

Abstract
Increased aortic stiffness is an early and independent biomarker of cardiovascular disease. Here we tested the hypothesis that vascular smooth muscle cells (VSMCs) contribute significantly to aortic stiffness and investigated the mechanisms involved. The relative contributions of VSMCs, focal adhesions (FAs), and matrix to stiffness in mouse aorta preparations at optimal length and with confirmed VSMC viability were separated by the use of small-molecule inhibitors and activators. Using biomechanical methods designed for minimal perturbation of cellular function, we directly quantified changes with aging in aortic material stiffness. An alpha adrenoceptor agonist, in the presence of NG-nitro-l-arginine methyl ester (l-NAME) to remove interference of endothelial nitric oxide, increases stiffness by 90–200% from baseline in both young and old mice. Interestingly, increases are robustly suppressed by the Src kinase inhibitor PP2 in young but not old mice. Phosphotyrosine screening revealed, with aging, a biochemical signature of markedly impaired agonist-induced FA remodeling previously associated with Src signaling. Protein expression measurement confirmed a decrease in Src expression with aging. Thus we report here an additive model for the in vitro biomechanical components of the mouse aortic wall in which 1) VSMCs are a surprisingly large component of aortic stiffness at physiological lengths and 2) regulation of the VSMC component through FA signaling and hence plasticity is impaired with aging, diminishing the aorta's normal shock absorption function in response to stressors.
Keywords: smooth muscle, focal adhesion, alpha agonist, Src, phosphotyrosine
cardiovascular disease (CVD) is the leading cause of death in the United States. Aortic stiffness, a biomechanical property of material and structure, is clinically assessed with in vivo observations of pulse wave velocity (PWV). Increased PWV, an early and independent biomarker of CVD, has been observed in a broad spectrum of individuals ranging from aged individuals with no other apparent disease (24, 46) to those with disorders such as hypertension (21), diabetes mellitus (10), emphysema (25), and end-stage renal failure (5). Elevated PWV in the large vessels leads to increased transmission of high pressures to small vessels downstream and is thought to cause remodeling of the microcirculation and subsequent adverse effects on end-organ function. Recently, increases in PWV in mice have been shown both to precede and to be augmented by hypertension (45). Additionally, mechanical stretch in mice can induce a self-perpetuating cycle of hypertrophy in small vessels (48). Collectively, these studies and other studies in humans indicate that factors that increase aortic PWV are prognostic, and may be causative, of CVD and mortality (3, 27).
It is far from clear how changes in blood vessel properties contribute to observed increases in PWV. A mathematical relationship between PWV and vessel stiffness has been described by the Moens-Korteweg equation, in which PWV is proportional to the square root of the elastic modulus, alternatively known as material stiffness (15), and also dependent on wall thickness and vessel diameter. However, the Moens-Korteweg equation is predicated on several simplifying and nonphysiological assumptions, including uniformity of vessel composition (9, 14), as well as dynamic values of vessel geometry that are difficult to quantify in vivo. Therefore, in order to understand more comprehensively the relationship between PWV and aortic stiffness, it is first necessary to directly determine how changes to in vitro material properties of the blood vessel wall and its subcomponents contribute to aging-induced increases in aortic stiffness.
While classical and recent studies of vascular mechanics attribute the majority of vessel wall stiffness largely to the extracellular matrix (ECM) (4, 44, 47), a few indicate that vascular smooth muscle (VSM) activation produces nonnegligible increases in stiffness (1). Many different structural and biochemical changes occur within the vessel wall with aging (20), and recent studies of cultured vascular smooth muscle cells (VSMCs) have shown an increase in cellular stiffness with aging (33). Thus VSMCs may play an important role in effecting and affecting aortic stiffness, although their contributions relative to that of ECM and the effects of aging have not been ascertained.
In the present study, aortic mechanical properties were directly measured to assess the VSMC component of stiffness in the aortic wall. In vitro biomechanical tissue properties were quantified in aortic samples confirmed to contain viable smooth muscle cells. We demonstrate that these properties are separable into quantifiable components distinguished by activation levels of VSMCs and that, unexpectedly, smooth muscle contributes up to half of total material stiffness. These measurements were performed for the first time in old mouse aortas by using direct mechanical distension to produce circumferential strain, in contrast to previous biomechanical studies of old aortas utilizing pressurization (29, 30). Furthermore, we show that with aging regulatory pathways related to focal adhesion signaling in the smooth muscle cell are lost, an effect expected to compromise the ability of the proximal aorta of an old mouse to perform its normal function as a hemodynamic shock absorber and maintain appropriate stiffness levels in the presence of in vivo challenges.
METHODS
Preparation of aortic samples.
Aortas were excised from euthanized male C57BL/6J mice (young, 3 ± 1 mo of age: Jackson Labs; old, 29 ± 1 mo of age: supported by National Institute of Aging) according to protocols approved by the Institutional Care and Use Committee (IACUC) of Boston University. Aortas were immediately placed in a physiological salt solution (PSS; in mM: 120 NaCl, 5.9 KCl, 11.5 dextrose, 25 NaHCO3, 1.2 NaH2PO4, 1.2 MgCl2, 2.5 CaCl2; pH = 7.4) equilibrated with 95% O2-5% CO2. Loose perivascular fat was removed from aortic samples. Segments of 4- to 5-mm axial length (unloaded) were isolated from the proximal end of the descending thoracic aorta. There was no significant difference in axial length from cut segments between young and old animals.
Measurements of aortic geometry.
