

Abstract
Skeletal muscle loading/overload stimulates the Ca2+-activated, serine/threonine kinase Ca2+/calmodulin-dependent protein kinase kinase-α (CaMKKα); yet to date, no studies have examined whether CaMKKα regulates muscle growth. The purpose of this study was to determine if constitutive activation of CaMKKα signaling could stimulate muscle growth and if so whether CaMKKα is essential for this process. CaMKKα signaling was selectively activated in mouse muscle via expression of a constitutively active form of CaMKKα using in vivo electroporation. After 2 wk, constitutively active CaMKKα expression increased muscle weight (∼10%) and protein content (∼10%), demonstrating that activation of CaMKKα signaling can stimulate muscle growth. To determine if active CaMKKα expression stimulated muscle growth via increased mammalian target of rapamycin complex 1 (mTORC1) signaling and protein synthesis, [3H]phenylalanine incorporation into proteins was assessed with or without the mTORC1 inhibitor rapamycin. Constitutively active CaMKKα increased protein synthesis ∼60%, and this increase was prevented by rapamycin, demonstrating a critical role for mTORC1 in this process. To determine if CaMKKα is essential for growth, muscles from CaMKKα knockout mice were stimulated to hypertrophy via unilateral ablation of synergist muscles (overload). Surprisingly, compared with wild-type mice, muscles from CaMKKα knockout mice exhibited greater growth (∼15%) and phosphorylation of the mTORC1 substrate 70-kDa ribosomal protein S6 kinase (Thr389; ∼50%), demonstrating that CaMKKα is not essential for overload-induced mTORC1 activation or muscle growth. Collectively, these results demonstrate that activation of CaMKKα signaling is sufficient but not necessary for activation of mTORC1 signaling and growth in mouse skeletal muscle.
Keywords: calcium, kinase, protein degradation, protein synthesis, synergist ablation
skeletal muscle plays a critical role in human health, as decreases in muscle mass dramatically increase the risk of disability, metabolic dysfunction (e.g., type 2 diabetes), and death (26, 30). Thus, the maintenance of muscle mass is a key factor determining the quality and duration of life. Skeletal muscle mass is regulated by the balance between anabolic and catabolic processes, and under steady-state conditions the rate of muscle protein synthesis equals the rate of degradation (25). Resistance exercise, muscle loading/overload, and stretch are potent stimulators of muscle growth and shift this balance to favor increased protein synthesis (2, 9, 10).
One of the key intracellular signaling molecules that controls muscle protein synthesis is the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) (18). Previous studies have shown that treatment of rodent muscle with the chemical mTORC1 inhibitor rapamycin prevents resistance exercise-, loading/overload-, and stretch-induced increases in muscle mass and protein synthesis (4, 13, 17), demonstrating that mTORC1 activity is critical for these processes. Unfortunately, although mTORC1 has clearly been implicated as a key regulator of muscle protein synthesis and growth, the upstream signaling mechanisms that link resistance exercise, loading/overload, and stretch to mTORC1 activation in muscle are less clear.
Intracellular Ca2+ plays a fundamental role in numerous processes in skeletal muscle, including excitation-contraction coupling, endo/exocytosis, carbohydrate and lipid metabolism, differentiation, etc. Given this large and diverse array of cellular functions, not surprisingly this has led many researchers to also propose a role for it in the regulation of muscle protein synthesis (16, 20, 31). Work in mouse plantaris muscle has shown that intracellular Ca2+ levels and mTORC1 signaling are increased by functional overload (15) and that intramuscular administration of the chemical Ca2+ chelator 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (BAPTA-AM) attenuates overload-induced muscle hypertrophy (15), collectively suggesting a positive relationship between intracellular Ca2+ and muscle growth/protein synthesis/mTORC1 signaling. However, stimulation of rodent muscle with a variety of chemical Ca2+-mobilizing agents has elicited varying and conflicting results regarding the role of intracellular Ca2+ in these processes, including an increase and decrease in protein synthesis rates (16, 20, 31) and an increase or no change in mTORC1 signaling (12, 15). Since the effects of the Ca2+-mobilizing agents were blocked by Ca2+ chelators (15, 16, 20, 31), the results demonstrate that the effects were dependent on Ca2+ signaling and suggest that the divergence in functional outcome may be dependent on the amplitude, duration, and/or source of the Ca2+ signal (sarcoplasmic reticulum vs. extracellular and/or type of Ca2+ channel). Consistent with this hypothesis, inhibition of stretch-activated Ca2+ channels with streptomycin attenuated eccentric contraction-induced mTORC1 signaling (28, 29), and loss of transient receptor potential vanilloid 1 (TRPV1) cation channels attenuated overload-induced muscle growth (15), whereas inhibition of dihydropyridine receptors or ryanodine receptors did not alter overload-induced mTORC1 signaling or growth (15). Thus, collectively, these results support the existence of an intracellular pathway linking intracellular Ca2+ to mTORC1 activation in skeletal muscle, with the activation of mTORC1 being dependent on the source of the Ca2+ signal.
The Ca2+/calmodulin-dependent protein kinase kinase-α (CaMKKα) is a serine/threonine kinase that is activated by increases in intracellular Ca2+ levels and Ca2+/calmodulin binding (19). Importantly, this enzyme does not possess autonomous (i.e., Ca2+/calmodulin-independent) activity (33); thus, activity levels return to baseline when Ca2+ levels decline. Endogenous mouse CaMKKα is a 505-amino acid, ∼68-kDa protein, and previous work has shown that 7 days of overload in mouse muscle induces an an approximately threefold increase in CaMKKα protein levels and an approximately fivefold increase in CaMKKα activity (21). Thus, collectively, these data suggest that CaMKKα could represent a novel protein kinase that links changes in intracellular Ca2+ levels to muscle growth/protein synthesis/mTORC1 signaling. However, to date, no studies have examined whether activation of CaMKKα signaling is sufficient to stimulate these processes in skeletal muscle, and, if so, whether CaMKKα plays an essential role in their regulation. Thus, the goal of this study was to determine whether CaMKKα plays a role in the regulation of mTORC1 signaling, protein synthesis, and growth in mouse skeletal muscle.
