

Abstract
We report a new inflammatory activity for extracellular d-dopachrome tautomerase (D-DT), the recruitment of neutrophils to the lung on D-DT intratracheal installation of C57BL/6J mice with an EC50 of 5.6 μg. We also find that D-DT and macrophage migration inhibitory factor (MIF) have additive effects in neutrophil recruitment. Although the tautomerase site of D-DT and its homologue MIF are biophysically very different, 4-iodo-6-phenylpyrimidine (4-IPP) forms a covalent bond with Pro-1 of both proteins, resulting in a 6-phenylpyrimidine (6-PP) adduct. Recruitment of neutrophils to the lung for the 6-PP adducts of D-DT and MIF are reduced by ∼50% relative to the apo proteins, demonstrating that an unmodified Pro-1 is important for this activity, but there is no cooperativity in inhibition of the proteins together. The differences in the binding mode of the 6-PP adduct for D-DT was determined by crystallographic studies at 1.13 Å resolution and compared to the structure of the MIF–6-PP complex. There are major differences in the location of the 6-PP adduct to the D-DT and MIF active sites that provide insight into the lack of cooperativity by 4-IPP and into tuning the properties of the covalent inhibitors of D-DT and MIF that are necessary for the development of therapeutic small molecules against neutrophil damage from lung infections such as Pseudomonas aeruginosa in cystic fibrosis and immunocompromised patients.—Rajasekaran, D., Zierow, S., Syed, M., Bucala, R., Bhandari, V., Lolis, E. J. Targeting distinct tautomerase sites of D-DT and MIF with a single molecule for inhibition of neutrophil lung recruitment.
Keywords: covalent inhibitor, 4-IPP
d-dopachrome tautomerase (D-DT) is a homologue of macrophage migration inhibitory factor (MIF), whose eponymous activity was described nearly 50 yr ago (1, 2), but is not nearly as well characterized as MIF. These proteins belong to the tautomerase family of proteins, which utilize a proline at the N terminus as an enzymatic base and have evolved from microbes to mammals (3). Pro-1 is found in a cleft between subunits that oligomerize into dimers (4), trimers (5), or hexamers (3) for various proteins in this family. D-DT and MIF are constitutively present in the cytosol and are exported from cells during infection or cellular stress, to activate receptors and initiate various biological responses. Extracellular D-DT and MIF bind to the receptor CD74 and recruit CD44, which is responsible for intracellular signaling (6–8). Recent studies have indicated that these two proteins have cooperative effects, including detrimental effects in cancer (7, 9, 10). These activities are decreased when either D-DT or MIF alone or both proteins together are reduced by siRNA. In a study on renal clear-cell carcinoma, D-DT and MIF showed additive protumorigenic effects, with D-DT exhibiting a more dramatic in vitro and in vivo effect (11). Knockdown of either D-DT or MIF did not have any inhibitory effect on Akt phosphorylation on the RCC4 renal carcinoma cell line. Only knockdown of both proteins resulted in inhibition of Akt phosphorylation, a phenomenon that is not observed in ERK-1/2 phosphorylation. These findings suggest that inhibiting both D-DT and MIF would prove superior for improving therapeutic efficacy in diseases associated with both proteins.
The physiological substrates for D-DT and MIF are not known, but two substrate mimics were inadvertently identified during experiments of the membrane enzyme dopachrome tautomerase, which converts l-dopachrome to 5,6-dihydroxyindole-2-carboxylic acid (DHICA) and is a critical activity in the melanogenesis pathway (12). The nonphysiological molecules d-dopachrome and d- and l-dopachrome methyl esters are decarboxylated to 5,6-dihydroxyindole (DHI) by D-DT, whereas MIF catalyzes the tautomerization of d-dopachrome and l-dopachrome methyl ester (13, 14). D-DT and MIF also share a keto-enol tautomerase activity for 3-(4-hydroxyphenyl) pyruvate (HPP). The use of similar ligands is notable, given the low (<30%) sequence identity, particularly at the catalytic site. The relationship between the catalytic site of MIF and receptor-mediated activities has been under intense scrutiny (15–18). Some complexes of MIF and small-molecule inhibitors of the active site function as CD74 antagonists, whereas others do not. There are no known inhibitors of D-DT.
The crystal structure of MIF with the substrate HPP (19) was used to design the MIF competitive inhibitor (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester (ISO-1; ref. 20). This inhibitor reduces inflammation and is an effective therapeutic in a variety of mouse models of disease (21, 22). Surprisingly, ISO-1 does not inhibit D-DT, even though HPP is a substrate. However, the MIF covalent inhibitor 4-iodo-6-phenylpyrimidine (4-IPP) also forms a covalent bond with Pro-1 of D-DT that leads to a 6-phenylpyrimidine (6-PP) adduct (23). We report on the kinetics of keto-enol HPP activity by D-DT and inhibition kinetics of 4-IPP. We also define a new biological activity for D-DT, neutrophil recruitment to lungs, which is additive with MIF. Neutrophil recruitment is reduced by ∼50% with the 6-PP adduct for either D-DT or MIF (compared to apo protein), indicating that unmodified Pro-1 contributes to this activity. Surprisingly, reduction in neutrophil recruitment remains at ∼50% when D-DT–6-PP and MIF–6-PP are used together. Structural studies reveal that the 6-PP adduct adopts a different conformation from MIF in the D-DT active site that correlates with inhibition kinetics and the biophysical property of each active site and also provides an explanation for the lack of cooperativity with the 6-PP adducts in lung neutrophil recruitment. 4-IPP is a potential lead compound for diseases such as Pseudomonas aeruginosa in critically ill patients who have lung damage due to neutrophil recruitment by D-DT and MIF (24–26).
MATERIALS AND METHODS
Cells and reagents
ISO-1 and HPP were purchased from Sigma-Aldrich (Milwaukee, WI, USA). 4-IPP was purchased from Specs (Delft, The Netherlands). All other chemical reagents were purchased from Sigma-Aldrich.
Expression and purification of D-DT
Cloning, expression, and purification were performed as described previously (7). Briefly, the cDNA for human or murine D-DT (hD-DT and mD-DT, respectively) was cloned into pET 22b(+), transformed, sequenced, and expressed in Escherichia coli BL21 DE3 cells. The cells were lysed in a buffer of 20 mM Tris and 20 mM NaCl, at pH 8.4 for hD-DT and at pH 7.4 for mD-DT, and purified by anion-exchange chromatography with a gradient of 20 mM to 1 M NaCl. The proteins were further purified on a C18 column with an acetonitrile gradient of 30–60% for hD-DT and 30–55% for mD-DT. The lyophilized proteins were refolded by using an established protocol for MIF and confirmed to be lipopolysaccharide (LPS) free (<0.1 EU/20 μg protein) (20).