Axial length and outer diameter of samples were measured under a light microscope (×4) during aortic dissection of unloaded segments in PSS. Small rings adjacent to both ends (proximal and distal) of the thoracic aorta ring to be studied were incubated for 30 min in nuclear stain (NucBlue from Life Technologies). After the incubation period, individual rings were imaged with fluorescence microscopy (×20) for autofluorescence of medial elastin fibers and the NucBlue stain via NIS-Elements software (Nikon Instruments). For each ring, 15–20 measurements were taken and averaged to obtain the average medial thickness of the wall. The measurements for both proximal and distal thoracic aortic rings were then averaged to obtain a single representative unloaded wall thickness measurement for the thoracic aorta segment to be studied.
Uniaxial stretching apparatus and normalization of force measurements.
Thoracic aorta segments were suspended in an organ bath, submerged in oxygenated PSS warmed to 37°C by a thermostat-controlled water bath and circulation system to maintain viability, and threaded with triangular pieces of wire. As shown in Fig. 1A, this allows the segments to be stretched uniaxially in a way that simulates circumferential distension. Stretch was monitored by the model 300C Dual-Mode Lever Arm System by Aurora Scientific (Aurora, ON, Canada) and performed by using an electronic input to the lever arm motor via a function generator. Chart software (AD Instruments) was used for data acquisition from the system. The extent of stretch performed to obtain a steady-state force response, known as the operating length and specific to each sample, was established as circumferential strain ε, a percentage of outer aortic diameter of that sample in the unloaded state.
Fig. 1.

In vitro steady-state stiffness methods. A: schematic of lever arm for stiffness measurements; steady-state force response measured from stretching the tissue sample (shown here with typical unloaded in vitro geometry) is that which is experienced in the circumferential direction at the center of the axial cross section (arrow). B: representative force trace with sample inset of electronic recordings for high-frequency, low-amplitude (HFLA) oscillatory stretch; sinusoidal length input (ΔL) and force output (ΔF) were used to calculate stiffness at optimal length L. Stiffness E is the ratio of change in stress (Δσ, equivalent to ΔF normalized to cross-sectional area A) to change in strain experienced by tissue (Δε).
The lever arm also served as a force transducer, allowing for measurements of a steady-state force F, the equilibrated response after stretching to the operating length. Dividing force by the axial length l of the tissue normalizes it to wall tension. Circumferential stress σ was obtained by normalizing F to cross-sectional area A of unloaded tissue: σ = F/A and A = 2hl, where h is the wall thickness and l is the axial length.
High-frequency, low-amplitude stretch for material stiffness measurements.
In vitro aortic stiffness is defined in this study as the material stiffness of aortic tissue, equivalent to the elastic modulus derived from directly measurable circumferential force responses to small-length oscillations (18). High-frequency (40 Hz), low-amplitude (+1% strain from optimal length) sinusoidal stretching was applied persistently to obtain a time course of force-response data. Based on our preliminary experiments and previous studies, these parameters resulted in negligible phase lag of the force response or breakage of actomyosin cross bridges in the tissue (8). Solution changes were conducted without disturbance to the ongoing movement of the lever arm or the force trace as monitored via software.
As shown in Fig. 1B, oscillatory sinusoidal input produced a force response from which the change in stress could be calculated as the amplitude of the force output waveform (ΔF) normalized to cross-sectional area A. Stiffness is defined as this change in stress divided by the change in strain (1%, ΔL) imposed atop the strain of the operating length (L). This strain was applied to equilibrated tissue at least 30 min after stretch to L. Effectively, the high-frequency, low-amplitude (HFLA) protocol produces a local stress-strain relationship in the neighborhood of the operating length L; the stiffness is the elastic modulus as calculated from this response.
Stress-strain curves determined from force responses to slow high-amplitude ramp stretch.
With the same stretching apparatus, stress-strain curves were obtained from force responses to slow, high-amplitude ramp stretches applied from the unloaded state, or slack length. The strain rate applied to aortic samples was 20%/s, up to a maximum of 400% or tissue yield and rupture. The force response corresponding to the loading curve was normalized to stress. Stiffness values for specific strains were obtained by plotting stress against strain and calculating the local slope, i.e., the elastic modulus, around specific strain levels. Tissues were maintained in calcium-free PSS (in mM: 120 NaCl, 5.9 KCl, 11.5 dextrose, 25 NaHCO3, 1.2 NaH2PO4, 1.2 MgCl2, 2 EGTA; pH = 7.4) to isolate passive mechanical properties of the aortic wall.
Confirmation of VSMC viability and agonist stimulation.
Prior to all steady-state force measurements made with the lever arm length controller, we used 51 mM KCl PSS (in mM: 75 NaCl, 51 KCl, 11.5 dextrose, 25 NaHCO3, 1.2 NaH2PO4, 5.35 MgCl2, 2.5 CaCl2; pH = 7.4) to confirm VSMC viability in all aortic samples. This solution depolarizes smooth muscle cells and opens voltage-gated calcium channels, increasing intracellular calcium levels and inducing muscle contraction. A stimulation period of 15 min enabled samples to approach contractile steady state.
After confirming VSMC viability, we washed out KCl with PSS and allowed samples an equilibration period of 30 min to return to an inactivated baseline. To maximally activate VSMCs in these samples, we then performed a two-step treatment. First, we added the alpha agonist phenylephrine (PE; 10 μM), which has previously been shown to increase contractility by both actin- and myosin-mediated mechanisms (49) maximally at the noted concentration (38). Equilibration occurred 15 min after addition of PE. Second, after equilibration, we followed with the addition of 300 μM NG-nitro-l-arginine methyl ester (l-NAME), a cell-permeant precursor of nitroarginine that inhibits nitric oxide synthase (NOS) (2, 36). Nitric oxide (NO), a product of the intimal endothelial monolayer in the mouse arterial wall, alters aortic smooth muscle tone and would be expected to alter stiffness. Equilibration was reached 20 min after addition of l-NAME.
