

Abstract
Cholesteryl ester storage disease (CESD) results from loss-of-function mutations in LIPA, the gene that encodes lysosomal acid lipase (LAL). Hepatomegaly and deposition of esterified cholesterol (EC) in multiple organs ensue. The present studies quantitated rates of synthesis, absorption, and disposition of cholesterol, and whole body cholesterol pool size in a mouse model of CESD. In 50-day-old lal−/− and matching lal+/+ mice fed a low-cholesterol diet, whole animal cholesterol content equalled 210 and 50 mg, respectively, indicating that since birth the lal−/− mice sequestered cholesterol at an average rate of 3.2 mg·day−1·animal−1. The proportion of the body sterol pool contained in the liver of the lal−/− mice was 64 vs. 6.3% in their lal+/+ controls. EC concentrations in the liver, spleen, small intestine, and lungs of the lal−/− mice were elevated 100-, 35-, 15-, and 6-fold, respectively. In the lal−/− mice, whole liver cholesterol synthesis increased 10.2-fold, resulting in a 3.2-fold greater rate of whole animal sterol synthesis compared with their lal+/+ controls. The rate of cholesterol synthesis in the lal−/− mice exceeded that in the lal+/+ controls by 3.7 mg·day−1·animal−1. Fractional cholesterol absorption and fecal bile acid excretion were unchanged in the lal−/− mice, but their rate of neutral sterol excretion was 59% higher than in their lal+/+ controls. Thus, in this model, the continual expansion of the body sterol pool is driven by the synthesis of excess cholesterol, primarily in the liver. Despite the severity of their disease, the median life span of the lal−/− mice was 355 days.
Keywords: cholesterol synthesis, esterified cholesterol, hepatomegaly, lysosomal storage disease, sterol balance
most organs continuously clear various lipoproteins from the circulation through receptor-mediated and bulk-phase endocytosis (4, 17, 33). These lipoproteins carry differing amounts of cholesterol, both esterified and unesterified. The initial site for processing these sterols is the late endosomal/lysosomal (E/L) compartment. Here, through the sequential actions of three proteins, lysosomal acid lipase (LAL), Niemann-Pick C2 (NPC2), and Niemann-Pick C1 (NPC1), lipoprotein-derived unesterified cholesterol (UC) is ultimately delivered into the cytosolic compartment where it mixes with cholesterol from other sources including that which is synthesized locally. Mutations in any of these three proteins cause profound changes in intracellular cholesterol homeostasis that lead to cell dysfunction and death. Niemann-Pick C disease is typically characterized by elevated UC levels in all the organs, liver disease, pulmonary dysfunction, and neurodegeneration (57).
The hydrolysis of esterified cholesterol (EC) and triacylglycerols by LAL generates unesterified cholesterol, free fatty acids, and glycerol for cellular utilization (20). In humans, loss-of-function mutations in LIPA, the gene that encodes LAL, result in Wolman disease or cholesteryl ester storage disease (CESD). Whereas Wolman disease is a severe, early-onset illness caused by complete absence of LAL activity, CESD is a milder, later-onset disease resulting from partial LAL deficiency (22). Hepatomegaly and a massive increase in tissue EC levels are hallmark features of both disorders. A spontaneous rat model for Wolman disease was documented in 1990 (25). In 1998 Du et al. (13) described the first mouse model for CESD. A more detailed description of the phenotype of the lal−/− mouse was published in 2001 (14). This model has since been used for developing an enzyme replacement therapy for Wolman disease and CESD (12, 15, 49).
Although CESD is considered a rare disorder, there is a growing consensus that it may be underdiagnosed (3, 35, 43, 47). There is also expanding research into the causes and treatment of various forms of nonalcoholic fatty liver disease (16, 27). These advances, together with evaluation of an enzyme replacement therapy for CESD in human subjects (1, 21), are driving interest in the type of animal model that is the focus of the present studies. Here we describe in quantitative terms how LAL deficiency in a mouse model impacts the major pathways that govern the entry and removal of cholesterol into and from the body sterol pool. Together, the data show that although essentially all organ systems are affected, events within the liver, particularly the rate of cholesterol synthesis, play a defining role in the progression of disease.
MATERIALS AND METHODS
Animals and diets.
Lal+/+ and lal−/− mice were generated from heterozygous breeding stock, all on an FVB/N strain background, that were kindly provided by Drs. Gregory Grabowski and Hong Du at the Children's Hospital Research Foundation, Cincinnati, OH. The litters were weaned at 21 days and genotyped by using an ear notch. Primer sequences for the PCR method used were supplied by Dr. Du. Unless studied on the day of weaning, all mice were fed ad libitum a basal low-cholesterol, low-fat rodent chow diet (no. 7001, Harlan Teklad, Madison, WI). This formulation had an inherent cholesterol content of 0.02% (wt/wt) and a crude fat content of not less than 4% (wt/wt). The age of the mice when studied varied widely depending on the objective of the study but the bulk of the data were obtained from mice that were ∼50 days old. This age point was selected partly because of our interest in comparing cholesterol metabolism data in a mouse model for CESD with those already documented for NPC1-deficient mice (npc1nih/nih) at that age (41, 42, 60). In nearly all experiments the mice were group housed in a light-cycled room and were studied in the fed state toward the end of the dark phase. In several experiments with mice before and after 50 days of age, body weights were recorded weekly. A subset of older mice was used for the measurement of life span. For these particular mice, in addition to weekly body weight measurement, their general physical condition, food intake and stool output were monitored daily. Together, these criteria were used to determine when euthanasia should be carried out. The total number of mice used for all studies combined was 343 (124 males, 219 females). All experiments were approved by the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center.
Rates of cholesterol synthesis in the liver, small intestine, other extrahepatic organs, and the whole animal.
These rates were measured in vivo by use of [3H]water as detailed elsewhere (46). In the first study, sterol synthesis was determined in multiple organs of 50-day-old mice. One hour after the mice were administered ∼40 mCi of [3H]water intraperitoneally, the mice were anesthetized with isoflurane and exsanguinated from the vena cava into a heparinized 1-ml syringe fitted with a 1-in., 23-G needle. The liver, small intestine, and other extrahepatic organs were removed, rinsed, blotted, and weighed. They were then saponified and the labeled sterols were extracted and quantitated as described (46). The rate of cholesterol synthesis in each organ was calculated as nanomoles of [3H]water incorporated into sterols per hour per gram wet weight of tissue. These rates were multiplied by organ weights to obtain synthesis in each organ (nmol·h−1·organ−1). In the second study, the rate of sterol synthesis in the whole liver, and in the entire remaining animal, was measured in mice at varying ages ranging from 21 to 165 days. For both the liver and the residual carcass, synthesis was expressed both per gram of tissue and also on a whole organ basis. These rates, expressed as nanomoles per hour per whole liver or per whole residual carcass, were summed to determine the rate of synthesis in the entire animal. These data were in turn used to quantitate the contribution of the liver to whole body cholesterol synthesis in the lal−/− mice as their disease progressed. Some of these data for [3H]water incorporation rates into sterol were converted to an equivalent milligram quantity of cholesterol generated, assuming that 0.69 hydrogen atoms from water were incorporated into the sterol molecule per carbon atom entering the biosynthetic pathway as acetyl CoA (9). In the sterol synthesis experiment with the 50-day-old mice, hepatic fatty acid synthesis was also measured as described for another model (44).