Enzyme kinetics and inhibition kinetics
For HPP keto-enol activity, HPP in 50 mM ammonium acetate (pH 6.0) was incubated overnight at 4°C to produce the keto form that is highly favored under this condition. To determine the appropriate D-DT concentration for steady-state kinetics, we first evaluated concentrations of 0.025–0.1 μM D-DT. The enzymatic measurements at various concentrations of the HPP were in a mixture of D-DT and 0.435 M boric acid (pH 6.2) and were measured by monitoring the increase in absorbance at 306 nm, due to the formation of a complex between borate and the enol form of HPP. Competitive inhibition studies of D-DT by ISO-1 were assayed as described for MIF, with HPP as the substrate (27). The half-life for covalent inhibition was determined after incubation of 4-IPP [100 nM in dimethyl sulfoxide (DMSO), or DMSO alone] with D-DT (50 nM in 20 mM NaCl and 20 mM Tris, pH 7.5) at room temperature. At different time points, an aliquot was removed and added to a mixture containing 1.2 mM HPP and 424 mM borate at pH 6.2 for measuring the initial velocity of the HPP tautomerase activity. All kinetics experiments were measured at 1-s intervals with a spectrophotometer (Infinite M200 Spectrophotometer; Tecan Systems, San Jose, CA, USA). Curve fitting and analysis were performed with Prism software (GraphPad, San Diego, CA, USA).
Murine in vivo recruitment of neutrophils to the lung
Neutrophil recruitment to the lung of C57BL/6J mice (8–12 wk old) was studied by dose response of recombinant D-DT (0.1–100 μg/ml) in 50 μl of saline via direct intratracheal administration (28). MIF (1 μg/mouse), D-DT (10 μg/mouse), the combination of MIF (1 μg/mouse) and D-DT (10 μg/mouse), MIF–6-PP (1 μg/mouse), D-DT–6-PP (10 μg/mouse), and the combination of MIF–6-PP (1 μg/mouse) and D-DT–6-PP (10 μg/mouse) were assayed for neutrophil recruitment. After 4 h of intratracheal instillation, differential cell counts were quantified from the bronchoalveolar lavage (BAL) with the HEMA 3 stain set (Thermo Scientific, Waltham, MA, USA). A minimum of 200 cells was used for the differential cell count (29). The use of mice for these experiments was approved by the Institutional Animal Care and Use Committee at Yale University.
Crystallization, data collection, and processing
To obtain the structure of the D-DT adduct with 6-PP (D-DT–6-PP), we mixed the protein with a 10-fold molar excess of 4-IPP and incubated the mixture overnight. The hD-DT–6-PP complex was purified from excess 4-IPP and crystallized at 8 mg/ml by the hanging-drop method with 1:1 ratio of the reservoir solution containing 0.2 M NaCl, 25% PEG 3350, and 0.1 M Tris (pH 8.5), for 3–4 d at 293 K. Crystals for the mD-DT–6-PP complex were obtained by mixing 8 mg/ml protein with reservoir solution of 0.2 M NaCl, 1.0 M sodium citrate, and 0.1 M Tris (pH 7.0) at 1:1 ratio by hanging drop for 2 d at 277 K before transferring the plate to 293 K. The hD-DT–6-PP and mD-DT–6-PP crystals diffracted to 1.13 and 2.78 Å, and belonged to the P3 and P63 space group, respectively. The data were collected at beamline X25 of the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY, USA). All the data sets were processed and scaled with HKL2000.
D-DT (8 mg/ml) was screened with a 5-fold molar excess of HPP for cocrystallization. The crystals were obtained from a crystallization solution containing 0.2 M sodium tartrate dibasic dehydrate (pH 7.3) and 20% PEG 3350. Instead of HPP, these crystals contained tartrate in the active site.
Structure determination and analysis
The structures of hD-DT and mD-DT–6-PP were solved by molecular replacement (software program Phaser; ref. 30) using the apo D-DT structure [Protein Data Bank (PDB) code 1DPT; ref. 31]. The D-DT-tartrate structure was solved with protein coordinates of D-DT–6-PP. The initial models were refined using Refmac (32) and CNS (with NCS averaging; ref. 33) before adding coordinates for the 6-PP adduct or tartrate and water molecules (using Coot; ref. 34) into the electron density maps, followed by further refinement. Superposition of structures used the secondary structure method (SSM) from the CCP4 package (35).
Accession codes
The PDB codes for hD-DT–6-PP and mD-DT–6-PP are 3KAN and 3KER, respectively. The code for D-DT tartrate is 4Q3F.
RESULTS
D-DT enzyme kinetics and inhibition of activity
D-DT and MIF are unique among cytokines and chemokines, in that they contain a conserved catalytic site (36). The major difference in activity between these two proteins is the product of the nonphysiological substrate l-dopachrome methyl ester. D-DT catalyzes a decarboxylase reaction, and MIF catalyzes a tautomerization reaction. The hallmark for catalysis by the tautomerase superfamily is an N-terminal proline that functions as a general base for transferring a proton between two atoms. The different physicochemical properties in the D-DT active site decrease the transition state energy for decarboxylation of l-dopachrome methyl ester vs. tautomerization. In contrast, both proteins catalyze the keto-enol activity using the substrate HPP. D-DT and MIF catalyze HPP reaction with a Km and kcat of 1.13 mM and 62.4 s−1, and 1.20 mM and 38 s−1, respectively. The kcat/Km values between the two proteins are comparable with a 12% increase for D-DT compared to MIF. The same catalytic reaction for HPP and the use of d-dopachrome (or l-dopachrome methyl ester) as a substrate by D-DT and MIF are surprising, given the low sequence identity between the active sites, with only Pro-1, Lys-32, and Ile-64 being identical (Fig. 1A, B). Eight of 11 active site residues were altered, leading to significant differences. Among these differences are Arg-36 (Tyr in MIF), Arg-97 (for Asn), and Lys-109 (for Trp-108), which result in an electropositive potential for the D-DT active site (vs. a hydrophobic active site for MIF). Based on similar substrates for these two proteins but a significant difference in the electrostatic potential of the active sites, it was not clear whether previously identified human MIF inhibitors would have any effect on D-DT activity (20, 37–43). However, the substrate HPP had been used to design the prototypical MIF inhibitor ISO-1 (19, 20), which was expected to inhibit D-DT, but surprisingly had no effect, even at a 1000-fold molar excess (Fig. 1C). Given the significant differences at the two active sites and the inability of ISO-1 to inhibit enzymatic activity, it did not seem likely that an MIF inhibitor would also inhibit D-DT. However, the MIF irreversible inhibitor 4-IPP decreases the keto-enol activity by 60% at 100-fold molar excess of hD-DT and >90% at an equimolar ratio for human MIF (Fig. 1D). We also examined the effects of ISO-1 and 4-IPP on mD-DT. There was no decrease with ISO-1 (Supplemental Fig. S1A), a 75% decrease in the HPP keto-enol activity at a 5-fold molar excess of 4-IPP, and no activity at 100-fold molar excess (Supplemental Fig. S1B).
Figure 1.