Immunoblots.
Tissues were quick-frozen in an acetone-dry ice slurry containing 10% trichloroacetic acid (TCA) and 10 mM dithiothreitol (DTT), homogenized, and processed at 4°C as described previously by our group (23). The homogenization buffer, designed to preserve native phosphoprotein levels, consisted of 20 mM MOPS, 4% SDS, 10% glycerol, 10 mM DTT, 20 mM β-glycerophosphate, 5.5 μM leupeptin, 5.5 μM pepstatin, 20 KIU aprotinin, 2 mM Na3VO4, 1 mM NaF, 100 μM ZnCl2, 20 μM AEBSF, and 5 mM EGTA. Densitometric analysis of immunoblots was performed with the Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Tyrosine phosphorylation (pTyr) densitometry was normalized to that of α-tubulin densitometry. pTyr was examined after 15-min exposure of the vascular tissue to PE. At this time point, VSMC activation has reached steady-state values, corresponding to the maximally effective dosage of 10 μM (38).
Statistics.
All results are reported as means ± SE. Unpaired Student's t-tests were performed to analyze single statistical comparisons between dimensional measurements and absolute values of stress and stiffness from young and old mouse aortas. Two-way ANOVA and Tukey's range test post hoc were used for all instances of multiple comparisons. Tests were performed in GraphPad Prism. Significance was defined at the P < 0.05 level.
RESULTS
Physiological optimal length in mouse aorta does not change with age.
As reviewed by Wagenseil and Mecham (43), the physiological range of circumferential stretch ratios experienced by mouse aorta in vivo during normal blood flow is between 1.5 and 1.8, i.e., a circumferential strain of 50–80%. To select the value within this range most appropriate for stiffness measurements reflective of physiological VSMC contributions, we also measured the physiological optimal length of the tissue, the value for which active force production is maximal, both in young and old mouse aortas (6).
At 20% increments of sustained circumferential strain, up to 200% total strain, the contraction induced by 15-min (steady state) equilibration with 51 mM KCl PSS was recorded. After washout with PSS and 30 min of equilibration, the tissue was stretched to the next increment of 20% strain. As shown in Fig. 2, the force production at 80% circumferential strain is not significantly different from maximal force production and is also within the in vivo range of circumferential strain measured by Wagenseil and Mecham. Furthermore, optimal length was determined not to differ between young and old mice. Thus 80% was used as the operating strain for all subsequent in vitro stiffness measurements.
Fig. 2.
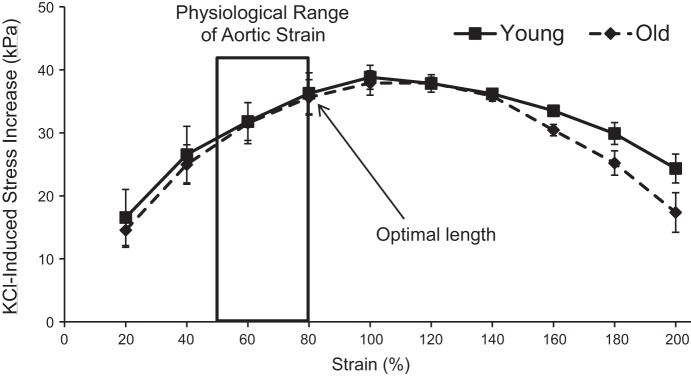
Physiological optimal length determination. Physiological optimal length, the strain level at which maximal contractile stress (kPa) occurs within physiological ranges of aortic strain, was determined to be 80% in both young and old mice (n = 3 each).
Baseline aortic wall tension increases with aging at optimal length, but stiffness decreases.
Measurements from 13 young and 12 old mice demonstrated significant increases in average outer vessel diameter at slack length (young: 755 ± 10 μm, old: 936 ± 17 μm; P < 0.001) and wall thickness (young: 56.9 ± 0.4 μm, old: 76.9 ± 0.9 μm; P < 0.001). Assuming cylindrical geometry of each wall at slack length, we infer that the inner diameter also increases with age and approximate this by subtracting twice the thickness from the outer diameter (641 μm in young vs. 782 μm in old).
We define baseline force as that experienced by an aortic tissue sample stretched to 80% strain and allowed to equilibrate. The baseline stiffness (elastic modulus) was measured as the resultant ratio of change in stress to change in strain in response to HFLA (1%) changes in length (see methods for details), as a sinusoidal oscillatory stretch superimposed upon optimal length. Changes in strain were kept small to cause essentially negligible perturbation of the tissue.
Baseline force, observed at steady state after equilibration at optimal length, increased significantly with aging (+34%, P < 0.001) in response to being stretched to the operating strain of 80% (Fig. 3A). In vivo, wall tension is an important parameter, particularly under hypertensive conditions, as it has been linked to changes in transforming growth factor-β (TGF-β) and NO release from the endothelium (32). When wall tension (Fig. 3B) is calculated by dividing the measured force by axial length, there is also a significant increase with aging (+32%, P < 0.001). However, when wall tension is normalized by cross-sectional area to calculate stress (Fig. 3C), no difference is seen, indicating that vessel geometry is the primary effector of increased wall tension.
Fig. 3.

Changes in baseline aortic mechanical parameters with aging. A: baseline steady-state force increases with aging. B: wall tension, equivalent to force normalized to axial length, increases with aging. C: stress, equivalent to tension normalized to wall thickness, does not change with aging. D: baseline aortic stiffness via HFLA measurements in the absence of agonist decreases with aging. Values are means ± SE; n = 13 young, 12 old. ***P < 0.001.