MATERIALS AND METHODS
Animals.
Experiments were performed in accordance with the East Carolina University Institutional Animal Care and Use Committee and the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Female CD-1 mice (6–8 wk) were obtained from Charles River Laboratories. Male and female whole body CaMKKα knockout mice were generated as previously described in Blaeser et al. (3). Briefly, exons 2 to 5 of the CaMKK1 gene were replaced with a neomycin gene cassette, and the resulting mutant DNA was transfected into RW4 embryonic stem cells. Heterozygous CaMKKα-deficient embryonic stem cells were injected into C57BL/6 blastocysts and reimplanted into psuedopregnant female mice. Chimeric male CaMKKα knockout mice were bred to C57BL/6 mice for 12 generations prior to arrival at the commercial laboratory animal vendor The Jackson Laboratory. For these studies, male and female whole body CaMKKα knockout mice were obtained from The Jackson Laboratory and bred to wild-type C57BL/6J mice to produce heterozygotes. Heterozygous mice were bred together to produce wild-type and CaMKKα knockout mice, which was confirmed in all mice by immunoblot analysis utilizing a commercially available CaMKKα antibody (R&D Systems). For the studies presented in this paper, only female CaMKKα knockout mice and their wild-type littermates (14–15 wk old) were utilized. Mice were housed in cages at 21–22°C with a 12:12-h light-dark cycle. Prolab RMH 3000 rodent chow diet (PMI Nutritional International) and water were available ad libitum.
Unilateral synergist ablation surgery.
Plantaris muscle overload was achieved by surgical ablation of the distal one-third of the gastrocnemius and soleus muscles, using methods adapted from Thomson and Gordon (32). Briefly, wild-type CD-1 mice (6–8 wk) or CaMKKα knockout mice and their wild-type littermates (14–15 wk) were anesthetized with isoflurane (2–3%), and an ∼1.0-cm incision was made through the skin to expose the gastrocnemius muscle. The plantaris muscle was separated from the gastrocnemius and soleus muscles at the Achilles tendon, and the distal one-third of the gastrocnemius and soleus was removed. The contralateral leg was sham operated as a control. Incisions were closed with 5-0 vicryl sutures. After 1, 3, 7, or 10 days, mice were euthanized by cervical dislocation, and plantaris muscles were collected and frozen in liquid N2. Muscles were weighed and homogenized in lysis buffer A, containing 20 mM Tris·HCl, pH 7.5, 5 mM EDTA, 10 mM Na4P2O7, 100 mM NaF, 2 mM NaVO4, 1% Tergitol Type NP-40, 0.01 mM leupeptin, 3 mM benzamidine, 1 mM phenylmethylsulfonyl fluoride, and 10 μg/ml aprotinin. Samples were rotated end over end at 4°C for 1 h and centrifuged at 14,000 g for 30 min. Protein concentrations of lysates were determined using the Bradford method.
Transfection of mouse muscle using in vivo electroporation.
Vectors containing constitutively active CaMKKα (amino acids 1–434) and the empty vector pCS2+ were generously donated by Thomas R. Soderling (Vollum Institute, Oregon Health and Science University) (6). CaMKKα was made constitutively active by removal of the COOH terminus, which contains the autoinhibitory and calmodulin-binding domains (6).
Plasmid DNA injections and in vivo muscle gene transfer/electroporation procedures were performed using methods adapted from Aihara and Miyazaki (1) and Hinkley et al. (11). Briefly, mice were anesthetized with isoflurane (2–3%), and an ∼1.0 cm incision was made in the skin between the tibialis anterior and gastrocnemius muscles. The extensor digitorum longus muscle was exposed by blunt dissection, and a stainless steel spatula was placed underneath the muscle to stabilize the muscle position. Plasmid DNA (40 μg; 10 μl of 4 μg/μl) was injected longitudinally into the muscle using a 31-gauge, 1.9-cm needle. Two stainless steel needle electrodes (27 gauge, 0.7 cm) connected to an electric pulse generator (model S48, Grass Instruments) were positioned ∼5 mm apart on either side of the muscle, and muscles were stimulated eight times as follows: train rate = 1 train/s; train duration = 500 ms; pulse rate = 1 pulse/s; duration = 20 ms; voltage = 80 V. Incisions were closed with 5-0 coated vicryl sutures. For all transfections, plasmid DNA for constitutively active CaMKKα was injected into the muscle of one leg, and empty vector DNA was injected into the contralateral muscle. Muscles were allowed 1 or 2 wk to express proteins prior to additional experimental perturbations. Importantly, in vivo electroporation does not result in plasmid delivery to all fibers contained within a muscle; thus, all whole muscle data obtained using this approach likely represent an underestimation of the magnitude of the effect.
Assessment of muscle mass, water, and total protein content.
Muscles were excised, frozen in liquid N2, and then quickly weighed using an analytical balance (model XS105DU, Mettler Toledo). Frozen muscles were dehydrated for 8 h using a FreeZone freeze-dry system (Labconco) and then reweighed. Muscle water content was calculated as the percent loss of muscle weight following dehydration. Dehydrated muscles were homogenized in lysis buffer A. Samples were rotated end over end at 4°C for 1 h and centrifuged at 14,000 g for 30 min. Lysate protein concentrations were determined using the Bradford method, and total muscle protein content was calculated by multiplying the sample protein concentration by the lysis buffer volume.
Ex vivo skeletal muscle protein synthesis and degradation assays.