Comparison of sequence, active site, and kinetics of inhibition in MIF and D-DT. A) Sequence alignment of hD-DT and hMIF. The 2 proteins have 30% identity, with a highly conserved secondary structure. B) Active site comparison. The enzymatic active site of D-DT is shown with green carbon atoms, with the active site of MIF in brown. The MIF active site, which contains many aromatic residues, is converted to an electropositive active site in D-DT (Arg-36, Arg-98, and Lys-109). The only aromatic group in the active site of D-DT is Phe-2, which replaces Met-2 in MIF. All the structural figures were made with PyMol (57). C) Effect of ISO-1 on tautomerase activity of MIF and D-DT. MIF was sensitive to ISO-1 and showed dose dependency, whereas D-DT was resistant to ISO-1 inhibition. ***P < 0.001. D) HPP tautomerization activity of MIF was reduced to <10% with an equimolar addition of 4-IPP to the reaction mix. The tautomerization activity of D-DT was reduced with 100-fold excess of 4-IPP to 40%. **P < 0.01, ***P < 0.001. E, F) Time-dependent inhibition by 4-IPP of D-DT (E) and MIF (F). The inhibitor was complexed with MIF or D-DT in a ratio of 1:2 and tested for its tautomerase activity at various time points. D-DT and MIF have half-lives of 63 and 34 min, respectively. Data represent means of 3 different experiments performed in triplicate.
To confirm that 6-PP was the covalent adduct, we used liquid chromatography/mass spectrometry after an overnight incubation of h- or mD-DT with an excess of 4-IPP. The increase in molecular mass was consistent with a nucleophilic attack on 4-IPP by the primary amine of Pro-1 to result in a 6-PP adduct with an iodo-leaving group (Supplemental Fig. S2). The half-life of the unmodified protein was determined by incubation of a 2-fold molar excess of 4-IPP with D-DT or MIF for various time points, followed by measurement of the remaining HPP tautomerase activity. MIF and D-DT were inhibited in a time-dependent fashion that fits a single exponential decay curve. The half-lives of unmodified hD-DT, hMIF, and mD-DT are 63, 34, and 60 min, respectively, reflecting the faster kinetics for forming a covalent bond with MIF (Fig. 1E, F and Supplemental Fig. S1C).
In vivo inhibition of D-DT-dependent neutrophil migration
MIF-induced neutrophil recruitment to the lung has been reported to be mediated by CD74 by a mechanism that involves activation of alveolar macrophages and the secretion of neutrophil-recruiting chemokines CXCL1 (KC) and CXCL2 (MIP-2) (18, 44). We examined whether D-DT is involved in this neutrophil recruitment to the lung. Intratracheal instillation of 0.1, 1.0, 10, and 100 μg of D-DT showed a dose-dependent response with an EC50 of 5.6 μg (Fig. 2A). We observed an additive effect when MIF and D-DT were used together at optimal concentrations (Fig. 2B). To compare the response of neutrophil recruitment to the lung in the presence of 4-IPP-modified D-DT and MIF, we prepared D-DT–6-PP and MIF–6-PP before intratracheal instillation. The use of a preformed covalent inhibitor would validate D-DT and MIF as therapeutic targets for neutrophil recruitment in lung disease, as there is no possibility of the off-target effect that might be observed with competitive inhibitors. Complete covalent formation of 10 μg D-DT–6-PP or 1 μg MIF–6-PP was validated by mass spectrometry after purification of the complexed proteins from excess 4-IPP. When each modified protein was used alone, there was a ∼50% reduction in lung neutrophil recruitment indicating that an uncomplexed Pro-1 is important for this biological activity (Fig. 2C). Interestingly, there was no cooperative decrease in neutrophil recruitment with the combination of D-DT–6-PP and MIF–6-PP instilled together (Fig. 2C).
Figure 2.
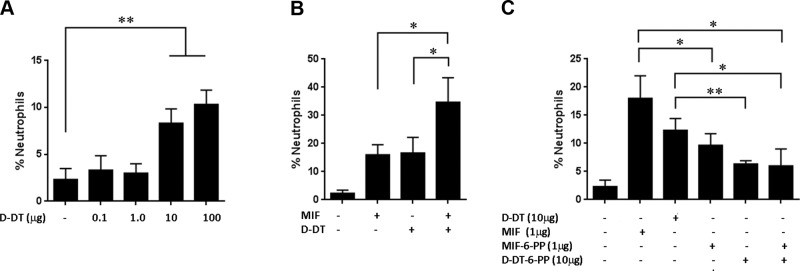
In vivo cell migration and inhibition by 4-IPP (or 6-PP protein adducts). A) D-DT-induced neutrophil recruitment in the lungs of mice was dose dependent. B) Additive effects by lung neutrophil recruitment of MIF and D-DT at 1 μg and 10 μg, respectively. C) Complex of 10 μg D-DT–6-PP and 1 μg MIF–6-PP each reduced neutrophil recruitment by 50%. Administration of both 10 μg D-DT–6-PP and 1 μg MIF–6-PP did not show cooperative effects. In vivo data represent mean ± se number of neutrophils from 6 female C57BL/6J mice, 8–10 wk of age. *P < 0.05, **P < 0.01; Student's t test.
Structural analysis of D-DT–6-PP
To better understand how 4-IPP modifies Pro-1 for two proteins with biophysically different active sites to inhibit enzymatic and functional activities, as well as to determine the specificity of 4-IPP over ISO-1, we cocrystallized D-DT–6-PP and compared the structure with the previously reported MIF–6-PP (23), MIF-ISO-1 (20), and MIF-HPP (19) structures. hD-DT and mD-DT–6-PP complexes were solved at resolutions of 1.13 and 2.78 Å and refined to R-factor/Rfree of 11.2/12.9% and 22/23.8%, respectively (Fig. 3A, Supplemental Fig. S3, and Table 1). The electron density for the 6-PP covalently bonded to Pro-1 is shown in Fig. 3B. The D-DT–6-PP complex superimposes on the apo D-DT structure (31) with a root mean square (RMS) difference of 0.2 Å and indicates that there are no significant global conformational changes. There are small local changes at the surface of the active site where 6-PP binds. Residues 33–36 that are part of the 30s loop connecting the first α helix to the second β strand moved closer to the helix, with the side-chain atoms of Cδ and Nε of Arg-36 moving away from the 6-PP adduct by 1.5 and 1.8 Å, respectively. Although atoms in the loop and Arg-36 side chain would not interfere with binding in their apo D-DT positions, the movement appeared to be a consequence of covalent bond formation with Pro-1. The RMS difference of the superposed Cα atoms for D-DT–6-PP and MIF–6-PP is 1.2 Å (Fig. 3C). The superposition indicates that 6-PP is in a different conformation with respect to the active site for the two proteins. The solvent-exposed surface of the superimposed proteins in Fig. 3C shows that the 6-PP adduct of D-DT does not bind within the active site, whereas the 6-PP group covalently bonded to Pro-1 occupies the active site (23) (Fig. 3D; for clarity, only the surface of D-DT is shown with 6-PP from both proteins). This difference may also explain the lack of cooperativity of D-DT–6-PP and MIF–6-PP in inhibition of neutrophil recruitment, because of the difference in affinity for CD74.