We performed small HFLA stretches (see methods for details) to calculate stiffness and found that the baseline value actually significantly decreased by 16% with aging (P < 0.001), as shown in Fig. 3D. This was surprising given previous reports of increased PWV in old mice (35). It is clear that baseline in vitro material stiffness, calculated as the elastic modulus, fails to account for aging-induced increases in PWV alone when applied to the Moens-Korteweg equation in conjunction with changes in wall geometry.
Increased passive aortic stiffness with aging occurs only beyond physiological strain levels.
To reconcile the apparent discrepancy between our steady-state baseline results and stiffness results from other studies in the literature (11), we constructed stress-strain curves for young and old mouse aortas by subjecting tissue samples to slow, high-amplitude ramps spanning a range of strains from 0 to 250%, well beyond physiological levels and beyond even the upper limit of previously acquired length-tension curves. Additionally, we eliminated any possible contributions from VSMCs by deactivating them in a calcium-free environment, mimicking the conditions established in similar studies (see methods for details) (12). Passive aortic stiffness values were calculated as the local slopes of the stress-strain curve at 25%-strain intervals, with the exception of replacing the 75% point with 80% in correspondence with optimal length.
As shown in Fig. 4, a significant increase with aging exists in stress and stiffness, beginning at strains of 250% and 200%, respectively. Between 100% and 200% strain, the stress-strain curve for old aorta appears to overtake that of young aorta, which leads to higher stiffness values at higher strains. Thus these results are compatible with increased passive stiffness attributed to old aortas in previous in vitro studies that evaluated stiffness at higher strains (11, 12, 29). Additionally, the stress-strain curve shows a downward trend in baseline stiffness with aging at physiological strain ranges, qualitatively comparable to stiffness measured via the HFLA protocol.
Fig. 4.
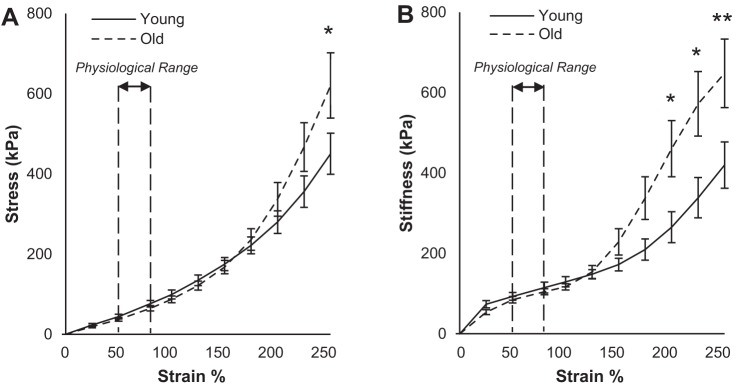
Passive aortic stiffness via mechanical stress-strain method increases with aging only beyond physiological strain range: stress (A) and stiffness (B) plotted against strain for young and old proximal thoracic aortas in calcium-free environment using ramp input from slack to failure. No significant difference is observed at low strains, including the physiological range of 50–80%. At higher strain levels, when collagen is heavily recruited because of large-amplitude stretching, old aortas are stiffer. Values are means ± SE; n = 6 young, 7 old. *P < 0.05, **P < 0.01.
VSMCs account for up to half of maximal total aortic stiffness.
To study the contributions of VSM to aortic stiffness under more physiologically correspondent conditions, we activated VSMCs in young and old aortas with PE and then l-NAME as detailed in methods. Steady-state stress and HFLA stiffness values were obtained after each equilibration period. We define the active stress and stiffness as the increases from baseline observed after addition of PE, reflecting the activation of the smooth muscle cells. Treatment with l-NAME induced no change in baseline stress or baseline stiffness values after 30 min [<2% change, not significant (ns)], indicating that the VSMCs must be activated with agonist in order for NOS inhibition to have an effect on stress and stiffness, further supporting the concept that the active component is separable and necessarily dependent upon agonist stimulation. We define maximal active stress and stiffness as the increases from baseline observed after addition of both PE and l-NAME, or, in purely quantitative terms, the sum of the active and NO-inhibited components.
No significant difference in the magnitude of the PE effects alone was seen between young and old mice (comparing open bars to corresponding filled bars in Fig. 5, A and B). After 15 min of stimulation with PE, we treated aortas with l-NAME for an additional 20 min, with PE still active. As a result, stress and stiffness significantly increased further, as shown in Fig. 5, A and B (P < 0.001). A smaller increase in stress resultant from NO inhibition was observed with aging as shown in Fig. 5A (P < 0.001), reflecting the probable decrease in endothelial NO production with aging, as has previously been reported in other studies (7, 42). On the other hand, no such difference in l-NAME-induced stiffness increases was seen (Fig. 5B).
Fig. 5.

Vascular smooth muscle (VSM) cells (VSMCs) contribute significantly to aortic stress and stiffness via HFLA method at optimal length. A and B: addition of phenylephrine (PE) increases aortic stress and stiffness from baseline values. Treatment with NG-nitro-l-arginine methyl ester (l-NAME) to remove effects of endothelial nitric oxide (NO) further augments these increases. Values are means ± SE; n = 4 for both young and old. C: representative smoothed stress trace for young mouse aorta with scale for time and typical levels of stress showing individually additive components of maximal stress and stiffness. D and E: maximally activated VSM accounts for approximately half of maximal total aortic stress (D) and stiffness (E). All stiffness values were obtained via HFLA protocol. ECM, extracellular matrix. Absolute values are means ± SE; n = 3 young, 4 old. ***P < 0.001 with aging; †††P < 0.001 with addition of l-NAME.
