, and apoptosis demonstrated by cleaved PARP1 and caspase-3 expression (Fig. 8C). To explore whether the loss of sEH contributes to decreased oxidative stress during UUO, we next evaluated lipid peroxidation in UUO kidneys in WT and sEH-KO mice. UUO increased lipid peroxidation as represented by the increased level of lipid hydroperoxide in WT kidneys at 3 and 10 days, whereas the loss of sEH ameliorated lipid peroxidation induced by UUO (Fig. 8D), suggesting that sEH upregulation induces oxidative stress in WT UUO-induced kidneys, potentially leading to the progression of tubular injury and cell death.
Fig. 8.

Loss of sEH suppresses tubular cell damage during interstitial fibrogenesis. A: periodic acid-Schiff (PAS) stain on kidney sections in WT or sEH-KO mice at 10 days after sham operation or UUO. Scale bars = 50 μm. B: tubular injury score represented by PAS stain in the kidneys. C: protein expression of poly ADP-ribose (PAR), poly(ADP-ribose) polymerase 1 (PARP1), cleaved PARP1, and cleaved caspase-3 in the kidneys using Western blot. The bands were quantified using Lab Works analysis software. D: lipid peroxidation indicated by lipid hydroperoxide level in the kidneys using lipid hydroperoxide assay kit. Error bars represent SD (n = 5). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
sEH is required for PPAR inactivation during renal fibrosis.
PPAR isoforms play a protective role in renal interstitial fibrosis (13, 23). The activity of PPAR isoforms was time-dependently decreased in WT mouse kidneys after UUO, but the loss of sEH prevented the decrease in activity of PPARs after UUO. PPARγ activity was reduced by UUO as early as 3 days and persisted at 10 days (Fig. 9), while PPARα and PPARβ/γ were reduced only at 10 days post-UUO. The reduced activity of all PPAR isoforms was significantly prevented by the loss of sEH (Fig. 9). To confirm whether the fibrogenic function of sEH is caused by inactivation of PPARs, we administered a mixture of PPAR isoform antagonists to UUO-subjected WT or sEH-KO mice. The administration of the antagonist mixture restored α-SMA, p-Smad3, and p-p65 expression at 10 days after UUO in sEH-KO kidneys, but not those in WT kidneys (Fig. 10). These data suggest that the loss of sEH prevents interstitial fibrosis and inflammation via the activation of PPARs.
Fig. 9.
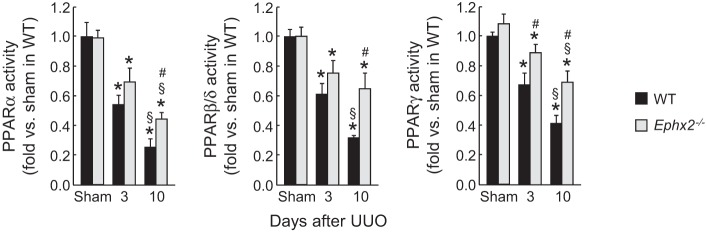
Loss of sEH restricts the reduction of peroxisome proliferator-activated receptors (PPARs) activity induced by UUO. The activity of PPAR isoforms in kidneys in WT or sEH-KO mice after sham operation or UUO was assayed using PPARα, β/δ, γ transcription factor assay kit. Error bars represent SD (n = 6). *P < 0.05 vs. sham. #P < 0.05 vs. WT. §P < 0.05 vs. 3 days.
Fig. 10.
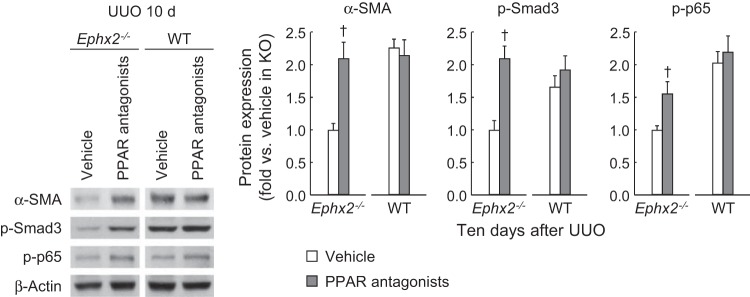
PPAR antagonism revives renal fibrosis and inflammation in sEH-KO mice. Expression of α-SMA, p-Smad3, and p-p65 in WT and sEH-KO mouse kidneys treated with either vehicle or mixture of PPAR isoform antagonists at 10 days after UUO using Western blot. The bands were quantified using Lab Works analysis software. Error bars represent SD (n = 4). †P < 0.05 vs. vehicle.