Figure 3.

Structure of D-DT–6-PP, comparison to MIF–6-PP, and structural models of ISO-1 and HPP in complex with D-DT. A) Representation of the 3-dimensional structure of trimeric hD-DT (green) in a covalent complex with 6-PP (pink). (See also Supplemental Fig. S3.) B) Electron density map (2Fo − Fc) of 6-PP covalently bonded to Pro-1 in D-DT contoured at 1σ. C) Superposition based on Cα atoms of human MIF–6-PP (gold–cyan) and hD-DT–6-PP (green–pink). The overall topology of the human MIF–6-PP complex superposed well with the hD-DT–6-PP complex. D) Different orientation of the 6-PP covalent adduct in D-DT (pink) and MIF (cyan) after superposition of the 2 proteins viewed down the 3-fold axis in the context of the solvent-exposed surface area. 6-PP of D-DT is outside the pocket, whereas the 6-PP of MIF is buried inside the active site. E, F) Electrostatic potential map of the D-DT (E) and MIF (F) active site complexed to 6-PP. The active site of D-DT has a greater positive potential compared to MIF (see Supplemental Fig. S4). G) Comparison of the active site atoms of D-DT (green) and MIF (cyan) with the 6-PP covalently bound form. MIF–6-PP aligned onto D-DT–6-PP showed different conformations of 6-PP buried inside the active site of MIF and at the surface of the pocket for D-DT. 6-PP was precluded from binding to the active site D-DT (green) structure due to close contacts by Phe-2 and Arg-98.
Table 1.
Summary of data collection and refinement statistics
| Parameter | Protein |
|
|---|---|---|
| hD-DT–6-PP | mD-DT–6-PP | |
| Data collection | ||
| Space group | P3 | P63 |
| Unit-cell parameters (Å) | ||
| a | 83.29 | 82.17 |
| b | 83.29 | 82.17 |
| c | 40.30 | 144.25 |
| Resolution (Å) | 50–1.13 (1.17-1.13) | 50–2.78 (2.88-2.78) |
| Unique reflections | 116,400 | 13,925 |
| Completeness (%) | 99.99 (94.2) | 99.8 (100) |
| Redundancy | 5.1 (9.1) | 4.3 (4.2) |
| Rmerge (%) | 12.5 (26.9) | 12.1 (51.4) |
| I/σ(I) | 20.9 (4.3) | 10 (2.1) |
| Solvent content (%) | 42 | 55 |
| Refinement | ||
| Reflections | 114,070 | |
| R factor (%) | 11.2 | 22.2 |
| Rfree (%) | 12.9 | 23.8 |
| Protein atoms | 2,655 | 3,632 |
| Water molecules | 496 | 8 |
| Ligand atoms | 38 | 64 |
| Average B factors (Å) | 7.09 | 42.72 |
| RMSD from ideal values | ||
| Bond lengths (Å) | 0.010 | 0.010 |
| Bond angles (deg) | 1.489 | 1.403 |
Values in the parentheses are for the highest resolution shell. Rmerge = ∑ |I(h,i) − 〈I(h)〉|/∑ I(h,i), where 〈I(h)〉 is the mean intensity of reflections. Rwork and Rfree were calculated from working and test set reflections. Bijvoet pairs are merged. RMSD, root mean square deviation.
As suggested by the differences in the residues in the active site, the electrostatic potential of D-DT was intensely positive and appeared to play a major role in the placement of the 6-PP outside the active site (Fig. 3E). MIF contained a positive potential at the surface of the active site due to Lys-32, but otherwise possessed aromatic and aliphatic residues that allowed aromatic 6-PP to bind within the active site (Fig. 3F). In addition to the role of the electrostatic potential, there were steric interactions precluding placement of 6-PP in the D-DT active site. Superposition of D-DT–6-PP and MIF–6-PP from Fig. 3C indicates there would be close contacts with Arg-98 and Phe-2 in the absence of a major conformational change if 6-PP bound within the D-DT active site (Fig. 3G). Consequently, the differences in the conformation of the 6-PP covalent adduct are associated with the biophysical properties of the 2 active sites (Fig. 3D–F and Supplemental Fig. S3), and the possibility of close contacts of 6-PP (or 4-IPP) with active site residues of D-DT.
The structural source of ISO-1 specificity for MIF and the use of HPP as a substrate for both proteins are important in understanding specificity at the tautomerase sites. Our attempt to cocrystallize D-DT with HPP was unsuccessful. Since we could not cocrystallize HPP, the Cα atoms of the structures of the MIF-HPP (19) and MIF-ISO-1 (20) complexes were superimposed on apo-D-DT, with RMS differences of 1.1 and 1.3 Å, respectively. Close examination of ISO-1 superimposed on D-DT revealed that the Arg-36 and Arg-98 side chains made contacts that were too close to ISO-1. Moreover, Phe-2 and ISO-1 occupied the same binding site and would require major rearrangements for ISO-1 for binding to D-DT (Fig. 4A), similar to that described for modeled 6-PP. In the model of HPP with D-DT there is close contact only with Phe-2, which required a conformational change of either HPP or Phe-2 in the active site (Fig. 4B). This change could be responsible for the 12% increase in kcat/Km for D-DT relative to MIF.
Figure 4.
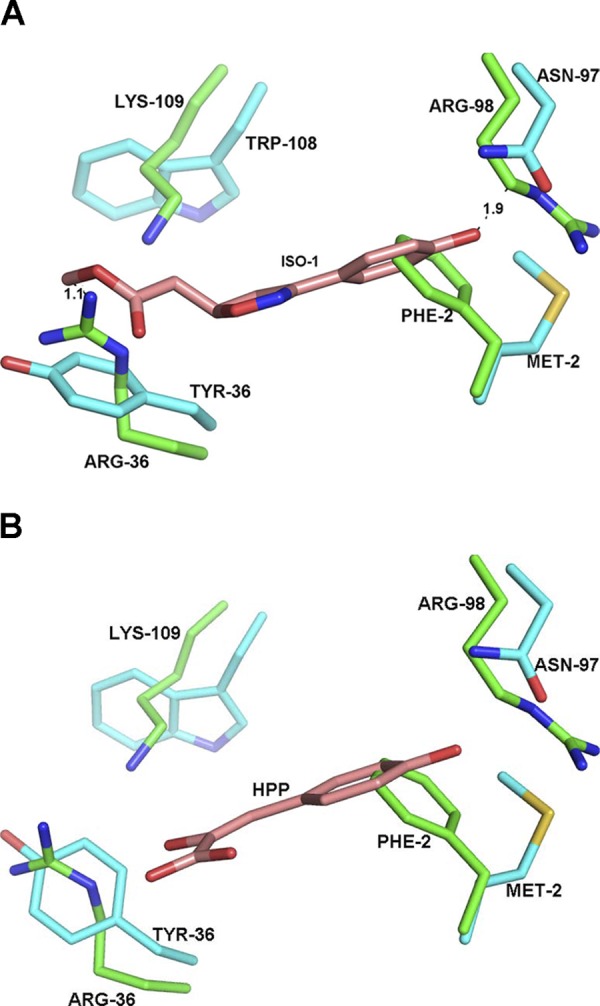
A) Active site of MIF (cyan) complexed with ISO-1 (brown) superposed onto the D-DT active site (green). There are close contacts between ISO-1 and Arg-36, Arg-98, and Lys-109 of D-DT. The modeled hydroxyphenyl group of ISO-1 occupies part of the same space as the phenyl group of Phe-2. B) Superposition of the MIF–HPP structure onto D-DT revealed no major clashes, except for Phe-2 in the active site. HPP and Phe-2 must undergo a conformational change to allow binding and catalysis to occur.