Liver histology, quantitation of hepatic triacylglycerol and total lipid content, and plasma alanine aminotransferase activity.
Sections of fresh liver were oriented, snap-frozen in isopentane cooled in liquid nitrogen, cryo-sectioned at 4-μm thicknesses and stained with hematoxylin and eosin and Oil Red O by standard procedures. The total lipid content of liver tissue was measured gravimetrically by the same method described for determination of fecal lipid content (46). Lipid content was expressed as a percentage of wet liver weight. Hepatic total triacylglycerol concentrations (mg/g) were measured by a method that combines column chromatography and an enzymatic colorimetric assay (2). The initial extracts of liver tissue contained [14C]triolein (American Radiolabeled Chemicals, St. Louis, MO) to correct for procedural losses. These values and liver weight were used to determine whole liver triacylglycerol content (mg/organ). Plasma alanine aminotransferase (ALT) activities (units/l) were measured by a commercial laboratory.
Tissue concentrations of esterified and unesterified cholesterol in liver and other organs.
These measurements were carried out by using a combination of column and gas chromatography as detailed previously (54). As described, procedural losses were corrected for using stigmastanol and two radiolabeled internal standards, [4-14C]cholesteryl oleate and [1,2-3H(N)]cholesterol (PerkinElmer Life Sciences, Boston, MA). For the study comparing the concentration of EC and UC in multiple organs, and another study that measured age-related changes in the levels of EC and UC in the small intestine, the data are expressed as milligrams per gram.
Intestinal cholesterol absorption and rates of fecal neutral sterol and bile acid excretion.
The methods used for measuring these parameters and also fecal lipid content have been described in detail previously (46). For the fractional cholesterol absorption measurements, the stools were collected from individually housed mice over 3 days immediately following the intragastric dosing with labeled tracer sterols. These were [5,6-3H]sitostanol and [4-14C]cholesterol. Similarly, the rates of fecal neutral sterol and bile acid excretion were determined via a 3-day stool collection. Additional details for the neutral sterol assay using gas chromatography were described earlier (46). The dominant neutral sterol was unmetabolized cholesterol together with small amounts of coporostanol, epicoprostanol, and cholestanone. Fecal bile acid excretion rates were measured by an enzymatic method that was initially developed for hamster stools (52) and subsequently amended for use in mice (53). The rates of neutral sterol and bile acid excretion were expressed as the equivalent mass of cholesterol appearing as neutral or acidic sterols per animal per day.
Relative mRNA expression analysis.
Aliquots of liver were quickly frozen in liquid nitrogen. mRNA levels were measured via a quantitative real-time PCR assay (55). The primer sequences used to measure RNA levels are given in several earlier publications (7, 28, 51, 54). All analyses were determined by the comparative cycle number at threshold method with cyclophilin as the internal control. The mRNA levels were normalized to cyclophilin and values for each lal−/− mouse were then expressed relative to that obtained for their matching lal+/+ controls, which in each case were arbitrarily set at 1.0.
Analysis of data.
All data are reported as means ± SE for the specified number of individual animals. GraphPad Prism 6 software (GraphPad, San Diego, CA) was used to perform all statistical analyses. Differences between means were tested for statistical significance (P < 0.05) by an unpaired Student's t-test. For the life span study, statistical differences among Kaplan-Meier survival curves were determined by the Gehan-Breslow-Wilcoxon test and log-rank analysis.
RESULTS
Age-related changes in body weight and liver mass are similar in male and female lal−/− mice but their life span is different.
As shown by the data for both males (Fig. 1A) and females (Fig. 1D), the body weight gain of the lal−/− mice did not become overtly different from that of their lal+/+ controls until ∼56 days after birth. Thereafter, the body weight curves for the two types of mice became increasingly divergent, with those deficient in LAL showing little or no weight gain up to 168 days of age. At the time of weaning there was no clear genotypic difference in liver weights, but by ∼50 days of age the mass of liver tissue in both male (Fig. 1B) and female (Fig. 1E) lal−/− mice was about double that in their lal+/+ controls. From that point the liver weight in the lal−/− mice continued to increase at a rapid rate in both the males and females. In the 168-day-old mutant mice, the relative liver weight in both the males (Fig. 1C) and females (Fig. 1F) was at least 4.5-fold greater than in lal+/+ mice at that age, with 20% or more of the body mass being accounted for by the liver. These data prompted the calculation of body weights, excluding the liver, of mice at different ages (Table 1). Unlike their lal+/+ counterparts, the lal−/− mice showed no gain in the mass of their nonhepatic tissues from ∼50 days onward. The massive hepatomegaly characteristic of lal−/− mice, previously documented (13, 14), is clearly seen in Fig. 1, G and H. In the 170-day-old male, the liver accounted for 22.5% of body weight, whereas in the 520-day-old female it represented almost 35% of body mass. The median life span for a total of 39 lal−/− mice was 355 days. However, as shown in Fig. 1I, not only did the life span of these mice vary over a wide range, but the median age at death for the females (377 days) significantly exceeded that of their male counterparts (308 days) (P < 0.05).
Fig. 1.

Age-related changes in the body and liver weights of lal+/+ and lal−/− mice and illustration of massive liver enlargement and measurement of life span in lal−/− mice. Body weight data (A and D) were obtained from large numbers of male and female mice over the age span of 21 to 168 days. Liver weight data (B, C, E, and F) were derived from mice at different ages that were used in various metabolic studies. Hepatomegaly characteristic of lal−/− mice shown in male (G) and female (H) specimens. Values are means ± SE of data from a minimum of 5 mice at every time point. For the life span study (I) there was a total of 39 lal−/− mice (18 females, 21 males).
Table 1.
Body weights of male and female lal+/+ and lal−/− mice at different ages after subtraction of liver weight
| Body Weight Less Liver Weight, g |
||||
|---|---|---|---|---|
| Male |
Female |
|||
| Age, days | lal+/+ | lal−/− | lal+/+ | lal−/− |
| 19–22 | 10.0 ± 1.16 (n = 6) | 10.3 ± 0.46 (n = 7) | 10.1 ± 0.34 (n = 6) | 9.4 ± 0.33 (n = 6) |
| 49–52 | 25.8 ± 0.65 (n = 6) | 23.1 ± 0.69 (n = 6) | 20.9 ± 0.3 (n = 6) | 19.7 ± 0.58 (n = 6) |
| 68–72 | 26.8 ± 0.66 (n = 5) | 22.6 ± 0.63 (n = 6) | 22.4 ± 0.4 (n = 9) | 21.0 ± 0.51 (n = 11) |
| 160–170 | 38.4 ± 0.7 (n = 3) | 24.7 ± 1.31 (n = 3) | 26.8 ± 1.25 (n = 5) | 21.8 ± 0.75 (n = 7) |
| 240–260 | 37.6 ± 1.64 (n = 3) | 21.5 ± 2.21 (n = 3) | 26.8 ± 0.42 (n = 3) | 20.3 ± 1.04 (n = 4) |
| 405–425 | 40.8 (n = 2) | 19.7 (n = 2) | – | – |
Young adult lal−/− mice exhibit marked histological changes and magnified levels of mRNA expression for CD68, cytokines, and cell surface proteins in their livers, as well as elevated plasma alanine aminotransferase activities.