DISCUSSION
Fibrosis is an essential process for survival; however, disregulated fibrosis leads to a variety of deleterious events. Thus, it is no surprise that the pathophysiological events that lead to renal interstitial fibrogenesis are complex and involve several independent and overlapping cellular and molecular signaling pathways. Endothelial cell activation, leukocyte infiltration, tubular cell injury, capillary rarefaction, and pericyte and myofibroblast activation all play important roles in the pathogenesis of renal fibrosis after an inciting insult to the renal parenchyma (2, 7, 30, 44). Traditionally, TGF-β signaling has been regarded as a central mediator of tubulointerstitial fibrosis (4, 5). Recently, TGF-β-independent signaling via other molecules including NF-κB and angiotensin II is also implicated in renal fibrosis (19, 39). However, the molecular signaling pathways that activate these overlapping pathways to elicit renal fibrogenesis are not defined.
In the present study, we tested the effect of the sEH deficiency on the progression of renal failure in mouse UUO model. Our data indicate that the loss of sEH is beneficial in attenuating the inflammation and interstitial fibrogenesis after UUO. UUO decreased the ratios of EETs to DHETs in WT mouse kidneys that were restored to normal levels by the loss of sEH. The loss of sEH significantly reduced the intrarenal levels of inflammatory cytokines and chemokines, and the infiltration of both neutrophils and macrophages into the renal parenchyma. The loss of sEH by a genetic approach prevented collagen, fibronectin, and α-SMA deposition in the UUO-induced kidneys. The loss of sEH also prevented histological damage, oxidative stress, PARP1 and NF-κB activation, and significantly increased the activities of PPARs, especially the level of PPARγ. Taken together, increasing the levels of EETs by the sEH deficiency prevents renal interstitial inflammation and fibrogenesis in the mouse UUO model. Our data suggest that the renal sEH activation decreases the EET levels to elicit pathologically relevant inflammatory responses and interstitial fibrogenesis, which are mediated by downregulation of PPAR activity.
PPARs are ligand-activated transcription factors. EETs bind to the ligand-binding domain of PPARγ with high affinity (32). Several independent reports have demonstrated that PPARγ is a negative regulator of profibrotic signaling and blocks matrix deposition and fibrogenesis in diabetic glomerulosclerosis (36) and CCl4-induced liver fibrosis (29). Our data indicate that PPAR levels are downregulated in UUO kidneys. The sEH deletion induces the levels of PPARs and prevents UUO-induced fibrosis. However, the molecular signaling pathways by which PPARs attenuate UUO-induced fibrosis are not well-characterized. A role for PPARγ in regulating reactive oxygen species (ROS) levels in the hypothalamus in high fat-fed mice was previously reported (9). The anti-inflammatory effect of PPARγ on LPS-induced pulpal inflammation is also due to decreasing ROS levels (28). Our data indicating that the sEH deficiency downregulates oxidative stress in UUO kidneys are consistent with these reports.
ROS generated during UUO contributes to oxidative damage to DNA (27). Under conditions of increased DNA damage, PARP1 is hyperactivated and can act as a promoter-specific coactivator of NF-κB in vivo independent of its enzymatic activity (14, 15). PARP1 binds directly to both subunits of NF-κB (p65 and p50) and promotes NF-κB-dependent transcription. Our data indicate that the sEH deficiency inhibits PARP1 activation in UUO-induced mice kidneys. Furthermore, the sEH deficiency prevents NF-κB activation and the inflammatory response. These data concur with earlier reports showing that sEH inhibition attenuated myocardial NF-κB activation in mice 6 wk after cardiac hypertrophy induced by thoracic aortic constriction (46). Similarly, NF-κB activation in the kidney was significantly attenuated in sEH-KO mice with DOCA-salt-induced hypertension (34). As we previously reported (25), in the absence of PARP1, NF-κB mediated inflammatory mediators TNF-α and ICAM-1, and attenuated infiltration of inflammatory cells and the production of cytokines leading to reduced tubulointerstitial lesion and fibrosis in UUO-induced mouse kidneys.