DISCUSSION
This study identified a new activity of D-DT, neutrophil recruitment to the lung, that is identical to the CD74-dependent activity of MIF (44). The mechanism of neutrophil recruitment for MIF involves activating CD74 on alveolar macrophages, phosphorylation of p44/p42, and secretion of the neutrophil-recruiting chemokines CXCL1 and CXCL2, as neutrophils do not express CD74 (44). Because of the absence of CD74 expression in all cells, this mechanism is likely to occur in other tissues with different kinetics (45), as MIF and D-DT can cause inflammation in most organ systems during infection. In the lung, however, a study has shown that CXCL1 and CXCL2 are released at their highest concentrations by MIF-activated alveolar macrophages within 3 h (44). Anti-MIF antibody reduces MIP-2 levels in the BAL of rats examined at 4 and 24 h after stimulation by LPS (46).
The in vivo D-DT potency in the lung is ∼5–10 fold less than MIF. However, we found that the in vivo lung neutrophil recruitment of D-DT and MIF are additive, similar to several other studies of D-DT. In non-small-cell lung carcinoma (NSCLC) cells, there is cooperativity in the negative regulation of AMP-activated protein kinase (AMPK) and cooperativity in the increase of CXCL8 and VEGF, human umbilical vascular endothelial cell (HUVEC) tube formation, and cell migration by D-DT and MIF (9, 10). In a study on renal clear-cell carcinoma, D-DT and MIF showed additive protumorigenic effects, with D-DT exhibiting a more dramatic in vitro and in vivo effect (11). Other overlapping features include the lethal effect of these proteins during sepsis, elevated serum concentrations during ovarian cancer (47), and inhibition of AMPK in human NSCLC cell lines with either wild-type or mutant tumor-suppressor kinase LKB1 (9). The present and previous studies demonstrating overlapping activities for both proteins and, in some cases, a cooperativity in the inhibition of D-DT and MIF as a more effective therapeutic mechanism, which provided the rationale for identifying a single molecule that inhibits both D-DT and MIF. Given that both D-DT and MIF use the same two model substrates in catalysis (7, 13), it was reasonable to postulate that the MIF competitive inhibitor ISO-1, which was designed based on the MIF-HPP crystal structure, would inhibit both proteins (19, 20). The inability of ISO-1 to inhibit D-DT led to experiments to more fully characterize the properties of D-DT for comparison to MIF.
The kcat/Km of the HPP keto-enol activity of D-DT and MIF are within 12% of each other, despite a sequence identity of less than 30% for the entire protein and the active site. The hallmark for catalysis by the tautomerase superfamily is an N-terminal proline that functions as a general base for transferring a proton between two atoms. pH-dependent NMR studies of the 15N proline peak for MIF produced a nonlinear least-squares fit with a pKa of 5.6, similar to the pKa values determined by Whitman and colleagues (48) using pH-dependent covalent inhibition of Pro-1 in catalytic assays. This pKa value is significantly lower than expected for a typical proline at the N terminus. The structural and chemical source for this decrease is the hydrophobic environment of Pro-1 in the presence of an aromatic pocket with a Lys-32, making another positively charged residue unfavorable without a counterion. An N-terminal positively charged Pro-1 for D-DT would be even more unfavorable because of the presence of Lys-32, Arg-36, Arg-98, and Lys-109 in the active site, supporting the presence of a lone electron that removes a proton from l-dopachrome methyl ester or HPP in their respective catalytic reactions. It is surprising, however, that both proteins can accept HPP and d-dopachrome as substrates, given that the D-DT active site has a high electrostatic potential, whereas the MIF active site is highly hydrophobic. To understand the structural source of catalytic activity, we attempted to cocrystallize D-DT with HPP but could produce only needles that did not diffract. In one crystallization condition, D-DT was cocrystallized with tartrate that was part of the crystallization solution (Supplemental Fig. S4A and Supplemental Table S1). Tartrate is an aliphatic, dicarboxylic acid, with 2 hydroxyl groups on adjacent carbon atoms, that has several interactions with the D-DT active site (Supplemental Fig. S4B), but at 50 μM has modest inhibition of HPP keto-enol activity. Tartrate could be used in a computational study to identify possible physiological metabolites that can undergo a tautomerase/isomerase activity or can be used for identifying more potent inhibitors. We also used D-DT (without tartrate) and superimposed MIF complexed to HPP and ISO-1. The modeling studies suggest that D-DT does not bind to ISO-1 because of 3 residues (Phe-2, Arg-36, and Arg-98) that block bind to the active site. Modeling with HPP suggests that a conformational change of Phe-2 should occur. In this regard, note that Met-2 of MIF is found in 2 different conformations (47) that may be an additional feature of the active site in human tautomerase superfamily.
Although ISO-1 does not inhibit D-DT, the MIF covalent inhibitor 4-IPP forms a covalent bond with Pro-1 of D-DT, leading to a 6-PP adduct. Kinetic studies of covalent inhibition by 4-IPP indicate that there are significant differences with the half-life of unmodified murine and hD-DT. The D-DT half-life is twice as long as that of hMIF. The structures of the 6-PP adducts are consistent with the kinetics, providing potential mechanisms to explain the slower kinetics for covalent bond formation to Pro-1 of D-DT. The 6-PP adduct of MIF is within the active site, whereas the adduct for D-DT is at the surface of the active site. If covalent bond formation occurs within the D-DT active site, conformational changes would be necessary for 4-IPP binding. Whether covalent bond formation occurs at the surface or within the D-DT active site, there is a higher energy state relative to MIF that leads to slower kinetics. It is also clear from the structure of D-DT–6-PP that if a covalent bond forms within the active site, the 6-PP adduct is ejected from the higher energy state within the active site to a lower energy conformation at the surface of the active site. Another interesting point for 6-PP adducts is that all interactions with 6-PP for both proteins are hydrophobic, with the exception of the covalent bond to Pro-1 (Supplemental Fig. S5). The structures of D-DT–6-PP and MIF–6-PP could be used to introduce chemical groups that increase affinity and specificity for D-DT and MIF by a modified 4-IPP relative to other proteins.