At the relatively young age of 50 days, marked histological changes were evident in the livers of the LAL-deficient mice (Fig. 2A). Hematoxylin and eosin-stained paraffin sections of the liver from an lal+/+ mouse (Fig. 2Aa) demonstrate well-preserved hepatocytes containing normal, amphophilic cytoplasm. No inflammatory infiltrates are present. Oil Red O stained cryostat sections of the wild-type liver (Fig. 2Ac) reveal scanty accumulations of lipid within the cytoplasm of hepatocytes, predominantly in the midzonal and periportal regions (arrow). In contrast, the cytoplasm of the hepatocytes from a matching lal−/− mouse (Fig. 2Ab) has a pale, vesicular appearance caused by lipid accumulation. In addition, the liver contains multiple collections of foamy, lipid-laden macrophages (arrows). Oil Red O-stained cryostat sections of the lal−/− liver (Fig. 2Ad) reveal a massive increase in cytoplasmic lipid throughout the organ. More detailed histology for the liver and other organs in lal−/− mice at different ages has been described earlier by Du et al. (13, 14). The histological changes in the liver of the lal−/− mice are consistent with the marked elevation found in their hepatic total lipid (Fig. 2B) and total triacylglycerol content (Fig. 2C), both of which were elevated more than threefold. Even more dramatic were the changes in the relative mRNA expression levels for a constellation of genes that serve as markers for macrophage presence, cell signaling, and inflammation (Fig. 2D). The mRNA level for CD68 (CD68 antigen, also known as macrosialin) was elevated 29-fold in the lal−/− livers, indicating a marked rise in the number of macrophages present. Among the cytokines and chemokines, the mRNA levels for MIP-1α [chemokine (c-c motif) ligand 3], TNF-α (tumor necrosis factor alpha), IL-12 (interleukin 12), and Cxcl1 [chemokine (c-x-c motif) ligand 1] showed the greatest increases ranging from 14- to 105-fold, but significant rises in the mRNA levels for IL-6 (interleukin 6) and Spp1 (secreted phosphoprotein 1) were also evident. For the cell surface proteins, modest but significant increases in mRNA levels were observed for CD69 (CD69 antigen), MAC1 (integrin alpha M), and TLR4 (Toll-like receptor 4) and pronounced increases for CD11c (integrin alpha x, 160-fold), a marker of type M1 macrophages, and CD14 (CD14 antigen, 43-fold), a marker of liver injury. Accompanying all of these changes in the lal−/− mice was a marked increase in their plasma ALT activity (Fig. 2E). At 50, 70, and 140 days of age this was elevated nearly 10-, 17- and 22-fold, respectively, in the mutant mice compared with matching lal+/+ controls.
Fig. 2.

Liver histology, lipid and triacylglycerol content, and mRNA expression levels for markers of macrophage presence, cytokines, and cell surface proteins in 49- to 52-day-old lal+/+ and lal−/− mice. These various parameters were measured as described. Additionally, plasma ALT activities were determined in mice of both genotypes at 50, 70, and 140 days of age. For the data in B and E, the measurements were in female mice. The liver histology (A), and triacylglycerol content (C) and mRNA expression analyses (D) were done in males. H&E, hematoxylin and eosin; ALT, alanine aminotransferase. Measurement bars in A equal 100 μm. The data in B, C, D, and E are means ± SE of data from 4–6 mice of each genotype. *Significantly different from value for matching lal+/+ controls (P < 0.05).
Young adult lal−/− mice manifest dramatic increases in tissue esterified cholesterol levels in their livers and multiple extrahepatic organs.
The data in Fig. 3A illustrate the pronounced increase in the EC concentrations in multiple organs of 50-day-old lal−/− mice. The magnitude of increase was most evident in the liver (∼100-fold) and spleen (∼35-fold) and least in the adrenals (1.9-fold), which normally contain much more EC than other organs. For the small intestine, testes, and lungs, the EC levels increased ∼15-, 8-, and 6-fold, respectively. In the kidneys the increase was ∼6-fold (data not shown). The total cholesterol concentration in the small intestine continued to rise in the lal−/− mice as they aged, with this reflecting almost entirely an expanding presence of EC (Fig. 3B). Comparable findings including age-related histological changes in the small intestine were described earlier by Du et al. (14).
Fig. 3.

Concentrations of esterified cholesterol (EC) and unesterified cholesterol in multiple organs of lal+/+ and lal−/− mice. The tissue cholesterol concentration data in various organs (A) are for 50-day-old male mice, whereas those specifically for the small intestine (B) are for female mice in the age range of 21–274 days. Values are means ± SE of data from 6 mice of each genotype (A), and from a minimum of 5 animals in each group (B), except at 52 days when there were 4 mice of each genotype, and also at 274 days when there were only 3 lal−/− mice. *Significantly different from value for matching lal+/+ controls (P < 0.05).
LAL deficiency leads to an increased rate of cholesterol synthesis in multiple organs, most particularly in the liver.
The data in Fig. 4 show the impact of LAL deficiency on the relative weights (Fig. 4A), cholesterol contents (Fig. 4B), and cholesterol synthesis rates (Fig. 4C) in multiple organs of 50-day-old female mice. In addition to marked hepatomegaly, the relative weights of the small intestine and spleen increased significantly in the lal−/− mice (Fig. 4A). In many of the organs, as anticipated from the tissue EC and UC concentration data for 50-day-old male mice (Fig. 3A), the total cholesterol content (mg/organ) was significantly elevated (Fig. 4B). The cholesterol synthesis rate (also expressed per whole organ) was significantly increased in the lal−/− mice in multiple organs, particularly the liver which was elevated 10.2-fold compared with the rate in the lal+/+ controls (Fig. 4C). This marked genotypic difference in whole liver synthesis resulted from the combination of a 4.6-fold higher rate of synthesis per gram of liver, together with 2.2-fold greater liver mass in the LAL-deficient mice. For the lungs, kidneys, and spleen, the corresponding fold differences for whole organ synthesis in the lal−/− vs. lal+/+ mice were 3.8, 2.7, and 7.3, respectively. Although the data are not presented, the cholesterol content and synthesis rate in the entire residual carcass were also measured, thus allowing for the calculation of both whole animal cholesterol content and rate of synthesis. Expressed as milligrams per mouse, the whole animal cholesterol content of the lal−/− and lal+/+ mice was 210 ± 9 and 50 ± 1, respectively (P < 0.05). The matching values for whole animal cholesterol synthesis (nmol·h−1·mouse−1) were 9,747 ± 320 vs. 2,897 ± 269, in the lal−/− and lal+/+ mice, respectively (P < 0.05). Unlike the rate of sterol synthesis, that for fatty acid synthesis in the liver (μmol·h−1·g−1) did not show a genotypic difference when expressed per gram of tissue (28.1 ± 1.8 in the lal−/− mice vs. 30.3 ± 2.9 in their lal+/+ controls). On a whole liver basis, fatty acid synthesis in the lal−/− mice exceeded that in their lal+/+ littermates by 2.1-fold (P < 0.05) (data not shown).
Fig. 4.

Relative weights (A), total cholesterol contents (B), and rates of cholesterol synthesis (C) for the liver and multiple other organs in 50-day-old female lal+/+ and lal−/− mice. Rates of cholesterol synthesis were measured in vivo as described. These rates (nmol·h−1·g−1) were multiplied by the respective whole organ weight to obtain synthesis in the entire organ. Similarly, whole organ cholesterol contents were calculated from the total cholesterol concentration (mg/g) multiplied by organ weight. Values are means ± SE of data for 6 mice of each genotype. *Significantly different from value for matching lal+/+ controls (P < 0.05).