In our data, sEH deficiency downregulated TGF-β1 level in the UUO kidneys. However, the EET-induced signaling pathways that lead to TGF-β1 downregulation remain undefined. A number of reports implicated a direct role for PPARγ in modulation of TGF-β/Smad3 signaling. The PPARγ agonist troglitazone attenuates UUO-induced renal interstitial fibrosis and inflammation through suppression of TGF-β1 expression (22). Ligand-activated PPARγ prevents TGF-β-induced collagen synthesis via sequestration of essential coactivator p300 from TGF-β-induced p-Smad complex on the collagen gene promoter (12). It remains to be determined whether EETs may attenuate TGF-β/Smad3 stimulation via PPARγ activation after UUO.
Our data are in contrast to a previous reported study in a hypertensive 5/6-Nx model of CKD where sEH inhibition did not delay disease progression but rather resulted in significant increase in glomerulosclerosis, tubulointerstitial damage index, and an increase in albuminuria (21). In previous reports, the antihypertensive effect of sEH inhibition was implicated for decreased renal inflammation and injury in rat and mouse models, like spontaneously hypertensive rats, angiotensin II-induced hypertension, and DOCA-induced and salt-sensitive hypertension (16, 20, 33). It should be emphasized that inflammation and fibrogenesis observed in our sEH-deficient UUO mouse model are independent of changes in blood pressure. The differences in the results could be due to the model and the strain of mice as the antihypertensive effect in sEH-KO mice was lost after backcrossing on the C57BL/6 background (10, 34). The effect of sEH inhibition on renal protection independently of its effect on lowering blood pressure was previously reported in streptozotocin-induced diabetic mice (11) and in DOCA-salt hypertension (34). Our data are also consistent with a recent report demonstrating the beneficial effects of sEH inhibitors on adverse cardiac remodeling in ischemic cardiomyopathy and pressure-overload hypertrophy (42). On the other hand, in a recent report, pharmacological inhibition of sEH attenuated collagen deposition in angiotensin II-treated heart ventricles, but genetic inhibition of sEH conversely aggravated the effect (31) showing differing effects of pharmacological and genetic inhibition of sEH on myocardial fibrosis. In our data, both pharmacological and genetic inhibition had the same preventive effect on collagen deposition in the UUO model of renal fibrosis. The discrepancy may be due to the different sEH pathways activated during fibrogenesis and inflammation in hearts and kidneys.
Collectively, these studies as summarized in Fig. 11 demonstrate that increased EETs after the sEH deficiency significantly prevent histological damage, oxidative stress, and increased the levels of PPAR, especially that of PPARγ. Furthermore, the increased levels of EETs prevented pathologically relevant inflammatory responses including reduced levels of inflammatory cytokines and chemokines, leukocyte infiltration, and adverse renal remodeling in the UUO kidneys. Our data demonstrate that EET promotes the anti-inflammatory and fibroprotective effects in UUO kidneys via activation of PPAR isoforms and downregulation of NF-κB, TGF-β1/Smad3, and inflammatory signaling pathways.
Fig. 11.