We used 4-IPP to introduce the 6-PP adduct on Pro-1 for D-DT and MIF and found that they are 50% as active in lung neutrophil recruitment as the unmodified versions. The 50% activity that remains in this biological activity is consistent with genetic experiments using a MIFP1G (proline to glycine) knock-in mutant that concluded that Pro-1 is important in protein-protein interactions, and the reduction in this biological activity is not due loss of catalytic function (16). Although the additive effects of D-DT and MIF are observed for neutrophil recruitment, no cooperativity in reduction of neutrophil recruitment was seen with D-DT–6-PP and MIF–6-PP. Although this result is confounding, it may be explained by the position of the 6-PP adduct on these two molecules. An emerging hypothesis for MIF disruption of CD74 interactions by MIF inhibitors is that these inhibitors have chemical moieties that protrude from the active site (17). Inhibitors that are entirely buried within the active site appeared to have no effect on CD74 binding and activity. If this hypothesis is proven to be correct and applies to D-DT, the D-DT–6-PP molecule should have a greater effect in disrupting interactions with CD74 than does MIF–6-PP, based on the X-ray structure (Fig. 3D). Although we observed such a trend in our experiments (Fig. 2C), additional experiments are needed that include in vitro CD74 binding and characterization of antagonism by the two 6-PP-modified proteins.
It is notable that MIF forms covalent bonds with other compounds, both within the active site and at the surface of the active site, that are dependent on the physicochemical properties of the compounds (49). Various isothiocyanates, which are cancer-preventive nutrients found in cruciferous vegetables, form covalent bonds with MIF, resulting in different conformations of the adduct. Pro-1 forms a covalent bond with the aromatic phenethyl isothiocyanate (PEITC) in the active site, whereas the alkyl l-sulforaphane forms a covalent bond that results in the adduct at the surface of the active site. The relative kinetics of inhibition are similar to those of 4-IPP inhibition of D-DT and MIF. PEITC within the hydrophobic MIF active site forms a cis-thiopeptide bond that is 2.5 times faster than the trans-thiopeptide of alkyl l-sulforaphane at the surface of the active site. These examples with MIF provide insight into the versatility of Pro-1, which is poised to form covalent bonds and has some flexibility that results in an adduct within the active site or at the surface. In addition to introducing chemical groups to increase affinity and specificity as noted above, the results with 4-IPP and isothiocyanates suggest different aromatic or aliphatic pharmacophores can be used to affect properties, such as affinity to CD74, antagonism, pharmacokinetics, and pharmacodynamics for therapeutics use in targeting D-DT and MIF. The use of covalent inhibitors as therapeutics is not favored because of off-target reactivity that may cause adverse effects. It is notable, however, that aspirin has been used as a covalent inhibitor of cyclooxygenases for >100 yr, and 3 of the 10 top-selling drugs in the United States in 2009 were covalent inhibitors (50). The off-target reactivity of 4-IPP is not known, but it has been used successfully in a mouse model of P. aeruginosa-induced keratitis (51). The lung is also a major target for P. aeruginosa infection that causes damage, in part, by recruitment of neutrophils and the damage these cells cause through inflammation (24–26). P. aeruginosa is an opportunistic, nosocomial, multidrug-resistant pathogen that increases mortality and length of stay of critically ill patients in intensive care units (52). Given the urgent need for new treatments and prevention of lung damage by neutrophils, 4-IPP is a leading compound with potential for development into a therapeutic.
The similarities in MIF and D-DT biological activities (7, 10, 11) are suggestive of the cytokine and chemokine superfamilies. Cytokines bind to a specific receptor subunit but share a signaling subunit expressed on different types of cells (53). In the chemokine superfamily, multiple chemokines bind and activate the same receptor due to tissue-dependent chemokine expression that requires the recruitment of specific immune cells expressing a specific chemokine receptor (54). D-DT and MIF function as cytokines by binding to CD74. Given the similar signaling mechanisms and overlapping biological activities, it is likely that the signaling subunit is CD44 for D-DT (8). However, there are D-DT biological activities that are not shared with MIF. For example, low levels of D-DT mRNA are observed in adipocytes from obese humans and from the db/db mouse model of obesity, diabetes, and dyslipidemia (55). Administration of recombinant D-DT in db/db mice improves glucose intolerance. D-DT is also involved in lipid metabolism in adipocytes. More recently, knockdown of D-DT in B16F10 melanoma cell lines led to decreased proliferation and increased apoptosis and treatment of a syngeneic tumor model with polyclonal antibodies resulted in lower tumor burden (56). These differences with MIF and the cooperativity of D-DT and MIF in various models of disease support additional mechanisms for D-DT.
Supplementary Material
Acknowledgments
The authors are grateful to Wibke Schulte and Melanie Merk for the data in Fig. 1C, D on the comparative effects of ISO-1 and 4IPP on MIF and DDT tautomerase activity. The authors thank the staff at the X25 beamline of the National Synchrotron Light Source (Brookhaven National Laboratory, Upton, NY, USA) for assistance in data collection and the Yale School of Medicine Keck Biotechnology Research Laboratory for mass spectrometry.