Ex vivo muscle protein synthesis rates were determined using methods adapted from Hornberger et al. (13). Briefly, mice were anesthetized with pentobarbital sodium (100 mg/kg ip) and euthanized by cervical dislocation. The extensor digitorum longus muscles were excised, weighed, attached to tissue mounts that held them at resting tension, and placed in continuously gassed (95% O2-5% CO2), 37°C Krebs-Ringer bicarbonate (KRB) solution: 117 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2·2H2O, 1.2 mM KH2PO4, 1.2 mM MgSO4·7H2O, and 24.6 mM NaHCO3 supplemented with 5 mM d-glucose, 2.5 mM l-phenylalanine, and either 150 nM rapamycin (mTORC1 inhibitor) or dimethylsulfoxide (DMSO, 0.006% vol/vol) for 90 min. Muscles were transferred to vials containing KRB, 5 mM glucose, 2.5 mM l-phenylalanine, and 20 μCi/ml [3H]phenylalanine with or without rapamycin for 30 min. Muscles were frozen in liquid N2 and homogenized in lysis buffer A minus Tergitol Type NP-40. For assessment of protein synthesis rates, 10% trichloroacetic acid was added to a portion of the homogenate, and the sample was vortexed and incubated on ice for 30 min. Samples were centrifuged at 4,500 g for 5 min. The acid-insoluble pellet was washed four times with 10% trichloroacetic acid and solubilized by the addition of 0.1 M NaOH and heated to 55°C for 1 h with intermittent vortexing. Aliquots were removed for scintillation counting of the 3H label, and [3H]phenylalanine incorporation into proteins was calculated as previously described (13). For immunoblot analysis, 1% Tergitol Type NP-40 was added to a portion of the homogenate, and samples were rotated end over end at 4°C for 1 h. Samples were centrifuged at 14,000 g for 30 min, and protein concentrations of lysates were determined using the Bradford method.
Ex vivo muscle protein degradation rates were determined using methods adapted from Fulks et al. (8) and Fang et al. (7). Briefly, mice were anesthetized with pentobarbital sodium (100 mg/kg ip) and euthanized by cervical dislocation. The extensor digitorum longus muscles were excised, weighed, and attached to tissue mounts that held them at resting tension. Muscles were placed in continuously gassed (95% O2-5% CO2) 37°C KRB solution supplemented with 5 mM glucose and 0.15 mM pyruvate for 30 min. Muscles were transferred to new vials containing KRB, 5 mM glucose, 0.15 mM pyruvic acid, and 0.5 mM cycloheximide (translation elongation inhibitor) for 2 h. Muscles were frozen in liquid N2, weighed, and processed for immunoblot analyses as described above.
For assessment of protein degradation rates, perchloric acid (1 N) was added to the KRB solutions, vortexed, and centrifuged at 5,000 g to precipitate proteins. The supernatant was removed, and amino acids present in the solution were derivatized using standard procedures from an AccQ Tag Ultra Derivatization kit (Waters). Concentrations of phenylalanine were determined using an Acquity Ultra Performance Liquid Chromatography H-class system (Waters), and the rates of protein degradation were calculated using the following equation: rate = amino acid amount ÷ muscle weight ÷ incubation time.
Assessment of intracellular signaling in muscle by immunoblot analysis.
Immunoblots were performed using standard methods as previously described (11). Briefly, muscle lysates (15–60 μg) were subjected to SDS-PAGE, proteins were transferred onto nitrocellulose membranes, and membranes were blocked with 5% bovine serum albumin or nonfat dry milk and then incubated with primary antibodies. The following primary antibodies were utilized in this study: p-Akt (Thr308; cat. no. 9275), p-Akt (Ser473; cat. no. 4058), pan-Akt (cat. no. 3063), p-AMPK (Thr172; cat. no. 2531), AMPKα (cat. no. 5831), p-TSC2 (Thr1462; cat. no. 3617), TSC2 (cat. no. 4308), p-mTOR (Ser2448; cat. no. 2971), mTOR (cat. no. 2983), p-p70 S6K (Thr389; cat. no. 9234), and p70 S6K (cat. no. 9202) from Cell Signaling Technology; and CaMKKα (cat. no. AF7899) from R&D Systems. Horseradish peroxidase-conjugated secondary antibodies were incubated with the membrane and detected using chemiluminescence reagents (PerkinElmer Life Sciences). Densitometric analysis of immunoblots was performed using Image Lab software (Bio-Rad Laboratories).
Statistical analysis.
Data are presented as means ± SE. Statistical significance was defined as P < 0.05 and determined by paired t-tests or two-way analysis of variance and Student-Newman-Keuls post hoc analysis. The numbers of mice or muscles utilized to determine statistical significance is indicated in the text or figure legends.
RESULTS
CaMKKα protein expression is increased early during muscle hypertrophy.
Previous work in mouse skeletal muscle had shown that 7 days of overload induced by muscle synergist ablation increased CaMKKα protein expression (∼3-fold) and activity (∼5-fold) (21), suggesting that CaMKKα may be a Ca2+-activated protein that regulates muscle growth. However, since CaMKKα was not examined until 7 days after the start of the overload, it is presently unknown how early CaMKKα protein expression increases during this process. Thus, the first goal of this study was to determine whether CaMKKα protein levels increase before or after the onset of measurable changes in muscle mass during overload-induced growth. Hypertrophy was induced in mouse plantaris muscles by unilateral ablation of synergist muscles for 1, 3, 7, or 10 days, and CaMKKα protein levels were assessed by immunoblot analysis. As shown in Fig. 1, overload led to a time-dependent increase in plantaris muscle weight (Fig. 1A) and CaMKKα protein expression (Fig. 1B) starting at 3 days and increasing up to 10 days, demonstrating that CaMKKα protein expression is stimulated early and concurrently with changes in muscle mass following overload.
Fig. 1.
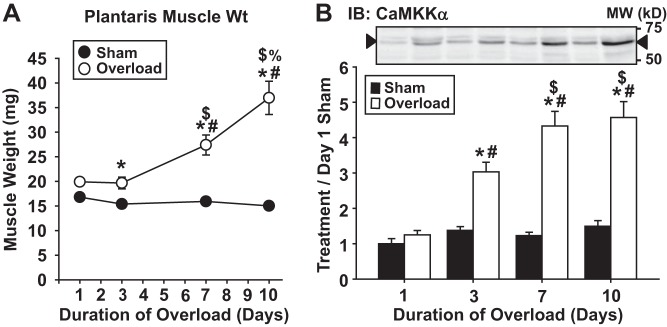
Functional overload induces Ca2+/calmodulin-dependent protein kinase kinase-α (CaMKKα) protein expression in a time-dependent manner in mouse skeletal muscle. Mouse plantaris muscles were stimulated to hypertrophy via unilateral functional overload. After 1, 3, 7, or 10 days, muscles were frozen, weighed, and processed for immunoblot analyses. A: muscle frozen wet weights. B: representative blots and quantification for CaMKKα; n = 7–8 muscles/group. P < 0.05 = * vs. sham, # vs. 1 day, $ vs. 3 days, % vs. 7 days.