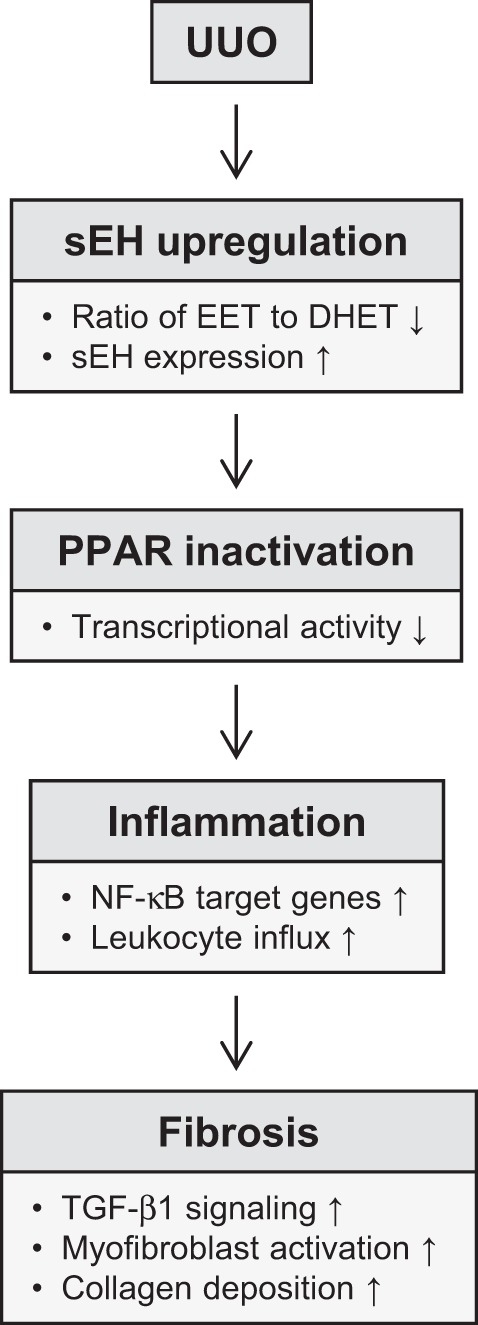
Flow diagram of sEH signaling pathway in UUO.
GRANTS
This work is supported by American Heart Association postdoctoral fellowship 13POST16350005 to J. Kim and partially supported by R01 ES002710 and P42 ES004699 to B. D. Hammock. B. J. Padanilam is supported by National Institutes of Health Grants DK-083291 and DK-090332.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.K. and B.J.P. conception and design of research; J.K. and J.Y. performed experiments; J.K. analyzed data; J.K., J.D.I., B.D.H., and B.J.P. interpreted results of experiments; J.K. prepared figures; J.K. drafted manuscript; J.K., B.D.H., and B.J.P. edited and revised manuscript; J.K. and B.J.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Youngsu Cho for technical assistance with the Western blot and immunohistochemistry, and Kelly E. Long and Sherry N. Westphal for mouse care.
REFERENCES
- 1.Bakris G, Vassalotti J, Ritz E, Wanner C, Stergiou G, Molitch M, Nesto R, Kaysen GA, Sowers JR. National Kidney Foundation consensus conference on cardiovascular and kidney diseases and diabetes risk: an integrated therapeutic approach to reduce events. Kidney Int 78: 726–736, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Basile DP, Fredrich K, Chelladurai B, Leonard EC, Parrish AR. Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol 294: F928–F936, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol 6: 643–656, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Border WA, Okuda S, Languino LR, Sporn MB, Ruoslahti E. Suppression of experimental glomerulonephritis by antiserum against transforming growth factor beta 1. Nature 346: 371–374, 1990 [DOI] [PubMed] [Google Scholar]
- 5.Borges FT, Melo SA, Ozdemir BC, Kato N, Revuelta I, Miller CA, Gattone VH, 2nd, LeBleu VS, Kalluri R. TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J Am Soc Nephrol 24: 385–392, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bottinger EP. TGF-β in renal injury and disease. Semin Nephrol 27: 309–320, 2007 [DOI] [PubMed] [Google Scholar]
- 7.Campanholle G, Ligresti G, Gharib SA, Duffield JS. Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. Am J Physiol Cell Physiol 304: C591–C603, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen JK, Capdevila J, Harris RC. Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol Cell Biol 21: 6322–6331, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, Belsham DD, Sarruf DA, Schwartz MW, Bennett AM, Shanabrough M, Mobbs CV, Yang X, Gao XB, Horvath TL. Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med 17: 1121–1127, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorrance AM, Rupp N, Pollock DM, Newman JW, Hammock BD, Imig JD. An epoxide hydrolase inhibitor, 12-(3-adamantan-1-yl-ureido)dodecanoic acid (AUDA), reduces ischemic cerebral infarct size in stroke-prone spontaneously hypertensive rats. J Cardiovasc Pharmacol 46: 842–848, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elmarakby AA, Faulkner J, Al-Shabrawey M, Wang MH, Maddipati KR, Imig JD. Deletion of soluble epoxide hydrolase gene improves renal endothelial function and reduces renal inflammation and injury in streptozotocin-induced type 1 diabetes. Am J Physiol Regul Integr Comp Physiol 301: R1307–R1317, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghosh AK, Bhattacharyya S, Wei J, Kim S, Barak Y, Mori Y, Varga J. Peroxisome proliferator-activated receptor-gamma abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J 23: 2968–2977, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han