This work was supported by the U.S. National Institutes of Health, R01 grants AI065029, AI082295, and AR049610.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 4-IPP
- 4-iodo-6-phenylpyrimidine
- 6-PP
- 6-phenylpyrimidine
- AMPK
- AMP-activated protein kinase
- BAL
- bronchoalveolar lavage
- D-DT
- d-dopachrome tautomerase
- DMSO
- dimethyl sulfoxide
- hD-DT
- human d-dopachrome tautomerase
- HPP
- 4-(hydroxyphenyl)pyruvate
- ISO-1
- (S,R)-3-(4-hydroxyphenyl)-4,5-dihydro-5-isoxazole acetic acid methyl ester
- LPS
- lipopolysaccharide
- MIF
- macrophage migration inhibitory factor
- mD-DT
- murine d-dopachrome tautomerase
- NSCLC
- non-small-cell lung carcinoma
- PDB
- Protein Data Bank
- PEITC
- phenethyl isothiocyanate
- RMS
- root mean square
REFERENCES
- 1. Bloom B. R., Bennett B. (1966) Mechanism of a reaction in vitro associated with delayed-type hypersensitivity. Science 153, 80–82 [DOI] [PubMed] [Google Scholar]
- 2. David J. R. (1966) Delayed hypersensitivity in vitro: its mediation by cell-free substances formed by lymphoid cell-antigen interaction. Proc. Natl. Acad. Sci. U. S. A. 56, 72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Subramanya H. S., Roper D. I., Dauter Z., Dodson E. J., Davies G. J., Wilson K. S., Wigley D. B. (1996) Enzymatic ketonization of 2-hydroxymuconate: specificity and mechanism investigated by the crystal structures of two isomerases. Biochemistry 35, 792–802 [DOI] [PubMed] [Google Scholar]
- 4. Almrud J. J., Kern A. D., Wang S. C., Czerwinski R. M., Johnson W. H., Murzin A. G., Hackert M. L., Whitman C. P. (2002) The crystal structure of YdcE, a 4-oxalocrotonate tautomerase homologue from Escherichia coli, confirms the structural basis for oligomer diversity. Biochemistry 41, 12010–12024 [DOI] [PubMed] [Google Scholar]
- 5. Sun H. W., Bernhagen J., Bucala R., Lolis E. (1996) Crystal structure at 2.6-A resolution of human macrophage migration inhibitory factor. Proc. Natl. Acad. Sci. U. S. A. 93, 5191–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leng L., Metz C. N., Fang Y., Xu J., Donnelly S., Baugh J., Delohery T., Chen Y., Mitchell R. A., Bucala R. (2003) MIF signal transduction initiated by binding to CD74. J. Exp. Med. 197, 1467–1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merk M., Zierow S., Leng L., Das R., Du X., Schulte W., Fan J., Lue H., Chen Y., Xiong H., Chagnon F., Bernhagen J., Lolis E., Mor G., Lesur O., Bucala R. (2011) The D-dopachrome tautomerase (DDT) gene product is a cytokine and functional homolog of macrophage migration inhibitory factor (MIF). Proc. Natl. Acad. Sci. U. S. A. 108, E577–E585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shi X., Leng L., Wang T., Wang W., Du X., Li J., McDonald C., Chen Z., Murphy J. W., Lolis E., Noble P., Knudson W., Bucala R. (2006) CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity 25, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brock S. E., Rendon B. E., Yaddanapudi K., Mitchell R. A. (2012) Negative regulation of AMP-activated protein kinase (AMPK) Activity by macrophage migration inhibitory factor (MIF) family members in non-small cell lung carcinomas. J. Biol. Chem. 287, 37917–37925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coleman A. M., Rendon B. E., Zhao M., Qian M.-W., Bucala R., Xin D., Mitchell R. A. (2008) Cooperative regulation of non-small cell lung carcinoma angiogenic potential by macrophage migration inhibitory factor and its homolog, D-dopachrome tautomerase. J Immunol. 181, 2330–2337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pasupuleti V., Du W., Gupta Y., Yeh I. J., Montano M., Magi-Galuzzi C., Welford S. M. (2014) Dysregulated D-dopachrome tautomerase, a hypoxia-inducible factor-dependent gene, cooperates with macrophage migration inhibitory factor in renal tumorigenesis. J. Biol. Chem. 289, 3713–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rosengren E., Bucala R., Aman P., Jacobsson L., Odh G., Metz C. N., Rorsman H. (1996) The immunoregulatory mediator macrophage migration inhibitory factor (MIF) catalyzes a tautomerization reaction. Mol. Med. 2, 143–149 [PMC free article] [PubMed] [Google Scholar]
- 13. Odh G., Hindemith A., Rosengren A. M., Rosengren E., Rorsman H. (1993) Isolation of a new tautomerase monitored by the conversion of D-dopachrome to 5,6-dihydroxyindole. Biochem. Biophys. Res. Commun. 197, 619–624 [DOI] [PubMed] [Google Scholar]
- 14. Zhang M., Aman P., Grubb A., Panagopoulos I., Hindemith A., Rosengren E., Rorsman H. (1995) Cloning and sequencing of a cDNA encoding rat D-dopachrome tautomerase. FEBS Lett. 373, 203–206 [DOI] [PubMed] [Google Scholar]
- 15. Swope M. D., Lolis E. (1999) Macrophage migration inhibitory factor: cytokine, hormone, or enzyme? Rev. Physiol. Biochem. Pharmacol. 139, 1–32 [DOI] [PubMed] [Google Scholar]
- 16. Fingerle-Rowson G., Kaleswarapu D. R., Schlander C., Kabgani N., Brocks T., Reinart N., Busch R., Schutz A., Lue H., Du X., Liu A., Xiong H., Chen Y., Nemajerova A., Hallek M., Bernhagen J., Leng L., Bucala R. (2009) A tautomerase-null MIF gene knock-in mouse reveals that protein interactions and not enzymatic activity mediate MIF-dependent growth regulation. Mol. Cell. Biol. 29, 1922–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho Y., Vermeire J. J., Merkel J. S., Leng L., Du X., Bucala R., Cappello M., Lolis E. (2011) Drug repositioning and pharmacophore identification in the discovery of hookworm MIF inhibitors. Chem. Biol. 18, 1089–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fan C., Rajasekaran D., Syed M. A., Leng L., Loria J. P., Bhandari V., Bucala R., Lolis E. J. (2013) MIF intersubunit disulfide mutant antagonist supports activation of CD74 by endogenous MIF trimer at physiologic concentrations. Proc. Natl. Acad. Sci. U. S. A. 110, 10994–10999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lubetsky J. B., Swope M., Dealwis C., Blake P., Lolis E. (1999) Pro-1 of macrophage migration inhibitory factor functions as a catalytic base in the phenylpyruvate tautomerase activity. Biochemistry 38, 7346–7354 [DOI] [PubMed] [Google Scholar]
- 20. Lubetsky J. B., Dios A., Han J., Aljabari B., Ruzsicska B., Mitchell R., Lolis E., Al-Abed Y. (2002) The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J. Biol. Chem. 277, 24976–24982 [DOI] [PubMed] [Google Scholar]
- 21. Al-Abed Y., VanPatten S. (2011) MIF as a disease target: ISO-1 as a proof-of-concept therapeutic. Future Med. Chem. 3, 45–63 [DOI] [PubMed] [Google Scholar]
- 22. Arjona A., Foellmer H. G., Town T., Leng L., McDonald C., Wang T., Wong S. J., Montgomery R. R., Fikrig E., Bucala R. (2007) Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J. Clin. Invest. 117, 3059–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Winner M., Meier J., Zierow S., Rendon B. E., Crichlow G. V., Riggs R., Bucala R., Leng L., Smith N., Lolis E., Trent J. O., Mitchell R. A. (2008) A novel, macrophage migration inhibitory factor suicide substrate inhibits motility and growth of lung cancer cells. Cancer Res. 68, 7253–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adamali H., Armstrong M. E., McLaughlin A. M., Cooke G., McKone E., Costello C. M., Gallagher C. G., Leng L., Baugh J. A., Fingerle-Rowson G., Bucala R. J., McLoughlin P., Donnelly S. C. (2012) Macrophage migration inhibitory factor enzymatic activity, lung inflammation, and cystic fibrosis. Am J. Respir. Crit Care Med. 186, 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bozza M., Satoskar A. R., Lin G., Lu B., Humbles A. A., Gerard C., David J. R. (1999) Targeted disruption of migration inhibitory factor gene reveals its critical role in sepsis. J. Exp. Med. 189, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plant B. J., Gallagher C. G., Bucala R., Baugh J. A., Chappell S., Morgan L., O'Connor C. M., Morgan K., Donnelly S. C. (2005) Cystic fibrosis, disease severity, and a macrophage migration inhibitory factor polymorphism. Am J. Respir. Crit. Care Med. 172, 1412–1415 [DOI] [PubMed] [Google Scholar]