Constitutively active CaMKKα expression increases muscle mass and protein content.
Intracellular Ca2+ levels regulate numerous metabolic processes in skeletal muscle, and previous work has shown that increases in intracellular Ca2+ can both stimulate and inhibit muscle growth, protein synthesis, and/or protein degradation rates (16, 20, 31). Given these conflicting and varied responses, it appears likely that Ca2+ regulates multiple proteins within muscle that can regulate muscle growth but with some exerting a positive and others exerting a negative effect. Thus, to determine the specific role of CaMKKα in the regulation of muscle growth, an approach was employed that allowed for the dissociation of CaMKKα activity and changes in intracellular Ca2+ levels. In this study and others previously described (6, 11, 34), this approach was expression of a truncated CaMKKα protein (amino acids 1–434) that possesses constitutive, Ca2+/calmodulin-independent kinase activity.
To determine whether selective activation of CaMKKα signaling was sufficient to stimulate muscle growth, mouse muscles were transfected with plasmids containing empty vector or truncated, constitutively active CaMKKα as described above. After 2 wk, muscle weights were measured pre- and postdehydration. As shown in Fig. 2, expression of the constitutively active form of CaMKKα significantly increased muscle weight (Fig. 2, A and B) without a change in muscle water content (Fig. 2C), demonstrating that the increase in mass was not due to edema. To determine whether the increase in weight was due to an increase in muscle protein, total muscle protein content was examined. As shown in Fig. 2D, muscles expressing constitutively active CaMKKα had significantly higher protein content, demonstrating that chronic activation of CaMKKα-dependent signaling is sufficient to stimulate growth in mouse muscle.
Fig. 2.
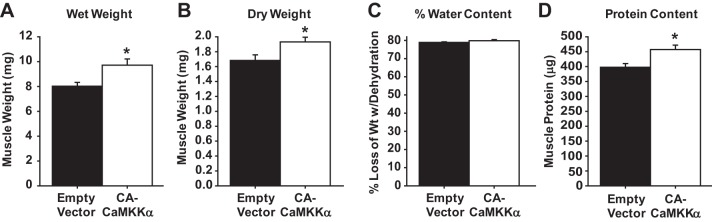
Expression of constitutively active CaMKKα (CA-CAMKKα) increases skeletal muscle mass and protein content. Mouse extensor digitorum longus muscles were transfected with empty vector or constitutively active CaMKKα using in vivo electroporation. Mice were euthanized 2 wk later and muscle collected. A: muscle wet weight. B: muscles were dehydrated for 8 h and reweighed (i.e., dry weight). C: percent loss of muscle weight with dehydration was calculated and is presented as percent muscle water content. D: dehydrated muscles were homogenized and muscle protein content determined; n = 8 muscles/group. P < 0.05 = * vs. empty vector.
Constitutively active CaMKKα expression increases muscle protein synthesis and does not decrease protein degradation.
Muscle protein content is regulated by the balance between protein synthesis and protein degradation rates. To determine whether the activation of CaMKKα signaling increases muscle mass via an increase in protein synthesis and/or a decrease in protein degradation, mouse muscles were transfected with empty vector or constitutively active CaMKKα for 1 or 2 wk. Protein synthesis was assessed by the rate of [3H]phenylalanine incorporation into muscle proteins; protein degradation was assessed by the loss of phenylalanine from muscle over time. As shown in Fig. 3, expression of constitutively active CaMKKα at both 1 and 2 wk significantly increased muscle protein synthesis rates by ∼60% (Fig. 3A), but not protein degradation rates (Fig. 3B), demonstrating that the increase in muscle protein content was due to an increase in protein synthesis. Protein degradation rates were significantly lower in both the empty vector- and active CaMKKα-transfected muscles at 2 wk posttransfection (Fig. 3B), suggesting an effect of the electroporation procedures on degradation rates.
Fig. 3.
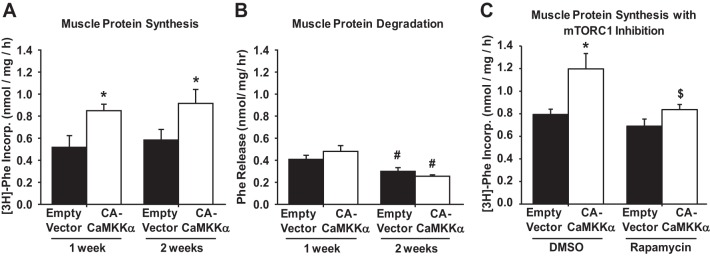
Expression of CA-CaMKKα increases skeletal muscle protein synthesis via mTOR complex 1 (mTORC1). Mouse extensor digitorum longus muscles were transfected with plasmids containing empty vector or constitutively active CaMKKα for 1 or 2 wk. A: protein synthesis was assessed as the rate of [3H]phenylalanine incorporation into protein for 30 min. B: protein degradation was assessed as the rate of phenylalanine loss from the muscle over 2 h. C: protein synthesis was assessed as the rate of [3H]phenylalanine incorporation into proteins in the presence of mTORC1 inhibitor rapamycin (150 nM) or vehicle (DMSO). Muscles were transfected for 2 wk; n = 6–9 muscles/group. P < 0.05 = * vs. empty vector, # vs. 1 wk, $ vs. DMSO.
Constitutively active CaMKKα expression increases muscle protein synthesis via mTORC1.
mTORC1 activity is critical for resistance exercise-, loading/overload-, and stretch-induced increases in muscle protein synthesis (4, 13, 17). Since expression of the constitutively active CaMKKα increased protein synthesis rates in skeletal muscle (Fig. 3), it was next important to determine whether the effect was dependent on signaling via mTORC1. To determine whether mTORC1 is essential for CaMKKα-induced protein synthesis, muscles expressing constitutively active CaMKKα for 2 wk were treated with the mTORC1 inhibitor rapamycin (150 nM) for 90 min and protein synthesis rates were assessed. Importantly, this concentration of rapamycin had previously been shown to prevent stretch-induced increases in muscle protein synthesis and mTORC1 signaling (13). As shown in Fig. 3C, rapamycin prevented the CaMKKα-induced increase in muscle protein synthesis, demonstrating that mTORC1 plays an essential role in the intracellular mechanism by which CaMKKα regulates muscle growth.