JY, Kim YJ, Kim L, Choi SJ, Park IS, Kim JM, Chu YC, Cha DR. PPARgamma agonist and angiotensin II receptor antagonist ameliorate renal tubulointerstitial fibrosis. J Korean Med Sci 25: 35–41, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO. The enzymatic and DNA binding activity of PARP-1 are not required for NF-κB coactivator function. J Biol Chem 276: 45588–45597, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Hassa PO, Hottiger MO. A role of poly (ADP-ribose) polymerase in NF-κB transcriptional activation. Biol Chem 380: 953–959, 1999 [DOI] [PubMed] [Google Scholar]
- 16.Imig JD. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol 289: F496–F503, 2005 [DOI] [PubMed] [Google Scholar]
- 17.Imig JD, Hammock BD. Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8: 794–805, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR. Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol 7: 2364–2370, 1996 [DOI] [PubMed] [Google Scholar]
- 19.Inoue T, Takenaka T, Hayashi M, Monkawa T, Yoshino J, Shimoda K, Neilson EG, Suzuki H, Okada H. Fibroblast expression of an IkappaB dominant-negative transgene attenuates renal fibrosis. J Am Soc Nephrol 21: 2047–2052, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I. Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 45: 759–765, 2005 [DOI] [PubMed] [Google Scholar]
- 21.Jung O, Jansen F, Mieth A, Barbosa-Sicard E, Pliquett RU, Babelova A, Morisseau C, Hwang SH, Tsai C, Hammock BD, Schaefer L, Geisslinger G, Amann K, Brandes RP. Inhibition of the soluble epoxide hydrolase promotes albuminuria in mice with progressive renal disease. PLos One 5: e11979, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N. PPAR-γ agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β. Lab Invest 89: 47–58, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Kawai T, Masaki T, Doi S, Arakawa T, Yokoyama Y, Doi T, Kohno N, Yorioka N. PPAR-γ agonist attenuates renal interstitial fibrosis and inflammation through reduction of TGF-β. Lab Invest 89: 47–58, 2009 [DOI] [PubMed] [Google Scholar]
- 24.Kim J, Long KE, Tang K, Padanilam BJ. Poly(ADP-ribose) polymerase 1 activation is required for cisplatin nephrotoxicity. Kidney Int 82: 193–203, 2012 [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Padanilam BJ. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am J Physiol Renal Physiol 301: F450–F459, 2011 [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Padanilam BJ. Renal nerves drive interstitial fibrogenesis in obstructive nephropathy. J Am Soc Nephrol 24: 229–242, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim J, Seok YM, Jung KJ, Park KM. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am J Physiol Renal Physiol 297: F461–F470, 2009 [DOI] [PubMed] [Google Scholar]
- 28.Kim JC, Lee YH, Yu MK, Lee NH, Park JD, Bhattarai G, Yi HK. Anti-inflammatory mechanism of PPARgamma on LPS-induced pulp cells: role of the ROS removal activity. Arch Oral Biol 57: 392–400, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Kon K, Ikejima K, Hirose M, Yoshikawa M, Enomoto N, Kitamura T, Takei Y, Sato N. Pioglitazone prevents early-phase hepatic fibrogenesis caused by carbon tetrachloride. Biochem Biophys Res Commun 291: 55–61, 2002 [DOI] [PubMed] [Google Scholar]