Age-related increase in whole animal cholesterol synthesis and content in lal−/− mice is accounted for almost entirely by the high rate of cholesterol synthesis and expanded cholesterol content in the liver.
The striking genotypic difference in whole liver cholesterol synthesis found in the 50-day-old mice (Fig. 4C) prompted the measurement of the synthesis rate in the livers of mice at 21, 98, and 165 days. These data, along with those for the 50-day-old mice, are shown in Fig. 5A. All values are presented as nanomoles per h our per gram wet weight of liver. The greatest genotypic difference, was seen at 21 days where the value for the lal−/− mice was ∼20-fold greater than in their lal+/+ littermates. Although the ingestion of milk over the preceding 21 days had a suppressive effect on hepatic sterol synthesis in the lal+/+ mice, this was clearly not the case in their LAL-deficient littermates. The rates of cholesterol synthesis in all the nonhepatic organs combined for these same mice are shown in Fig. 5B. Compared with those for the liver, these rates, particularly in the lal−/− mice, were much lower. Nevertheless, at all four ages, the rate in the LAL-deficient mice was significantly higher than in their matching lal+/+ controls. The data in Fig. 5, A and B, together with the corresponding weights for the liver and remaining organs combined, were used to calculate the rate of whole animal cholesterol synthesis and the fraction of it that occurred in the liver (Fig. 5C). This fraction is denoted by the cross hatching within each histogram. In the 21-day-old, newly weaned lal−/− mice, the liver accounted for 44.8 ± 1.6% of whole animal synthesis compared with only ∼6% in their lal+/+ littermates. At 50 and 165 days, the proportion of whole body synthesis occurring in the liver of the mutants increased to 68.2 ± 1.3 and 79.6 ± 0.3%, respectively. In the lal+/+ mice, whole body cholesterol synthesis after weaning stayed relatively constant, with the bulk of cholesterol synthesis occurring in the peripheral organs. Whole animal cholesterol content and the proportion of it within the liver were determined in the same mice used for the sterol synthesis measurements. As shown by the data in Fig. 5D, the age-related changes and genotypic differences in these contents followed a remarkably similar pattern to that seen for the cholesterol synthesis data. For example, in the 50-day and 165-day-old mutants, the proportion of whole body cholesterol contained within the liver was 63.3 ± 1.4 and 74.0 ± 0.9%, respectively. At an additional time point of 384 days, almost 80% of the cholesterol was in the liver. In contrast, across all ages, the proportion of whole animal cholesterol content present in the livers of the lal+/+ mice remained constant at ∼5 to 7%.
Fig. 5.

Rate of cholesterol synthesis in the liver vs. all extrahepatic organs combined and contribution of the liver to whole body cholesterol synthesis and content in lal+/+ and lal−/− mice at different ages. Cholesterol synthesis was measured in vivo as described in female mice at 21, 50, 98, and 165 days of age. The rate of synthesis in the liver and whole residual carcass was expressed 2 ways. In one, the rate was normalized per gram wet weight of tissue (nmol·h−1·g−1) (A and B), whereas in the other this value was multiplied by the respective whole organ weight to obtain synthesis in the entire liver vs. the remainder of the animal. The summation of these 2 values yielded synthesis in the whole mouse (nmol·h−1·animal−1) (C). Similarly, whole organ cholesterol contents were calculated from the total cholesterol concentration (mg/g) multiplied by organ weight (D). Values are means ± SE of data from 4–6 mice of each genotype. At 384 days there were 4 mice (2 males and 2 females) of each genotype. *Significantly different from value for matching lal+/+ controls (P < 0.05). nm, Not measured.
Young adult lal−/− mice manifest a moderate increase in fecal neutral sterol excretion but no change in fractional cholesterol absorption, fecal lipid content, or bile acid excretion.
In separate groups of 50-day-old male and female mice, various other parameters of cholesterol metabolism were measured that focused on determining how LAL deficiency might impact the main pathways that dictate the entry and disposition of cholesterol through the gastrointestinal tract. As shown in Fig. 6, within sex, there were no discernible differences between the lal−/− and lal+/+ mice in body weight (Fig. 6A), daily stool output (Fig. 6B), fecal lipid content (Fig. 6C), or fractional cholesterol absorption (Fig. 6D). However, fecal neutral sterol excretion rates in the lal−/− mice were significantly higher (P < 0.05) than in their sex-matched lal+/+ controls (Fig. 6E). Fecal bile acid excretion rates did not show a genotypic difference (Fig. 6F). These data were in turn used to address the critical question of what happened to all the additional cholesterol synthesized in the LAL-deficient mice. From the whole animal body cholesterol content data for the 50-day-old mice (Fig. 5D), it can be calculated that, since birth, the whole body cholesterol pool in the lal−/− and lal+/+ mice expanded at a rate of 4.2 and 1.0 mg per animal per day, respectively. Thus the daily net sequestration rate of cholesterol in the mutant mice averaged ∼3.2 mg per animal per day. This was not attributable to a genotypic difference in the amount of cholesterol entering the body pool through the intestinal absorption pathway because fractional cholesterol absorption was essentially the same in the lal−/− and lal+/+ mice (Fig. 6D). To obtain a broad estimate of the daily rate of whole body cholesterol synthesis, we used the data for the 21- and 50-day-old mice (Fig. 5C), together with the conversion factors given in materials and methods for determining amounts of cholesterol generated from the rates of incorporation of [3H]water into sterols. The difference in the rate of synthesis between the lal−/− and lal+/+ mice, based on the data for these two time points, amounted to 3.7 mg·day−1·animal−1. This exceeds the amount of cholesterol being sequestered (3.2 mg·animal−1·day−1) in the LAL-deficient mice by ∼0.5 mg·animal−1·day−1. However, this is the same amount of extra sterol excreted daily in the feces by the female lal−/− mice (Fig. 6E).
Fig. 6.

Fecal lipid content, intestinal cholesterol absorption, and rates of fecal neutral sterol and bile acid excretion in 50-day-old lal+/+ and lal−/− mice. Two matching sets of mice were used for these measurements, one for fractional cholesterol absorption (D) and fecal neutral sterol excretion (E), the other for quantitation of fecal lipid content (C) and the rate of fecal bile acid excretion (F). Values are means ± SE of data for 6 mice of each genotype. *Significantly different from value for matching lal+/+ controls (P < 0.05). M, male; F, female.
LAL deficiency results in marked changes in the mRNA expression level for multiple genes involved in regulating hepatic lipid metabolism.
The metabolic data for the liver prompted the measurement of the relative level of expression of mRNA for a constellation of genes that play key roles in intrahepatic sterol and fatty acid metabolism (Fig. 7). These measurements were made in 50-day-old male lal−/− mice and their matching lal+/+ controls. As anticipated, there was essentially no detectable mRNA for LIPA (Fig. 7A). The mRNA level for NPC2 (Fig. 7B) was elevated nearly fourfold whereas that for NPC1 (Fig. 7C) was unchanged. Consistent with the high rate of hepatic cholesterol synthesis in the lal−/− mice, their livers had moderate increases in mRNA levels for SREBP2 (Fig. 7D), HMG CoA SYN (Fig. 7E), and HMG CoA RED (Fig. 7F). In the case of ABCA1 (Fig. 7G), the mRNA level was unchanged whereas for both ABCG5 (Fig. 7H) and ABCG8 (Fig. 7I) it was reduced by at least 50%. SOAT2 (Fig. 7J) mRNA levels were unchanged. The marked increases in mRNA for SOAT1 (Fig. 7K) and ABCG1 (Fig. 7L) are consistent with the elevated level of mRNA for the macrophage marker, CD68 (Fig. 2D). For the transcription factor SREBP1c (Fig. 7M), the mRNA level was reduced by ∼60% in the lal−/− mice, whereas there was no discernable change in the level for either ACCa (Fig. 7N) or SCD1 (Fig. 7O).