Constitutively active CaMKKα expression stimulates mTORC1 signaling independently of known upstream regulators Akt and AMPK.
Activation of mTORC1 stimulates the phosphorylation of two well-established mTOR substrates, the 70-kDa ribosomal protein S6 kinase (p70 S6K, Thr389) and the eukaryotic initiation factor 4E-binding protein-1 (4E-BP1, Thr36/47) (see review in Ref. 18). Since rapamycin completely prevented the active CaMKKα-induced increase in protein synthesis (Fig. 3C), this would suggest that constitutively active CaMKKα expression stimulates mTORC1 activity and thus phosphorylation of downstream substrates in skeletal muscle. To assess this, immunoblot analyses were performed to examine p70 S6K and 4E-BP1 phosphorylation and expression. As shown in Fig. 4, expression of constitutively active CaMKKα increased the phosphorylation of p70 S6K (Thr389) ∼50%, and 4E-BP1 (Thr36/47) ∼35%, with no change in p70S6K or 4E-BP1 protein levels, consistent with mTORC1 activation in skeletal muscle.
Fig. 4.
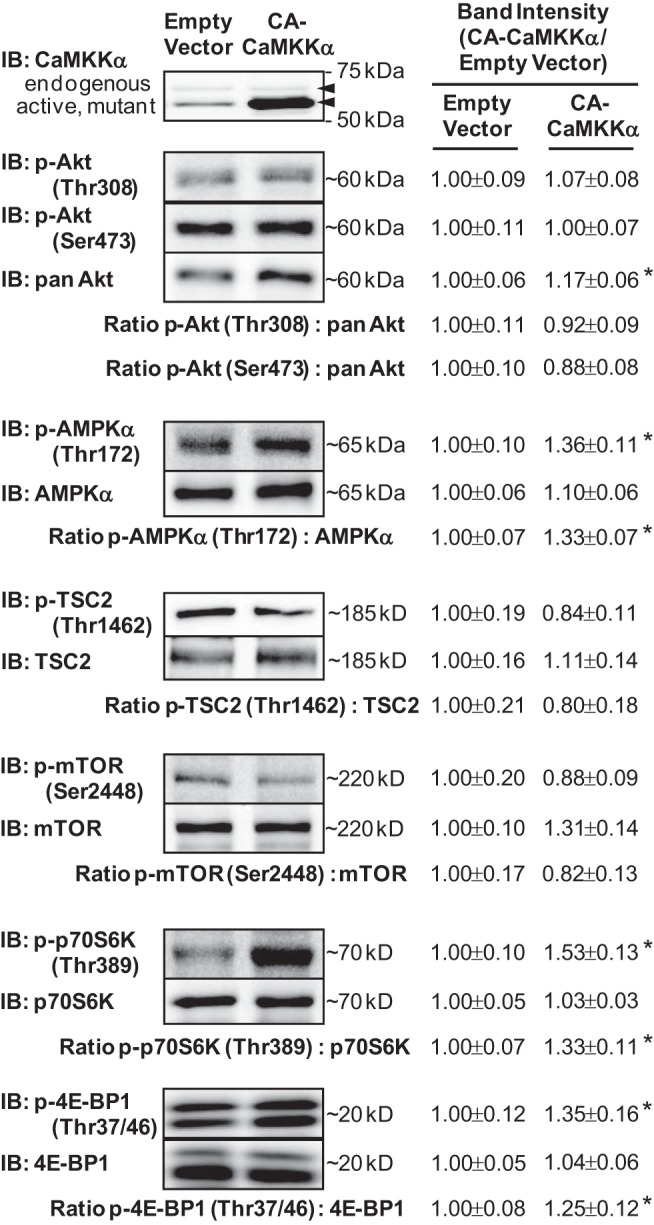
Expression of CA-CaMKKα stimulates mTORC1 signaling in skeletal muscle. Mouse extensor digitorum longus muscles were transfected with plasmids containing empty vector or constitutively active CaMKKα for 2 wk. Representative blots and quantification provided above for key signaling proteins implicated in the regulation of mTORC1 signaling; n = 7–9 muscles/group. P < 0.05 = * vs. empty vector.
The serine/threonine kinase Akt (also known as protein kinase B) is a positive upstream regulator of mTORC1 signaling, and previous work has shown that Akt can phosphorylate the tuberous sclerosis complex 2 (TSC2) on multiple sites (e.g., Thr1462), releasing its inhibitory action on mTOR (14) and thus stimulating mTOR kinase activity. In addition, Akt can directly phosphorylate mTOR on Ser2448 (22, 24, 27), although the effect of increased mTOR Ser2448 phosphorylation on mTORC1 activity is less clear. To determine whether constitutively active CaMKKα expression increases protein synthesis via stimulation of Akt signaling, immunoblot analyses were performed to examine the phosphorylation and protein levels of Akt, TSC2, and mTOR. As shown in Fig. 4, expression of constitutively active CaMKKα did not increase the phosphorylation of Akt (Thr308), Akt (Ser473), TSC2 (Thr1462), or mTOR (Ser2448), demonstrating that activation of Akt signaling is not the mechanism by which CaMKKα regulates mTORC1 and muscle protein synthesis.
mTORC1 is inhibited by alterations in cellular energetics (i.e., increased AMP:ATP), and the activation of the serine/threonine kinase AMPK. Although previous work from our laboratory has shown that expression of constitutively active CaMKKα in mouse muscle increases AMPK (Thr172) phosphorylation (11, 34) and activity (34), these studies were performed in mouse tibialis anterior muscle and thus there could be muscle-specific differences in response to active CaMKKα expression. Thus, to determine whether constitutively active CaMKKα expression stimulates protein synthesis via inhibition of AMPK signaling in mouse extensor digitorum longus muscles, immunoblot analyses were performed to examine AMPK phosphorylation and protein content. As shown in Fig. 4, muscles expressing constitutively active CaMKKα exhibited a significant increase in the phosphorylation of AMPK (Thr172; ∼40%) with no change in AMPK protein levels, demonstrating that inhibition of AMPK signaling is not the mechanism by which CaMKKα regulates mTORC1 and muscle protein synthesis.