- 27. Cho Y., Crichlow G. V., Vermeire J. J., Leng L., Du X., Hodsdon M. E., Bucala R., Cappello M., Gross M., Gaeta F., Johnson K., Lolis E. J. (2010) Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc. Natl. Acad. Sci. U. S. A. 107, 11313–11318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rajasekaran D., Keeler C., Syed M. A., Jones M. C., Harrison J. K., Wu D., Bhandari V., Hodsdon M. E., Lolis E. J. (2012) A model of GAG/MIP-2/CXCR2 interfaces and its functional effects. Biochemistry 51, 5642–5654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bhandari V., Choo-Wing R., Harijith A., Sun H., Syed M. A., Homer R. J., Elias J. A. (2012) Increased hyperoxia-induced lung injury in nitric oxide synthase 2 null mice is mediated via angiopoietin 2. Am. J. Respir. Cell Mol. Biol. 46, 668–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sugimoto H., Taniguchi M., Nakagawa A., Tanaka I., Suzuki M., Nishihira J. (1999) Crystal structure of human D-dopachrome tautomerase, a homologue of macrophage migration inhibitory factor, at 1.54 A resolution. Biochemistry 38, 3268–3279 [DOI] [PubMed] [Google Scholar]
- 32. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 33. Brunger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J. S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warren G. L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921 [DOI] [PubMed] [Google Scholar]
- 34. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 35.(1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 36. Swope M., Sun H. W., Blake P. R., Lolis E. (1998) Direct link between cytokine activity and a catalytic site for macrophage migration inhibitory factor. EMBO J. 17, 3534–3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McLean L. R., Zhang Y., Li H., Li Z., Lukasczyk U., Choi Y.-M., Han Z., Prisco J., Fordham J., Tsay J. T., Reiling S., Vaz R. J., Li Y. (2009) Discovery of covalent inhibitors for MIF tautomerase via cocrystal structures with phantom hits from virtual screening. Bioorg. Med. Chem. Lett. 19, 6717–6720 [DOI] [PubMed] [Google Scholar]
- 38. Cournia Z., Leng L., Gandavadi S., Du X., Bucala R., Jorgensen W. L. (2009) Discovery of human macrophage migration inhibitory factor (MIF)-CD74 antagonists via virtual screening. J. Med. Chem. 52, 416–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ouertatani-Sakouhi H., El-Turk F., Fauvet B., Cho M.-K., Pinar Karpinar D., Le Roy D., Dewor M., Roger T., Bernhagen J., Calandra T., Zweckstetter M., Lashuel H. A. (2010) Identification and characterization of novel classes of macrophage migration inhibitory factor (MIF) inhibitors with distinct mechanisms of action. J. Biol. Chem. 285, 26581–26598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McLean L. R., Zhang Y., Li H., Choi Y.-M., Han Z., Vaz R. J., Li Y. (2010) Fragment screening of inhibitors for MIF tautomerase reveals a cryptic surface binding site. Bioorg. Med. Chem. Lett. 20, 1821–1824 [DOI] [PubMed] [Google Scholar]
- 41. Hare A. A., Leng L., Gandavadi S., Du X., Cournia Z., Bucala R., Jorgensen W. L. (2010) Optimization of N-benzyl-benzoxazol-2-ones as receptor antagonists of macrophage migration inhibitory factor (MIF). Bioorg. Med. Chem. Lett. 20, 5811–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jorgensen W. L., Gandavadi S., Du X., Hare A. A., Trofimov A., Leng L., Bucala R. (2010) Receptor agonists of macrophage migration inhibitory factor. Bioorg. Med. Chem. Lett. 20, 7033–7036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jorgensen W. L., Trofimov A., Du X., Hare A. A., Leng L., Bucala R. (2011) Benzisothiazolones as modulators of macrophage migration inhibitory factor. Bioorg. Med. Chem. Lett. 21, 4545–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi K., Koga K., Linge H. M., Zhang Y., Lin X., Metz C. N., Al-Abed Y., Ojamaa K., Miller E. J. (2009) Macrophage CD74 contributes to MIF-induced pulmonary inflammation. Respir. Res. 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rovai L. E., Herschman H. R., Smith J. B. (1998) The murine neutrophil-chemoattractant chemokines LIX, KC, and MIP-2 have distinct induction kinetics, tissue distributions, and tissue- specific sensitivities to glucocorticoid regulation in endotoxemia. J. Leukoc. Biol. 64, 494–502 [DOI] [PubMed] [Google Scholar]
- 46. Makita H., Nishimura M., Miyamoto K., Nakano T., Tanino Y., Hirokawa J., Nishihira J., Kawakami Y. (1998) Effect of anti-macrophage migration inhibitory factor antibody on lipopolysaccharide-induced pulmonary neutrophil accumulation. Am. J. Respir. Crit. Care Med. 158, 573–579 [DOI] [PubMed] [Google Scholar]
- 47. Merk M., Mitchell R. A., Endres S., Bucala R. (2012) D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine 59, 10–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stamps S. L., Fitzgerald M. C., Whitman C. P. (1998) Characterization of the role of the amino-terminal proline in the enzymatic activity catalyzed by macrophage migration inhibitory factor. Biochemistry 37, 10195–10202 [DOI] [PubMed] [Google Scholar]
- 49. Crichlow G. V., Fan C., Keeler C., Hodsdon M., Lolis E. J. (2012) Structural interactions dictate the kinetics of macrophage migration inhibitory factor inhibition by different cancer-preventive isothiocyanates. Biochemistry 51, 7506–7514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh J., Petter R. C., Baillie T. A., Whitty A. (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317 [DOI] [PubMed] [Google Scholar]
- 51. Gadjeva M., Nagashima J., Zaidi T., Mitchell R. A., Pier G. B. (2010) Inhibition of macrophage migration inhibitory factor ameliorates ocular Pseudomonas aeruginosa-induced keratitis. PLoS Pathog. 6, e1000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sydnor E. R. M., Perl T. M. (2011) Hospital epidemiology and infection control in acute-care settings. Clin. Microbiol. Rev. 24, 141–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang X., Lupardus P., Laporte S. L., Garcia K. C. (2009) Structural biology of shared cytokine receptors. Annu. Rev. Immunol. 27, 29–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fernandez E. J., Lolis E. (2002) Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42, 469–499 [DOI] [PubMed] [Google Scholar]
- 55. Iwata T., Taniguchi H., Kuwajima M., Taniguchi T., Okuda Y., Sukeno A., Ishimoto K., Mizusawa N., Yoshimoto K. (2012) The action of D-dopachrome tautomerase as an adipokine in adipocyte lipid metabolism. PloS One 7, e33402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kobold S., Merk M., Hofer L., Peters P., Bucala R., Endres S. (2014) The macrophage migration inhibitory factor (MIF)-homologue D-dopachrome tautomerase is a therapeutic target in a murine melanoma model. Oncotarget 5, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. DeLano W. L. (2002) The PyMOL molecular graphics system. DeLano Scientific, Palo Alto, CA, USA; http://www.pymol.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.