Fig. 7.
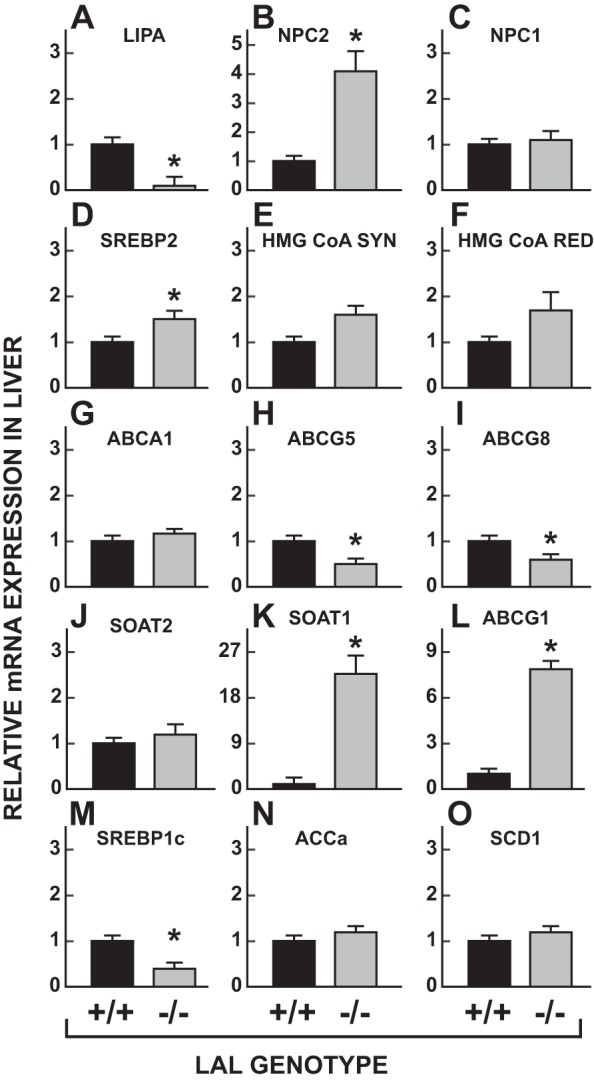
Relative mRNA levels for multiple genes in the livers of 50-day-old male lal+/+ and lal−/− mice. The livers used for these analyses were derived from the same mice used for the measurement of unesterified cholesterol (UC) and EC levels in multiple organs (Fig. 3A). The mRNA levels were normalized against the housekeeping gene cyclophilin and arithmetically adjusted to yield a unit of 1.0 for lal+/+ controls. The names of the genes studied are as follows: LIPA, lysosomal acid lipase (LAL); NPC2, Niemann-Pick type C2; NPC1, Niemann-Pick type C1; SREBP2, sterol regulatory element-binding protein-2; HMG CoA SYN, hydroxymethylglutaryl-CoA synthase; HMG CoA RED, hydroxymethylglutaryl-CoA reductase; ABCA1, ATP-binding cassette A1; ABCG5, ATP-binding cassette G5; ABCG8, ATP-binding cassette G8; ABCG1, ATP-binding cassette G1; SOAT1, sterol O-acyltransferase 1 (ACAT1); SOAT2, sterol O-acyltransferase 2 (ACAT2); SREBP1c, sterol regulatory element-binding protein-1c; ACCa, acetyl-CoA carboxylase; SCD1, steroyl-CoA desaturase 1. Values are means ± SE of data for 6 mice of each genotype. *Significantly different from value for matching lal+/+ controls (P < 0.05).
DISCUSSION
The present experiments extend the earlier work of Du et al. (13, 14), who described the development and major features of this model for CESD. Those formative studies were conducted on mice that were of a mixed 129/Sv:CF-1 background, whereas for the present experiments all mice were of the FVB/N strain. A comparison of the features of the lal−/− mice described here with those reported for the mutants on the 129/Sv:CF-1 background shows their phenotypes to be essentially identical. In each case the most striking feature of the mutants was their massive hepatomegaly and profound increase in liver EC content. The detailed pathology described by Du et al. for multiple organs in this model, particularly the liver, is complemented by the extensive mRNA data presented here (Figs. 2D and 7, K and L), which together attest to the marked level of inflammation and macrophage presence in the livers of 50-day-old lal−/− mice. The hepatocellular injury stemming from all of those adverse changes was reflected in a pronounced rise in plasma ALT activity, which continued to increase as the mutants aged (Fig. 2E).
Two other notable features of this mouse model, irrespective of strain background, are noteworthy. One is its dramatic change in body composition, apart from the enlargement of the liver, spleen, and small intestine. The other is its remarkable longevity. As documented by Du et al. (14), lal−/− mice show increasing loss of adipose tissue as they age, even though their caloric intake, relative to body weight, was found to marginally exceed that of their lal+/+ controls, at least at 4 mo of age. Despite the severity of their disease, the LAL-deficient mice have a surprisingly long life span. In the present studies, the lal−/− mice for both sexes combined had a median life span of 355 days (range was from 169 to 621 days). Our finding that the lal−/− females had a significantly longer life span than lal−/− males was unexpected because there were no clear sex-related differences in the measures of disease severity, in particular liver dysfunction, that were assessed. The life span of lal−/− mice far exceeds that of various mouse models for NPC1 deficiency (34, 39, 42). This in itself defines how different the consequences are when nearly all the excess cholesterol sequestered in the E/L compartment of every cell is esterified vs. when it is unesterified. In the latter case, the entrapment of UC in the central nervous system leads to a critical loss of Purkinje cells, progressive neurodegeneration, and early death (57). Thus, although enzyme replacement therapy is effective in CESD and Wolman disease, a study in a mouse model for NPC2 disease given enzyme replacement therapy showed that, despite a marked reduction in tissue UC content in several organs, there was little to no change in a number of regions of the brain (37). An alternate approach using 2-hydroxypropyl-β-cyclodextrin for UC mobilization continues to be explored (56).
In evaluating the biochemical and metabolic data generated in the present studies, a point regarding how they were normalized warrants emphasis. Traditionally, whole body cholesterol metabolism measurements in most models, and certainly in humans, are normalized per kilogram body weight. For LAL-deficient mice, this approach can be used up until they reach ∼50 days of age. However, after that the increasing divergence in the body weights of the mutant and wild-type mice means quantitative comparisons of metabolic parameters between them could be distorted if data were normalized per kilogram body weight instead of being expressed per whole animal. This applied particularly to the data from old lal−/− mice and their age-matched lal+/+ controls. For individual organ systems, most of the data were expressed on a whole organ basis and not per gram of tissue largely because of the genotypic differences in organ weight, particularly for the liver.