These components, defined and separable under our experimental conditions, lead to an additive model of stress and stiffness. This model is presented visually in Fig. 5C in the context of stress, incorporating timescales as well as force responses to stretches that are normalized to wall geometry. We define the sum of baseline and active components of stress and stiffness as intact total and the sum of baseline and maximal active components as maximal total. When baseline and maximal active components of aortic stress and stiffness are plotted to display different fractions of the total, as shown in Fig. 5, D and E, it becomes evident that activated aortic smooth muscle (the source of maximal active components) accounts for more than half of maximal total stress and approximately half of maximal stiffness.
Tyrosine phosphorylation of focal adhesion proteins is impaired with aging because of decreased Src expression.
Increases in aortic stiffness resulting from VSM activity give rise to the question of what molecular mechanisms underlie these contributions. Our group has previously shown that focal adhesions play a key role in regulating aortic stiffness (39). The Src kinase inhibitor PP2 suppresses focal adhesion dynamics as measured by pTyr and endosomal recycling of focal adhesion proteins in aortic smooth muscle (26, 31). This specifically alters cortical stiffness of VSMCs as measured by magnetic microneedle technology (39). In contractile VSMCs, focal adhesion signaling during cell adhesion events is a major source of pTyr (37). The majority of remaining phosphorylation events can be attributed to Ser/Thr phosphorylations (22). Thus we performed screening with phosphotyrosine immunoblots as an indicator of focal adhesion signaling in young and old mouse aorta homogenates of vessels quick-frozen under the same conditions as the aortic rings used for stress and stiffness measurements.
pTyr levels of several bands in PE-stimulated young aortas increased significantly in the presence of the agonist (P < 0.05) (Fig. 6A). Past work has identified the tyrosine phosphorylated bands in aorta as containing Cas (130 kDa), FAK (125 kDa), and paxillin (68 kDa) (26). This increase was readily inhibited in the presence of PP2, an inhibitor of Src kinase (Fig. 6A). In contrast, in old aortas PE produced no significant increase in pTyr and PP2 produced no statistically significant decrease in phosphorylation under the same conditions (Fig. 6B).
Fig. 6.

Agonist-induced, PP2-sensitive focal adhesion signaling is impaired in old aortas. A and B: mean ± SE of densitometry for tyrosine phosphorylation (pTyr) for young and old mouse aortas, with typical blots shown. Typical gel image brightness was adjusted for visual clarity, but densitometry was based on raw data. n = 10 young unstimulated, 4 young +PE and +PE+PP2, 5 old for each treatment. *P < 0.05, **P < 0.01, ***P < 0.001. C: densitometry of c-Src immunoblots of young and old aortic homogenates (n = 3 each). Typical c-Src blot shown in inset; bands are from the same gel but have been digitally moved adjacent to each other. n = 3 young, 3 old. *P < 0.05.
To investigate further the ineffectiveness of Src inhibition in old aortas, we directly assayed expression levels of c-Src by immunoblot analysis. Densitometry of c-Src immunoblots of aortic homogenates, as shown in Fig. 6C, revealed a significant decrease in c-Src levels in aged aortas compared with young aortas, thus at least in part providing an explanation for the impaired regulation of FA signaling in the aortas from old mice. Since tubulin was used in these studies for normalization, the possibility arises that tubulin levels could differ with aging. To test for this possibility, we also quantified tubulin levels in young and old mouse aortas normalized to another “housekeeping” protein, GAPDH, and no significant difference was detectable (young: 0.646 ± 0.149, old: 0.509 ± 0.122; n = 12 each, P = 0.48).
Aging impairs action of a Src small-molecule inhibitor on aortic stiffness and stress.
Since we have previously shown that focal adhesion signaling regulates stiffness in young aortas, the biochemical results above predict that the aortas from old mice should have impaired stress and stiffness responses to PP2. Thus we directly investigated the impact of inhibition of Src and focal adhesion signaling on stress and stiffness in the context of aging. After equilibration at baseline, aortas were treated with PP2 (10 μM), which induced no significant change in stress or stiffness after 30 min of exposure in either young or old aortas (<2% change, ns). After this pretreatment, we activated the samples with PE for 15 min.
As previously reported (39) and confirmed here, PP2 reduced active stress and stiffness in young aortas, but, in marked contrast, the Src inhibitor produced no significant change in the maximal active stress of old aortas (−2%, ns) and a slight decrease in maximal active stiffness (−14%, P < 0.05). In comparing young and old samples we found that, in the presence of PP2, maximal active stress (Fig. 7A) and stiffness (Fig. 7B) of old aortas are more than twice those of young aortas. Thus the in vitro removal of the regulatory mechanism of focal adhesion signaling that normally is able to decrease stiffness in young vessels leads to a high stiffness state in old vessels, reminiscent of the increased PWV measurement in vivo in old vessels (35).
Fig. 7.

Agonist-induced increases in stress and stiffness with Src kinase inhibitor pretreatment are higher with aging. Thoracic aortas pretreated with 10 μM PP2 contract and stiffen less in young but not old mice in the presence of maximal activation (PE + l-NAME). As a result, maximal active stress (A) and stiffness (B) increase >2-fold with aging under these conditions with HFLA measurements. Also shown for comparison are control maximal active data in the absence of PP2 (PP2−), reproduced from Fig. 5, A and B. Absolute values are means ± SE; n = 3 young, 4 old. ***P < 0.001 with aging; ‡P < 0.05, ‡‡‡P < 0.001 with addition of PP2.
DISCUSSION
Components of in vitro stress and stiffness are attributable to distinct tissue-level biological constituents of the aortic wall.