Loss of CaMKKα does not impair overload-induced muscle growth or mTORC1 signaling.
Use of a constitutively active form of CaMKKα revealed that activation of CaMKKα signaling is sufficient to increase mTORC1 signaling, muscle protein synthesis rates, and muscle mass in the mouse. To determine whether CaMKKα plays an essential role in muscle growth, global CaMKKα knockout mice and their wild-type littermates were subjected to unilateral synergist ablation to induce functional overload of the plantaris muscle, as described above. Loss of CaMKKα did not significantly affect mouse body weight (Fig. 5A). Functional overload for 7 days induced an ∼35% increase in plantaris muscle weight in the wild-type mice and an ∼50% increase in the muscle of CaMKKα knockout mice (FIGS 5, B–D), demonstrating that loss of CaMKKα expression enhances overload-induced muscle hypertrophy.
Fig. 5.
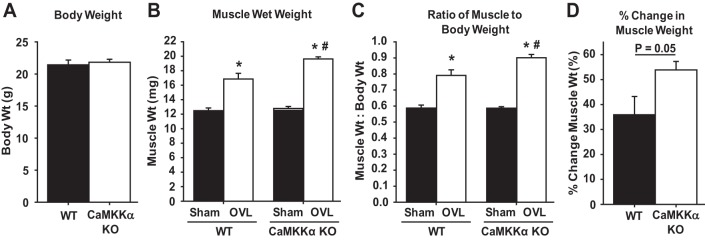
Loss of CaMKKα enhances functional overload-induced increases in skeletal muscle mass. Plantaris muscles from CaMKKα knockout mice and their wild-type littermates were stimulated to hypertrophy via unilateral functional overload (OVL). After 7 days, muscles were frozen and weighed. A: mouse body weights at the time of overload surgery. B: muscle frozen wet weight. C: ratio of mouse muscle weight to body weight. D: percent change in muscle weight in response to overload; n = 9–11 mice or muscles/group. P < 0.05 = * vs. sham, # vs. wild type.
The observation that muscles from CaMKKα knockout mice exhibited enhanced overload-induced muscle growth was surprising, since expression of the constitutively active form of CaMKKα led to an increase in muscle mass. Thus, these data could suggest that loss of CaMKKα resulted in the hypercompensation of a positive growth regulatory pathway (e.g., Akt signaling) or loss of a negative growth regulatory pathway (e.g., AMPK signaling) in these muscles. Thus, to determine whether the increase in muscle growth found in the CaMKKα knockout mice was due to increased signaling via Akt, or decreased signaling via AMPK, control and overloaded muscles from the wild-type and CaMKKα knockout mice were subjected to immunoblot analyses. As shown in Fig. 6, muscles from CaMKKα knockout mice subjected to overload exhibited a significantly greater amount of phosphorylated p70 S6K (Thr389) and no change in the phosphorylated amount of Akt (Thr308 or Ser473), AMPK (Thr172), TSC2 (Thr1462), or mTOR (Ser2448) when considered relative to their protein levels (i.e., phospho-to-total ratio). Collectively, these data show that signaling through mTORC1 is upregulated but Akt signaling and AMPK signaling is unaltered in muscles from CaMKKα knockout mice undergoing hypertrophic growth.
Fig. 6.

Loss of CaMKKα enhances overload-induced activation of mTORC1 signaling in mouse skeletal muscle. Plantaris muscles from CaMKKα knockout mice and their wild-type littermates were stimulated to hypertrophy via functional overload (OVL). After 7 days, muscles were frozen and processed for immunoblot analyses. Representative blots and quantification provided above; n = 6–9 muscles/group. P < 0.05 = * vs. sham, # vs. wild type.
DISCUSSION
The data presented in this study are the first to demonstrate that activation of signaling via the Ca2+/calmodulin-activated, serine/threonine kinase, CaMKKα, is sufficient but not necessary for mTORC1 activation and growth in mouse skeletal muscle. We show that in muscle CaMKKα protein expression is induced early and in a time-dependent manner during functional overload and that transient expression of a constitutively active form of CaMKKα is sufficient to increase muscle mass, protein content, protein synthesis rates, and mTORC1 signaling. We also show that chronic loss of CaMKKα expression, via a global CaMKKα knockout mouse, did not prevent overload-induced muscle growth but instead enhanced it, suggesting a complex regulation of protein synthesis and mTORC1 signaling by CaMKKα.
Expression of constitutively active CaMKKα significantly increased muscle protein synthesis rates following 1 and 2 wk of transfection (∼40%; Fig. 3A). This amount of increase in protein synthesis rate is consistent with that observed following 15 days of overload in rat skeletal muscle (23) and 90 min of static stretch in the mouse (13). The constitutively active CaMKKα-induced increase in protein synthesis rate was prevented by acute treatment with the mTORC1 inhibitor rapamycin (150 nM, 90 min; Fig. 3C). These results are consistent with those observed for stretch-induced increases in muscle protein synthesis rate (13) and suggest that the intracellular mechanism by which CaMKKα regulates muscle protein synthesis is completely dependent on signaling via the mTORC1 complex.
In mouse skeletal muscle, expression of constitutively active CaMKKα did not affect protein degradation rates (Fig. 3B), demonstrating that the mechanism underlying CaMKKα-induced increases in muscle mass is not due to decreases in muscle protein breakdown. Interestingly, muscle protein degradation rates were significantly higher in both the empty vector- and constitutively active CaMKKα-transfected muscles at 1 wk posttransfection compared with 2 wk posttransfection, suggesting an acute rise in muscle protein breakdown induced by the in vivo electroporation procedures. Although this does not affect our overall conclusion that expression of constitutively active CaMKKα is sufficient to increase muscle mass, we do speculate that this may be part of the reason underlying the modest increase in muscle mass (∼10%) observed in muscles expressing active CaMKKα. Future studies will examine the effects of chronic CaMKKα activation at longer time points (i.e., >2 wk posttransfection).