The tissue cholesterol content and synthesis data presented here provide important new insights into how the global deficiency of LAL and the consequent sequestration of unbridled amounts of EC by tissues throughout the body result in a dramatic compensatory upregulation of cholesterol synthesis, mainly in the liver, that in turn drives disease progression. For the tissue cholesterol concentration data (mg/g) in the lal−/− mice, several points should be made in comparing the degree of increase in EC levels among the different organs (Fig. 3A). One is that in LAL-deficient mice, the liver might reasonably be expected to show the greatest increase in EC levels given that, not only is it the principal site of clearance of low-density lipoproteins (LDL) and very low density lipoprotein remnants, but it also receives all of the chylomicron cholesterol that originates in the small intestine (8, 10, 17). In these various classes of lipoprotein particles, the bulk of the cholesterol is esterified (5). Hence, in the absence of functional LAL, large amounts of EC would be expected to become sequestered in the E/L compartment of hepatocytes, in addition to the EC contained within macrophages reaching the liver from other sites. In one case of a young adult CESD patient, the hepatic EC concentration was 94.6 mg/g vs. only ∼1 mg/g in healthy subjects (26, 48). In the 50-day-old lal−/− mice it was 48.4 mg/g (Fig. 3A).
Just as the liver and spleen manifest the greatest increase in UC concentration in NPC1 deficiency (32, 59), this was also the case for EC sequestration in the lal−/− mice at 50 days of age. Although the increase in EC levels in the small intestine of these mice also was very pronounced (Fig. 3B), the amount of EC present in this organ at any age cannot be taken as a true measure of its cumulative entrapment because of continual sloughing of cells from the mucosal surface (31). The fold increase in the EC concentration in the testes of the lal−/− mice was much greater than it was in their adrenal glands, but this largely reflected the fact that the bulk of adrenal cholesterol is ordinarily esterified as shown by the values for the lal+/+ mice. Although the lung phenotype in the LAL-deficient mouse has been described in detail (29, 30), the EC concentration data for this organ (Fig. 3A) appear to be the first quantitative measure of how much the cholesterol level in pulmonary tissue rises in the young adult lal−/− mouse. The studies by Du et al. found that the plasma total cholesterol concentration did not a show a genotypic difference (13, 14). Nevertheless, the mutant mice did manifest a shift in lipoprotein composition, with an increase in the level of cholesterol in the LDL fraction (14). That finding was consistent with the dyslipidemia that has been documented in patients with CESD (43).
The cholesterol synthesis data show that just as the highest degree of EC sequestration resulting from LAL deficiency was seen in the liver, this was also the organ where most of the compensatory increase in sterol biosynthesis occurred. The dramatic elevation in hepatic cholesterol synthesis, which was not accompanied by any change in the rate of fatty acid synthesis per gram of liver, was clearly driven by a profound deficit in the supply of lipoprotein-derived cholesterol to the metabolically active pool in the cytosolic compartment of the hepatocytes. The markedly lower mRNA expression levels of ABCG5 (Fig. 7H) and ABCG8 (Fig. 7I) are, in themselves, a clear measure of this deficit. Nevertheless, the additional cholesterol synthesized within the liver was sufficient to maintain normal rates of bile acid synthesis given that fecal acidic sterol excretion in the lal−/− mice was the same as in their lal+/+ controls (Fig. 6F). In fact, there was a modest overproduction of cholesterol judging by the elevated levels of fecal neutral sterol excretion in the mutants (Fig. 6E). However, this extra cholesterol in fecal sterols was far less than the quantity of newly synthesized cholesterol that became entrapped daily as ester in the E/L compartment. On the basis of the rates of whole animal cholesterol synthesis and sequestration that were determined in the mutants up to 50 days of age, ∼80% of newly synthesized cholesterol ultimately became sequestered.
The finding that in the small intestine of the lal−/− mice there was only a modest increase in sterol synthesis, but a marked rise in tissue EC content, warrants comment. Ordinarily, most of the cholesterol requirement of enterocytes is met through the uptake of sterol from luminal sources via the NPC1L1-mediated pathway, which does not involve the E/L compartment (23, 58). When this pathway is disrupted by the sterol absorption inhibitor, ezetimibe, there is a marked compensatory increase in intestinal cholesterol synthesis (45). In the mouse at least, comparatively little cholesterol in the small intestine is derived from the clearance of LDL (38). Hence, in LAL deficiency one would expect only a small increase in intestinal cholesterol synthesis to compensate for the entrapment of LDL-derived EC. Indeed, this was found to be the case (Fig. 4C). The origin of the EC in the small intestine of lal−/− mice as they age becomes less clear because of the increasing presence of EC-laden macrophages (14).
Several of the findings for the LAL-deficient mouse parallel those already documented for a mouse model of NPC1 deficiency. However, in that disorder neither the level of sequestration of cholesterol nor the compensatory increase in hepatic cholesterol synthesis is as pronounced as in lal−/− mice. For example, in npc1−/− mice (on a BALB/c background) at 49 days of age, the whole animal sterol pool expands to 107 ± 3 mg/animal compared with 52 ± 2 mg/animal in their npc1+/+ controls (42). This represents a daily sequestration rate in the NPC1 mutants of ∼1.1 mg per animal per day (32, 59). In the 50-day-old lal−/− mice, the entrapment rate of EC amounts to 3.2 mg of sterol per animal per day. The substantially higher rates of cholesterol sequestration in the LAL- vs. NPC1-deficient mice cannot be attributed to strain differences because we have made such measurements recently in lal−/− mice on a BALB/c background and found essentially the same cholesterol pool sizes in the liver and whole animal as were obtained for the LAL-deficient mice on an FVB background (A. Lopez, K. Posey, and S. Turley, unpublished observations). In npc1−/− mice, compared with matching npc1+/+ controls, the rate of cholesterol synthesis in the whole liver and whole animal is elevated by twofold at most (32, 59). In lal−/− vs. lal+/+ mice at 50 days of age, the increase in sterol synthesis is substantially more than twofold, especially for the liver (Fig. 5A). This difference is consistent with the greater level of sterol entrapment and perceived cellular cholesterol deficit in the lal−/− mice than in their npc1−/− counterparts. As previously discussed, the greater sequestration rates in the lal−/− mice might reflect the fact that EC is contained within the lysosomal lumen, whereas in the case of UC its ability to interact with other lipids in the membranes of the E/L compartment, and to diffuse through such membranes, may facilitate the exit of some UC (41). The possibility of such leakage has been previously raised by Peake and Vance (40).
The exceptionally high rates of sterol synthesis in the lal−/− mice raise the question of how they might respond to statin treatment. Unfortunately, given the evidence that statins increase the rate of hepatic cholesterol synthesis in mice (7), little would be gained from such a study. There are reports of CESD patients being given statin therapy alone and in combination with ezetimibe primarily for management of their dyslipidemia (18, 19, 43, 50). In the mouse model described here, ezetimibe treatment alone markedly reduced hepatic EC levels and improved liver function (6). This finding raises the question of how genetic deletion of the intestinal sterol transporter Niemann-Pick C1 Like 1, which is the target of ezetimibe, or of the cholesterol esterifying enzyme, sterol O-acyltransferase 2, in the LAL-deficient mouse might impact disease progression in this model (24, 36). The ultimate strategy, however, for treatment of CESD is enzyme replacement therapy that has been developed (1, 21). Several studies in the mouse CESD model have demonstrated remarkable efficacy of enzyme replacement therapy including in animals with advanced disease (12, 15, 49).