A major finding of the present study is that the smooth muscle cell is a major source and regulator of vascular stiffness, in contrast with the often-assumed dominance of ECM in effecting changes in wall stiffness with aging (15). Also, activated VSM has previously been incorporated into mathematical models of vessel stiffness derived from pressurization studies (13, 34); here we quantify directly the contributions from VSMCs as percentages of total stiffness in an additive model established from real-time observations of force responses to mechanical uniaxial stretch and subsequent normalization to elastic modulus. These findings establish a new perspective in evaluating the relative importance of different biomechanical components in the aortic wall.
The baseline components reflect the biomechanical properties of the in vitro aortic sample without agonist activation of the VSMCs and thus are largely reflective of ECM; this is consistent with the lack of change in baseline mechanical properties to both PP2 and l-NAME. Acute agonist-induced effects are attributed solely to aortic VSMC contributions to stress and stiffness and can be further parsed into those that occur with and without the presence of endothelial NO production, respectively. Establishing such discrete quantities provides a way to compare the relative importance of different biological constituents of the aortic wall in their contributions to aortic stress and stiffness. Our results demonstrate that, when activated, VSMCs are undeniably significant effectors of both total aortic stress and vascular stiffness and, interestingly, are strongly subjected to persistent regulation by endothelial NO production.
Impaired focal adhesion regulation is a source of increased aortic stiffness with aging.
In old aortas, the inefficacy of PP2 implicates focal adhesions as a site of defective cellular signaling with aging. It has previously been shown that Src-dependent aortic focal adhesion signaling and focal adhesion cycling play a critical role in regulating stress and stiffness induced by activation of VSMCs in young mice (31, 39). We show here that old mice lose the response to the inhibitory effects of the Src inhibitor PP2 on agonist-induced stress, stiffness, and phosphotyrosine increases.
These results can be framed within the context of total aortic VSMC biomechanics as shown in Fig. 8. The attachment of myosin cross bridges to actin in activated smooth muscle cells is a major component of agonist-induced stress and stiffness increases in VSMCs, while focal adhesions are known to play the important role of linking the contractile filaments, through the nonmuscle cytoskeleton, to the ECM and the vessel wall via integrins. Alpha agonists as shown here and in past studies regulate both the contractile filaments and the focal adhesions (19, 40). Normal focal adhesions in young aortas provide the capacity to regulate their maturation cycles and their connections to the nonmuscle cytoskeleton to accommodate changes in vessel stress and stiffness and provide, in essence, shock absorption against in vivo hemodynamic events. A similar mechanism has been proposed for airway smooth muscle (50). This regulatory pathway and the consequent potential for shock absorption function are lost in old mice.
Fig. 8.

Shock absorption via Src-mediated focal adhesion/nonmuscle cytoskeleton regulation is defective in old aortas: model of how changes in stiffness generated by cross-bridge attachment and remodeling of focal adhesions and nonmuscle cortical cytoskeleton can be regulated by a PP2-sensitive mechanism present in young but not old aortas. FA, focal adhesion; NMC, nonmuscle cytoskeleton; MLCK, myosin light chain kinase.
HFLA-based measurements of stiffness combine reductionism with physiological relevance.
The traditional engineering approach of large-scale stress-strain analysis, prevalent in mechanical analysis of materials, typically uses a high-amplitude ramp input that produces a stress-strain curve through loading and unloading a tissue sample with large stretches from the unloaded state, otherwise known as slack length or 0% strain. Stiffness in this protocol is defined as the incremental slope (stress-strain ratio) at a particular strain level. In this study, we used stress-strain curves to contextualize our observations of aortic stiffness with aging at specific strain levels. At high strain levels, old aortas have higher passive material stiffness consistent with greater collagen content and recruitment (43). On the other hand, we confirmed the lack of a stiffness increase in old aortas at physiological strain levels, which we ascertained with the HFLA protocol.
While stress-strain curves define a large pool of stiffness data across a wide range of strain values, the high-amplitude protocol used to collect these data has several disadvantages. The large stretches disrupt actomyosin cross bridges, decreasing contractile responses in the VSM and removing a major source of cellular stiffness. In addition, VSMC force output depends critically on the level of stretch it experiences (28). Therefore, large-amplitude stretches disturb the system whose properties we are trying to measure. Furthermore, the stress-strain curve protocol stipulates the slack or unloaded state as the equilibration point before and after stretch; this prohibits the acquisition of steady-state stress data at any physiologically relevant aortic strain or during the response to a contractile agonist, which is more relevant to the in vivo biomechanical environment of the aorta.
The HFLA protocol, in contrast, uses low-amplitude stretches and ample equilibration time after perturbations and treatments, in correspondence with traditional isometric studies of smooth muscle cell contractility, to cause minimal perturbation of the system (8, 18). Additionally, we are interested specifically in the mechanical properties of the aorta under physiological conditions. Thus HFLA allowed us in the present study to determine both steady-state stress and local stiffness via incremental stress-strain measurements at optimal length that preserves smooth muscle cell activity, functionality, and relevance.
Applications and limitations of in vitro results for in vivo predictions.
The present results are an important first step in fully understanding the mechanisms of changes in aortic stiffness with aging and identifying potential molecular targets for the prevention or reversal of aging-induced increases in stiffness. Geometric considerations in vivo will modify the in situ stiffness of the aorta. These factors are challenging to measure accurately, since experimental animals are generally anesthetized for PWV measurements. In addition to geometric considerations, another notable factor in vivo that is not taken into consideration in an in vitro environment is the presence of multiple ill-defined neurohumoral mediators. We have shown the capacity of NO to dramatically modify the material properties of the aorta, but knowing how much NO is released at any point in time in vivo by blood shear forces is technically challenging (17, 41). This reinforces the notion that the stiffness measurements made here are representative of mechanical properties that are inherent to the material, which is only one aspect of aortic function in vivo.