- 30.LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med 19: 1047–1053, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Li N, Pang W, Zhang X, Hammock BD, Ai D, Zhu Y. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLos One 9: e94092, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y, Zhang Y, Schmelzer K, Lee TS, Fang X, Zhu Y, Spector AA, Gill S, Morisseau C, Hammock BD, Shyy JYJ. The antiinflammatory effect of laminar flow: the role of PPARÎ3, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc Natl Acad Sci USA 102: 16747–16752, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loch D, Hoey A, Morisseau C, Hammock BO, Brown L. Prevention of hypertension in DOCA-salt rats by an inhibitor of soluble epoxide hydrolase. Cell Biochem Biophys 47: 87–98, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manhiani M, Quigley JE, Knight SF, Tasoobshirazi S, Moore T, Brands MW, Hammock BD, Imig JD. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am J Physiol Renal Physiol 297: F740–F748, 2009. 19553349 [Google Scholar]
- 35.Masaki T, Chow F, Nikolic-Paterson DJ, Atkins RC, Tesch GH. Heterogeneity of antigen expression explains controversy over glomerular macrophage accumulation in mouse glomerulonephritis. Nephrol Dial Transplant 18: 178–181, 2003 [DOI] [PubMed] [Google Scholar]
- 36.McCarthy KJ, Routh RE, Shaw W, Walsh K, Welbourne TC, Johnson JH. Troglitazone halts diabetic glomerulosclerosis by blockade of mesangial expansion. Kidney Int 58: 2341–2350, 2000 [DOI] [PubMed] [Google Scholar]
- 37.Mehal W, Imaeda A. Cell death and fibrogenesis. Semin Liver Dis 30: 226–231, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohmori Y, Fukumoto S, Hamilton TA. Two structurally distinct kappa B sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. J Immunol 155: 3593–3600, 1995 [PubMed] [Google Scholar]
- 39.Rodriguez-Vita J, Sanchez-Lopez E, Esteban V, Ruperez M, Egido J, Ruiz-Ortega M. Angiotensin II activates the Smad pathway in vascular smooth muscle cells by a transforming growth factor-β-independent mechanism. Circulation 111: 2509–2517, 2005 [DOI] [PubMed] [Google Scholar]
- 40.Schmelzer KR, Kubala L, Newman JW, Kim IH, Eiserich JP, Hammock BD. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc Natl Acad Sci USA 102: 9772–9777, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simpkins AN, Rudic RD, Schreihofer DA, Roy S, Manhiani M, Tsai HJ, Hammock BD, Imig JD. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am J Pathol 174: 2086–2095, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sirish P, Li N, Liu JY, Lee KS, Hwang SH, Qiu H, Zhao C, Ma SM, Lopez JE, Hammock BD, Chiamvimonvat N. Unique mechanistic insights into the beneficial effects of soluble epoxide hydrolase inhibitors in the prevention of cardiac fibrosis. Proc Natl Acad Sci USA 110: 5618–5623, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, Fukushima J, Kawamoto S, Ishigatsubo Y, Okubo T. NF-κB and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol 153: 2052–2063, 1994 [PubMed] [Google Scholar]
- 44.Venkatachalam MA, Griffin KA, Lan R, Geng H, Saikumar P, Bidani AK. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Widmer U, Manogue KR, Cerami A, Sherry B. Genomic cloning and promoter analysis of macrophage inflammatory protein (MIP)-2, MIP-1α, and MIP-1β, members of the chemokine superfamily of proinflammatory cytokines. J Immunol 150: 4996–5012, 1993 [PubMed] [Google Scholar]
- 46.Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N. Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci USA 103: 18733–18738, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang J, Schmelzer K, Georgi K, Hammock BD. Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem 81: 8085–8093, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res 87: 992–998, 2000 [DOI] [PubMed] [Google Scholar]