Finally, of the many potential uses for the LAL-deficient mouse, one might be to apply it to learning more about the pathogenesis of lipid malabsorption in this disorder. In the present studies fecal lipid content was unchanged in the lal−/− mice. However, these measurements were made in mice at a relatively early stage of disease that had been maintained on a low-fat chow diet. A case report of lipid malabsorption in a CESD patient (11) suggests that detailed lipid absorption measurements in older LAL-deficient mice fed lipid-rich diets are warranted.
GRANTS
This research was supported by National Institutes of Health Grants R01HL009610 (S. D. Turley), R01DK078592 (J. J. Repa), and T32-GM-007062 (A. M. Taylor).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.A., K.S.P., J.J.R., and S.D.T. conception and design of research; A.A., A.M.L., K.S.P., A.M.T., D.K.B., and S.D.T. performed experiments; A.A., A.M.L., K.S.P., A.M.T., J.J.R., and S.D.T. analyzed data; A.A., A.M.L., K.S.P., A.M.T., J.J.R., D.K.B., and S.D.T. interpreted results of experiments; A.A., A.M.L., K.S.P., and S.D.T. prepared figures; A.A., A.M.L., K.S.P., J.J.R., and S.D.T. drafted manuscript; A.A., A.M.L., K.S.P., A.M.T., J.J.R., D.K.B., and S.D.T. edited and revised manuscript; A.A., A.M.L., K.S.P., A.M.T., J.J.R., D.K.B., and S.D.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We are indebted to Drs. Gregory Grabowski and Hong Du for the generous gift of LAL heterozygous breeding stock, and also for providing us with the genotyping protocol for LAL mice as well as helpful information regarding their reproduction and phenotype. Mario Saucedo, Carolyn Crumpton, Jennifer Burg, Stephen Ostermann, and Monti Schneiderman provided excellent technical assistance.
Present Address for A. M. Taylor: Department of Physiology, University of Texas Health Science Center San Antonio, San Antonio, Texas, United States.
REFERENCES
- 1.Balwani M, Breen C, Enns GM, Deegan PB, Honzik T, Jones S, Kane JP, Malinova V, Sharma R, Stock EO, Valayannopoulos V, Wraith JE, Burg J, Eckert S, Schneider E, Quinn AG. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology 58: 950–957, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beltroy EP, Richardson JA, Horton JD, Turley SD, Dietschy JM. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology 42: 886–893, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an underdiagnosed disease. J Hepatol 58: 1230–1243, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Brown MS, Goldstein JL. Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci USA 76: 3330–3337, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman MJ. Comparative analysis of mammalian plasma lipoproteins. Methods Enzymol 128: 70–143, 1986 [DOI] [PubMed] [Google Scholar]
- 6.Chuang JC, Lopez AM, Posey KS, Turley SD. Ezetimibe markedly attenuates hepatic cholesterol accumulation and improves liver function in the lysosomal acid lipase-deficient mouse, a model for cholesteryl ester storage disease. Biochem Biophys Res Commun 443: 1073–1077, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chuang JC, Valasek MA, Lopez AM, Posey KS, Repa JJ, Turley SD. Sustained and selective suppression of intestinal cholesterol synthesis by Ro 48-8071, an inhibitor of 2,3-oxidosqualene:lanosterol cyclase, in the BALB/c mouse. Biochem Pharmacol 88: 351–363, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooper AD. Hepatic uptake of chylomicron remnants. J Lipid Res 38: 2173–2192, 1997 [PubMed] [Google Scholar]
- 9.Dietschy JM, Spady DK. Measurement of rates of cholesterol synthesis using tritiated water. J Lipid Res 25: 1469–1476, 1984 [PubMed] [Google Scholar]
- 10.Dietschy JM, Turley SD, Spady DK. Role of liver in the maintenance of cholesterol and low density lipoprotein homeostasis in different animal species, including humans. J Lipid Res 34: 1637–1659, 1993 [PubMed] [Google Scholar]
- 11.Drebber U, Andersen M, Kasper HU, Lohse P, Stolte M, Dienes HP. Severe chronic diarrhea and weight loss in cholesteryl ester storage disease: a case report. World J Gastroenterol 11: 2364–2366, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Du H, Cameron TL, Garger SJ, Pogue GP, Hamm LA, White E, Hanley KM, Grabowski GA. Wolman disease/cholesteryl ester storage disease: efficacy of plant-produced human lysosomal acid lipase in mice. J Lipid Res 49: 1646–1657, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du H, Duanmu M, Witte D, Grabowski GA. Targeted disruption of the mouse lysosomal acid lipase gene: long-term survival with massive cholesteryl ester and triglyceride storage. Hum Mol Genet 7: 1347–1354, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. Lysosomal acid lipase-deficient mice: depletion of white and brown fat, severe hepatosplenomegaly, and shortened life span. J Lipid Res 42: 489–500, 2001 [PubMed] [Google Scholar]
- 15.Du H, Heur M, Witte DP, Ameis D, Grabowski GA. Lysosomal acid lipase deficiency: correction of lipid storage by adenovirus-mediated gene transfer in mice. Hum Gene Ther 13: 1361–1372, 2002 [DOI] [PubMed] [Google Scholar]
- 16.Erickson SK. Nonalcoholic fatty liver disease. J Lipid Res 50 Suppl: S412–S416, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foley EM, Gordts PL, Stanford KI, Gonzales JC, Lawrence R, Stoddard N, Esko JD. Hepatic remnant lipoprotein clearance by heparan sulfate proteoglycans and low-density lipoprotein receptors depend on dietary conditions in mice. Arterioscler Thromb Vasc Biol 33: 2065–2074, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fouchier SW, Defesche JC. Lysosomal acid lipase A and the hypercholesterolaemic phenotype. Curr Opin Lipidol 24: 332–338, 2013 [DOI] [PubMed] [Google Scholar]
- 19.Ginsberg HN, Le NA, Short MP, Ramakrishnan R, Desnick RJ. Suppression of apolipoprotein B production during treatment of cholesteryl ester storage disease with lovastatin. Implications for regulation of apolipoprotein B synthesis. J Clin Invest 80: 1692–1697, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem 250: 8487–8495, 1975 [PubMed] [Google Scholar]
- 21.Grabowski G. Therapy for lysosomal acid lipase deficiency: replacing a missing link. Hepatology 58: 850–852, 2013 [DOI] [PubMed] [Google Scholar]
- 22.Grabowski GA, Du H. Lysosomal acid lipase deficiencies: the Wolman disease/cholesteryl ester storage disease spectrum [Online]. McGraw-Hill. http://ommbid.mhmedical.com/content.aspx?bookid=474§ionid=45374143. [July 1, 2014]. [Google Scholar]
- 23.Jia L, Betters JL, Yu L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol 73: 239–259, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jia L, Ma Y, Rong S, Betters JL, Xie P, Chung S, Wang N, Tang W, Yu L. Niemann-Pick C1-Like 1 deletion in mice prevents high-fat diet-induced fatty liver by reducing lipogenesis. J Lipid Res 51: 3135–3144, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuriyama M, Yoshida H, Suzuki M, Fujiyama J, Igata A. Lysosomal acid lipase deficiency in rats: lipid analyses and lipase activities in liver and spleen. J Lipid Res 31: 1605–1612, 1990 [PubMed] [Google Scholar]
- 26.Kwiterovich PO, Jr, Sloan HR, Fredrickson DS. Glycolipids and other lipid constituents of normal human liver. J Lipid Res 11: 322–330, 1970 [PubMed] [Google Scholar]