Additionally, the results presented here are definitive only for the experimental parameters used. The protocol applies only circumferential strain and does not account for the axial strain experienced by aorta in vivo (16). Optimal length was determined to be at the same strain level—80%—in both young and old aortas and used to set vessel diameter in our studies, but it is unknown whether optimal length corresponds to a diameter characteristic of the predominant in vivo operating strain of either, especially where aging-associated hypertension is expected to increase diameter. The physiological range of strain defined by Wagenseil and Mecham (44) does not specify which values occur most commonly in mice of different ages during normal blood flow. Furthermore, the HFLA protocol is designed to minimize perturbations to cross bridges, but in vivo aortas undergo significantly larger deformation during the cardiac cycle, which may in itself alter cross bridge formation.
Despite these characteristics of an in vitro environment, we attempted to mimic in vivo conditions as closely as possible. At physiological mean strain and optimal length, in oxygenated salt solutions containing extracellular calcium, maintaining body temperature, and with fully viable VSMCs, we measured material stiffness of young and old mouse aorta with high reproducibility. We conclude from our results that the smooth muscle focal adhesions represent a potential therapeutic target in the context of preventing or reversing increases in aortic stiffness. The reasons are threefold: first, the smooth muscle cell is a major regulator of stiffness at physiological lengths, as shown here; second, a Src small-molecule inhibitor is sufficient to counteract stiffness increases at the tissue level; and third, this capacity is diminished with aging. Therefore, an understanding of this mechanism may lead to an approach to reverse this aging-induced deficiency.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants P01 HL-086655 to K. G. Morgan and HL-098976 to B. Suki and by the Evans Center for Interdisciplinary Biomedical Research Affinity Research Collaborative (ARC) on Molecular, Biomechanical, and Genetic Mechanisms of Aortic Stiffness at Boston University.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: Y.Z.G., R.J.S., B.S., and K.G.M. conception and design of research; Y.Z.G. and R.Y. performed experiments; Y.Z.G. and K.G.M. analyzed data; Y.Z.G., R.J.S., and K.G.M. interpreted results of experiments; Y.Z.G. prepared figures; Y.Z.G. and R.J.S. drafted manuscript; Y.Z.G., R.J.S., B.S., and K.G.M. edited and revised manuscript; Y.Z.G., R.J.S., R.Y., B.S., and K.G.M. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Cynthia Gallant, Qian Qian Lin, and Susanne Vetterkind for their technical assistance as well as Dr. Richard A. Cohen (Boston University) and Dr. Gary Mitchell (Cardiovascular Engineering, Inc.) for scientific input and discussions.
REFERENCES
- 1.Barra JG, Armentano RL, Levenson J, Fischer EI, Pichel RH, Simon A. Assessment of smooth muscle contribution to descending thoracic aorta elastic mechanics in conscious dogs. Circ Res 73: 1040–1050, 1993 [DOI] [PubMed] [Google Scholar]
- 2.Barton M, Haudenschild CC, D'Uscio LV, Shaw S, Münter K, Lüscher TF. Endothelin ETA receptor blockade restores NO-mediated endothelial function and inhibits atherosclerosis in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA 95: 14367–14372, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ben-Shlomo Y, Spears M, Boustred C, May M, Anderson SG, Benjamin EJ, Boutouyrie P, Cameron J, Chen CH, Cruickshank JK, Hwang SJ, Lakatta EG, Laurent S, Maldonado J, Mitchell GF, Najjar SS, Newman AB, Ohishi M, Pannier B, Pereira T, Vasan RS, Shokawa T, Sutton-Tyrell K, Verbeke F, Zoungas S, McEniery CM, Cockcroft JR, Wilkinson IB. Aortic pulse wave velocity improves cardiovascular event prediction: an individual participant meta-analysis of prospective observational data from 17,635 subjects. J Am Coll Cardiol 63: 636–646, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry CL, Greenwald SE, Rivett JF. Static mechanical properties of the developing and mature rat aorta. Cardiovasc Res 9: 669–678, 1975 [DOI] [PubMed] [Google Scholar]
- 5.Blacher J, Guerin AP, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness on survival in end-stage renal disease. Circulation 99: 2434–2439, 1999 [DOI] [PubMed] [Google Scholar]
- 6.Blaustein MP, Kao JP, Matteson DR. Dynamics of smooth muscle contraction differ markedly from those of skeletal and cardiac muscle. In: Cellular Physiology and Neurophysiology. Philadelphia, PA: Elsevier, 2012 [Google Scholar]
- 7.Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res 66: 286–294, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Brozovich FV, Morgan KG. Stimulus-specific changes in mechanical properties of vascular smooth muscle. Am J Physiol Heart Circ Physiol 257: H1573–H1580, 1989 [DOI] [PubMed] [Google Scholar]
- 9.Clark JM, Glagov S. Transmural organization of the arteria media. The lamellar unit revisited. Arterioscler Thromb Vasc Biol 5: 19–34, 1985 [DOI] [PubMed] [Google Scholar]
- 10.Cruickshank K, Rise L, Anderson SG, Wright JS, Dunn G, Gosling RG. Aortic pulse-wave velocity and its relationship to mortality in diabetes and glucose intolerance: an integrated index of vascular function? Circulation 106: 2085–2090, 2002 [DOI] [PubMed] [Google Scholar]
- 11.Fleenor BS, Eng JS, Sindler AL, Pham BT, Kloor JD, Seals DR. Superoxide signaling in perivascular adipose tissue promotes age-related artery stiffness. Aging Cell 13: 576–578, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleenor BS, Sindler AL, Eng JS, Nair DP, Dodson RB, Seals DR. Sodium nitrite de-stiffening of large elastic arteries with aging: role of normalization of advanced glycation end-products. Exp Gerontol 47: 588–594, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gleason RL, Dye WW, Wilson E, Humphrey JD. Quantification of the mechanical behavior of carotid arteries from wild-type, dystrophin-deficient, and sarcoglycan-δ knockout mice. J Biomech 41: 3213–3218, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gosling RG, Budge MM. Terminology for describing the elastic behavior of arteries. Hypertension 41: 1180–1182, 2003 [DOI] [PubMed] [Google Scholar]