Previous work has shown that CaMKKα can phosphorylate Akt on Thr308 in a cell-free system (35), in COS-7 cells (35), and in HEK-293 cells (5), suggesting that CaMKKα-dependent phosphorylation and activation of Akt could be at least part of the mechanism underlying the ability of the constitutitvely active CaMKKα to stimulate skeletal muscle growth. However, muscles expressing constitutively active CaMKKα for 2 wk exhibited an increase in p70 S6K (Thr389) phosphorylation but no change in Akt (Thr308) or (Ser473) phosphorylation (Fig. 4), suggesting that in mouse muscle CaMKKα activates mTORC1 signaling downstream of Akt. These results are consistent with previous work in mouse muscle that showed that constitutively active CaMKKα expression does not stimulate the phosphorylation of Akt (Thr308) (11, 34) or Akt (Ser473) (11). The reason behind the discrepancy in findings between the activation of Akt by CaMKKα in the cell-free system and kidney cell culture models compared with the mouse muscle is currently not known but could be due to tissue-specific differences, a lack of necessary cofactors, and/or protein localization.
Consistent with previous work, muscles expressing constitutively active CaMKKα exhibited increased phosphorylation of AMPK (Thr172) (11, 34), a known CaMKK substrate (Fig. 4). This finding is intriguing in the context of previous work linking activation of AMPK with suppression of mTORC1 signaling and protein synthesis, and it suggests that the stimulatory effects of constitutively active CaMKKα on protein synthesis rates may be attenuated by the simultaneous activation of AMPK. However, loss of CaMKKα did not impair overload-induced increases in AMPK (Thr172) phosphorylation (Fig. 6), demonstrating that CaMKKα is not required for overload-induced AMPK activation in mouse skeletal muscle. Thus, the role of CaMKKα-AMPK signaling in the regulation of muscle growth is still in question, and future studies using mouse models of AMPK inactivation and expression of constitutively active CaMKKα expression would need to be performed to fully test this interaction.
This is the first study to examine skeletal muscle hypertrophic responses in a mouse model of CaMKKα ablation. In the mice utilized in this study, we did not observe a significant difference in mouse body weight at any age studied (6–14 wk; only data for 14 wk shown in Fig. 5A). Thus, in the basal state, loss of CaMKKα expression had no significant effect on mouse growth or size. In response to overload, plantaris muscles from CaMKKα knockout mice exhibited enhanced growth compared with overloaded muscles from wild-type littermate controls (Fig. 5), suggesting that CaMKKα is a negative regulator of muscle hypertrophic growth. However, since expression of the constitutively active form of CaMKKα was sufficient to stimulate muscle growth, an alternative explanation for these findings is that another growth-regulatory pathway is upregulated in the muscles from the knockout mice that overcompensates for the loss of CaMKKα. Consistent with this hypothesis, overloaded muscles from the CaMKKα knockout mice exhibited a significantly greater phosphorylation of p70 S6K (Thr389). Future studies will investigate the possible mechanism for the hyper-activation of p70 S6K and its connection with loss of CaMKKα expression.
In summary, in the present study we found that CaMKKα protein expression is induced early and in a time-dependent manner during functional overload-induced skeletal muscle hypertrophy and that expression of a constitutively active form of CaMKKα is sufficient to increase muscle mass, protein content, protein synthesis rates, and mTORC1 signaling. In addition, we also found that, paradoxically, loss of CaMKKα expression enhanced overload-induced muscle growth and mTORC1 signaling, collectively suggesting a complex regulation of muscle growth by CaMKKα.
GRANTS
This project was supported by Grant No. R00 AR-056298 (C. A. Witczak) from the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Arthritis and Musculoskeletal and Skin Diseases or the National Institutes of Health. Additional funds to support this project were provided by East Carolina University in the form of laboratory start-up funds to C. A. Witczak, as well as J. J. Brault.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.L.F., J.J.B., C.A.S., and C.A.W. performed experiments; J.L.F., J.J.B., C.A.S., and C.A.W. analyzed data; J.L.F., J.J.B., C.A.S., and C.A.W. interpreted results of experiments; J.L.F., J.J.B., C.A.S., and C.A.W. edited and revised manuscript; J.L.F., J.J.B., C.A.S., and C.A.W. approved final version of manuscript; C.A.W. conception and design of research; C.A.W. prepared figures; C.A.W. drafted manuscript.