- 27.Lambert JE, Ramos-Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 146: 726–735, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Repa JJ, Valasek MA, Beltroy EP, Turley SD, German DC, Dietschy JM. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J Neuropathol Exp Neurol 64: 323–333, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Lian X, Yan C, Qin Y, Knox L, Li T, Du H. Neutral lipids and peroxisome proliferator-activated receptor-γ control pulmonary gene expression and inflammation-triggered pathogenesis in lysosomal acid lipase knockout mice. Am J Pathol 167: 813–821, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lian X, Yan C, Yang L, Xu Y, Du H. Lysosomal acid lipase deficiency causes respiratory inflammation and destruction in the lung. Am J Physiol Lung Cell Mol Physiol 286: L801–L807, 2004 [DOI] [PubMed] [Google Scholar]
- 31.Lipkin M. Proliferation and differentiation of gastrointestinal cells in normal and disease states. In: Physiology of the Gastrointestinal Tract, edited by Johnson LR. New York: Raven, 1981, p. 145–167 [Google Scholar]
- 32.Liu B, Ramirez CM, Miller AM, Repa JJ, Turley SD, Dietschy JM. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J Lipid Res 51: 933–944, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu B, Xie C, Richardson JA, Turley SD, Dietschy JM. Receptor-mediated and bulk-phase endocytosis cause macrophage and cholesterol accumulation in Niemann-Pick C disease. J Lipid Res 48: 1710–1723, 2007 [DOI] [PubMed] [Google Scholar]
- 34.Maue RA, Burgess RW, Wang B, Wooley CM, Seburn KL, Vanier MT, Rogers MA, Chang CC, Chang TY, Harris BT, Graber DJ, Penatti CA, Porter DM, Szwergold BS, Henderson LP, Totenhagen JW, Trouard TP, Borbon IA, Erickson RP. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum Mol Genet 21: 730–750, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Muntoni S, Wiebusch H, Jansen-Rust M, Rust S, Seedorf U, Schulte H, Berger K, Funke H, Assmann G. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol 27: 1866–1868, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Nguyen TM, Sawyer JK, Kelley KL, Davis MA, Rudel LL. Cholesterol esterification by ACAT2 is essential for efficient intestinal cholesterol absorption: evidence from thoracic lymph duct cannulation. J Lipid Res 53: 95–104, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen GK, Dagnaes-Hansen F, Holm IE, Meaney S, Symula D, Andersen NT, Heegaard CW. Protein replacement therapy partially corrects the cholesterol-storage phenotype in a mouse model of Niemann-Pick type C2 disease. PLoS One 6: e27287, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osono Y, Woollett LA, Herz J, Dietschy JM. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J Clin Invest 95: 1124–1132, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parra J, Klein AD, Castro J, Morales MG, Mosqueira M, Valencia I, Cortes V, Rigotti A, Zanlungo S. Npc1 deficiency in the C57BL/6J genetic background enhances Niemann-Pick disease type C spleen pathology. Biochem Biophys Res Commun 413: 400–406, 2011 [DOI] [PubMed] [Google Scholar]
- 40.Peake KB, Vance JE. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett 584: 2731–2739, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Ramirez CM, Liu B, Aqul A, Taylor AM, Repa JJ, Turley SD, Dietschy JM. Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J Lipid Res 52: 688–698, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramirez CM, Liu B, Taylor AM, Repa JJ, Burns DK, Weinberg AG, Turley SD, Dietschy JM. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr Res 68: 309–315, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reiner Z, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, Jones S, Coric M, Calandra S, Hamilton J, Eagleton T, Ros E. Lysosomal acid lipase deficiency: an under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis 235: 21–30, 2014 [DOI] [PubMed] [Google Scholar]
- 44.Repa JJ, Lund EG, Horton JD, Leitersdorf E, Russell DW, Dietschy JM, Turley SD. Disruption of the sterol 27-hydroxylase gene in mice results in hepatomegaly and hypertriglyceridemia. Reversal by cholic acid feeding. J Biol Chem 275: 39685–39692, 2000 [DOI] [PubMed] [Google Scholar]
- 45.Repa JJ, Turley SD, Quan G, Dietschy JM. Delineation of molecular changes in intrahepatic cholesterol metabolism resulting from diminished cholesterol absorption. J Lipid Res 46: 779–789, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Schwarz M, Russell DW, Dietschy JM, Turley SD. Marked reduction in bile acid synthesis in cholesterol 7α-hydroxylase-deficient mice does not lead to diminished tissue cholesterol turnover or to hypercholesterolemia. J Lipid Res 39: 1833–1843, 1998 [PubMed] [Google Scholar]
- 47.Scott SA, Liu B, Nazarenko I, Martis S, Kozlitina J, Yang Y, Ramirez C, Kasai Y, Hyatt T, Peter I, Desnick RJ. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology 58: 958–965, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sloan HR, Fredrickson DS. Enzyme deficiency in cholesteryl ester storage disease. J Clin Invest 51: 1923–1926, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Y, Xu YH, Du H, Quinn B, Liou B, Stanton L, Inskeep V, Ran H, Jakubowitz P, Grilliot N, Grabowski GA. Reversal of advanced disease in lysosomal acid lipase deficient mice: a model for lysosomal acid lipase deficiency disease. Mol Genet Metab 112: 229–241, 2014 [DOI] [PubMed] [Google Scholar]
- 50.Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegele RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis 4: 26, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor AM, Liu B, Mari Y, Liu B, Repa JJ. Cyclodextrin mediates rapid changes in lipid balance in Npc1-/- mice without carrying cholesterol through the bloodstream. J Lipid Res 53: 2331–2342, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turley SD, Daggy BP, Dietschy JM. Effect of feeding psyllium and cholestyramine in combination on low density lipoprotein metabolism and fecal bile acid excretion in hamsters with dietary-induced hypercholesterolemia. J Cardiovasc Pharmacol 27: 71–79, 1996 [DOI] [PubMed] [Google Scholar]
- 53.Turley SD, Schwarz M, Spady DK, Dietschy JM. Gender-related differences in bile acid and sterol metabolism in outbred CD-1 mice fed low- and high-cholesterol diets. Hepatology 28: 1088–1094, 1998 [DOI] [PubMed] [Google Scholar]
- 54.Turley SD, Valasek MA, Repa JJ, Dietschy JM. Multiple mechanisms limit the accumulation of unesterified cholesterol in the small intestine of mice deficient in both ACAT2 and ABCA1. Am J Physiol Gastrointest Liver Physiol 299: G1012–G1022, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Valasek MA, Repa JJ. The power of real-time PCR. Adv Physiol Educ 29: 151–159, 2005 [DOI] [PubMed] [Google Scholar]
- 56.Vance JE, Karten B. Niemann-Pick C disease and mobilization of lysosomal cholesterol by cyclodextrin. J Lipid Res 55: 1609–1621, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 5: 16, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang DQ. Regulation of intestinal cholesterol absorption. Annu Rev Physiol 69: 221–248, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Xie C, Turley SD, Pentchev PG, Dietschy JM. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol Endocrinol Metab 276: E336–E344, 1999 [DOI] [PubMed] [Google Scholar]
- 60.Xie X, Brown MS, Shelton JM, Richardson JA, Goldstein JL, Liang G. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc Natl Acad Sci USA 108: 15330–15335, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]