ACKNOWLEDGMENTS
We thank T. R. Soderling for the generous donation of the expression vectors. C. A. Witczak is the guarantor of this work, and as such has full access to all of the data in the study and takes full responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.Aihara H, Miyazaki J. Gene transfer into muscle by electroporation in vivo. Nat Biotechnol 16: 867–870, 1998 [DOI] [PubMed] [Google Scholar]
- 2.Biolo G, Maggi SP, Williams BD, Tipton KD, Wolfe RR. Increased rates of muscle protein turnover and amino acid transport after resistance exercise in humans. Am J Physiol Endocrinol Metab 268: E514–E520, 1995 [DOI] [PubMed] [Google Scholar]
- 3.Blaeser F, Sanders MJ, Truong N, Ko S, Wu LJ, Wozniak DF, Fanselow MS, Zhuo M, Chatila TA. Long-term memory deficits in Pavlovian fear conditioning in Ca2+/calmodulin kinase kinase alpha-deficient mice. Mol Cell Biol 26: 9105–9115, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019, 2001 [DOI] [PubMed] [Google Scholar]
- 5.Chen BC, Wu WT, Ho FM, Lin WW. Inhibition of interleukin-1beta-induced NF-kappa B activation by calcium/calmodulin-dependent protein kinase kinase occurs through Akt activation associated with interleukin-1 receptor-associated kinase phosphorylation and uncoupling of MyD88. J Biol Chem 277: 24169–24179, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Enslen H, Tokumitsu H, Stork PJ, Davis RJ, Soderling TR. Regulation of mitogen-activated protein kinases by a calcium/calmodulin-dependent protein kinase cascade. Proc Natl Acad Sci USA 93: 10803–10808, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fang CH, Li BG, Wang JJ, Fischer JE, Hasselgren PO. Treatment of burned rats with insulin-like growth factor I inhibits the catabolic response in skeletal muscle. Am J Physiol Regul Integr Comp Physiol 275: R1091–R1098, 1998 [DOI] [PubMed] [Google Scholar]
- 8.Fulks RM, Li JB, Goldberg AL. Effects of insulin, glucose, and amino acids on protein turnover in rat diaphragm. J Biol Chem 250: 290–298, 1975 [PubMed] [Google Scholar]
- 9.Goldberg AL. Protein synthesis during work-induced growth of skeletal muscle. J Cell Biol 36: 653–658, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldspink DF. The influence of immobilization and stretch on protein turnover of rat skeletal muscle. J Physiol 264: 267–282, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinkley JM, Ferey JL, Brault JJ, Smith CS, Gilliam LAA, Witczak CA. Constitutively active CaMKKalpha stimulates skeletal muscle glucose uptake in insulin resistant mice in vivo. Diabetes 63: 142–151, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hornberger TA, Chu WK, Mak YW, Hsiung JW, Huang SA, Chien S. The role of phospholipase D and phosphatidic acid in the mechanical activation of mTOR signaling in skeletal muscle. Proc Natl Acad Sci USA 103: 4741–4746, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornberger TA, Stuppard R, Conley KE, Fedele MJ, Fiorotto ML, Chin ER, Esser KA. Mechanical stimuli regulate rapamycin-sensitive signalling by a phosphoinositide 3-kinase-, protein kinase B- and growth factor-independent mechanism. Biochem J 380: 795–804, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4: 648–657, 2002 [DOI] [PubMed] [Google Scholar]
- 15.Ito N, Ruegg UT, Kudo A, Miyagoe-Suzuki Y, Takeda S. Activation of calcium signaling through Trpv1 by nNOS and peroxynitrite as a key trigger of skeletal muscle hypertrophy. Nat Med 19: 101–106, 2013 [DOI] [PubMed] [Google Scholar]
- 16.Kameyama T, Etlinger JD. Calcium-dependent regulation of protein synthesis and degradation in muscle. Nature 279: 344–346, 1979 [DOI] [PubMed] [Google Scholar]
- 17.Kubica N, Bolster DR, Farrell PA, Kimball SR, Jefferson LS. Resistance exercise increases muscle protein synthesis and translation of eukaryotic initiation factor 2Bepsilon mRNA in a mammalian target of rapamycin-dependent manner. J Biol Chem 280: 7570–7580, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 149: 274–293, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee KH, Lee S, Lee WY, Yang HW, Heo WD. Visualizing dynamic interaction between calmodulin and calmodulin-related kinases via a monitoring method in live mammalian cells. Proc Natl Acad Sci USA 107: 3412–3417, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lewis SE, Anderson P, Goldspink DF. The effects of calcium on protein turnover in skeletal muscles of the rat. Biochem J 204: 257–264, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGee SL, Mustard KJ, Hardie DG, Baar K. Normal hypertrophy accompanied by phosphoryation and activation of AMP-activated protein kinase alpha1 following overload in LKB1 knockout mice. J Physiol 586: 1731–1741, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nave BT, Ouwens M, Withers DJ, Alessi DR, Shepherd PR. Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem J 344: 427–431, 1999 [PMC free article] [PubMed] [Google Scholar]
- 23.Noble EG, Tang Q, Taylor PB. Protein synthesis in compensatory hypertrophy of rat plantaris. Can J Physiol Pharmacol 62: 1178–1182, 1984 [DOI] [PubMed] [Google Scholar]
- 24.Reynolds TH, Bodine SC, Lawrence JC, Jr. Control of Ser2448 phosphorylation in the mammalian target of rapamycin by insulin and skeletal muscle load. J Biol Chem 277: 17657–17662, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Rothman S. How is the balance between protein synthesis and degradation achieved? Theor Biol Med Model 7: 25, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seguin R, Nelson ME. The benefits of strength training for older adults. Am J Prev Med 25: 141–149, 2003 [DOI] [PubMed] [Google Scholar]
- 27.Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 60: 3504–3513, 2000 [PubMed] [Google Scholar]
- 28.Sonobe T, Inagaki T, Poole DC, Kano Y. Intracellular calcium accumulation following eccentric contractions in rat skeletal muscle in vivo: role of stretch-activated channels. Am J Physiol Regul Integr Comp Physiol 294: R1329–R1337, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Spangenburg EE, McBride TA. Inhibition of stretch-activated channels during eccentric muscle contraction attenuates p70S6K activation. J Appl Physiol (1985) 100: 129–135, 2006 [DOI] [PubMed] [Google Scholar]
- 30.Srikanthan P, Karlamangla AS. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the third National Health and Nutrition Examination Survey. J Clin Endocrinol Metab 96: 2898–2903, 2011 [DOI] [PubMed] [Google Scholar]
- 31.Sugden PH. The effects of calcium ions, ionophore A23187 and inhibition of energy metabolism on protein degradation in the rat diaphragm and epitrochlearis muscles in vitro. Biochem J 190: 593–603, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thomson DM, Gordon SE. Diminished overload-induced hypertrophy in aged fast-twitch skeletal muscle is associated with AMPK hyperphosphorylation. J Appl Physiol 98: 557–564, 2005 [DOI] [PubMed] [Google Scholar]
- 33.Tokumitsu H, Iwabu M, Ishikawa Y, Kobayashi R. Differential regulatory mechanism of Ca2+/calmodulin-dependent protein kinase kinase isoforms. Biochemistry 40: 13925–13932, 2001 [DOI] [PubMed] [Google Scholar]
- 34.Witczak CA, Fujii N, Hirshman MF, Goodyear LJ. Ca2+/calmodulin-dependent protein kinase kinase-alpha regulates skeletal muscle glucose uptake independent of AMP-activated protein kinase and Akt activation. Diabetes 56: 1403–1409, 2007 [DOI] [PubMed] [Google Scholar]
- 35.Yano S, Tokumitsu H, Soderling TR. Calcium promotes cell survival through CaM-K kinase activation of the protein-kinase-B pathway. Nature 396: 584–587, 1998 [DOI] [PubMed] [Google Scholar]