- 15.Greenwald S. Ageing of the conduit arteries. J Pathol 211: 157–172, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Humphrey JD, Eberth JF, Dye WW, Gleason RL. Fundamental role of axial stress in compensatory adaptations by arteries. J Biomech 42: 1–8, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irace C, Carallo C, De Franceschi MS, Scicchitano F, Milano M, Tripolino C, Scavelli F, Gnasso A. Human common carotid wall shear stress as a function of age and gender: a 12-year follow-up study. Age (Dordr) 34: 1553–1562, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawai M, Brandt PW. Sinusoidal analysis: a high resolution method for correlating biochemical reactions with physiological processes in activated skeletal muscles of rabbit, frog and crayfish. J Muscle Res Cell Motil 1: 279–303, 1980 [DOI] [PubMed] [Google Scholar]
- 19.Kim HR, Appel S, Vetterkind S, Gangopadhyay S, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med 12: 2165–2180, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. III. Cellular and molecular clues to heart and arterial aging. Circulation 107: 490–497, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, Ducimetiere P, Benetos A. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension 37: 1236–1241, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Li Y, Reznichenko M, Tribe RM, Hess PE, Taggart M, Kim H, DeGnore JP, Gangopadhyay S, Morgan KG. Stretch activates human myometrium via ERK, caldesmon and focal adhesion signaling. PLoS One 4: e7489, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marganski WA, Gangopadhyay SS, Je HD, Gallant CM, Morgan KG. Targeting of a Ca2+/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res 97: 541–549, 2005 [DOI] [PubMed] [Google Scholar]
- 24.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, Witteman JC. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam study. Circulation 113: 657–663, 2006 [DOI] [PubMed] [Google Scholar]
- 25.McAllister DA, Maclay JD, Mills NL, Mair G, Miller J, Anderson D, Newby DE, Murchison JT, MacNee W. Arterial stiffness is independently associated with emphysema severity in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 176: 1208–1214, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Min J, Reznichenko M, Poythress RH, Gallant CM, Vetterkind S, Li Y, Morgan KG. Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J Cell Physiol 227: 3585–3592, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell GF, Guo CY, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, Vasan RS, Levy D. Cross-sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation 115: 2628–2636, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Peiper U, Laven R, Regnat K, Schmidt E. Mechanical response to stretch of depolarized vascular smooth muscle fibres. Basic Res Cardiol 69: 1–10, 1974 [DOI] [PubMed] [Google Scholar]
- 29.Pezet M, Jacob MP, Escoubet B, Gheduzzi D, Tillet E, Perret P, Huber P, Quaglino D, Vranckx R, Li DY, Starcher B, Boyle WA, Mecham RP, Faury G. Elastin haploinsufficiency induces alternative aging processes in the aorta. Rejuvenation Res 11: 97–112, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Philibert C, Bouillot S, Huber P, Faury G. Protocadherin-12 deficiency leads to modifications in the structure and function of arteries in mice. Pathol Biol (Paris) 60: 34–40, 2012 [DOI] [PubMed] [Google Scholar]
- 31.Poythress RH, Gallant CM, Vetterkind S, Morgan KG. Vasoconstrictor-induced endocytic recycling regulates focal adhesion protein localization and function in vascular smooth muscle. Am J Physiol Cell Physiol 305: C215–C227, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prado CM, Rossi MA. Circumferential wall tension due to hypertension plays a pivotal role in aortic remodeling. Int J Exp Pathol 87: 425–436, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Rusuello RR, Natividad FF, Hunter WC, Genin GM, Elson EL, Vatner DE, Meininger GA, Vatner SF. Vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res 107: 615–619, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rachev A, Hayashi K. Theoretical study of the effects of vascular smooth muscle contraction on strain and stress distributions in arteries. Ann Biomed Eng 27: 459–468, 1999 [DOI] [PubMed] [Google Scholar]
- 35.Reddy AK, Li YH, Pham TT, Ochoa LN, Treviño MT, Hartley CJ, Michael LH, Entman ML, Taffet GE. Measurement of aortic input impedance in mice: effects of age on aortic stiffness. Am J Physiol Heart Circ Physiol 285: H1464–H1470, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Rees DD, Palmer RM, Schulz R, Hodson HF, Moncada S. Characterization of three inhibitors of endothelial nitric oxide synthesis in vitro and in vivo. Br J Pharmacol 101: 746–752, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res 98: 606–616, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Russell A, Watts S. Vascular reactivity of isolated thoracic aorta of C57BL/6J mouse. J Pharmacol Exp Ther 294: 598–604, 2000 [PubMed] [Google Scholar]
- 39.Saphirstein RJ, Gao YZ, Jensen MH, Gallant CM, Vetterkind S, Moore JR, Morgan KG. The focal adhesion: a regulated component of aortic stiffness. PLoS One 8: e62461, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saphirstein RJ, Morgan KG. The contribution of vascular smooth muscle to aortic stiffness across length scales. Microcirculation 21: 201–207, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soucy KG, Ryoo S, Benjo A, Lim HK, Gupta G, Sohi JS, Elser J, Aon MA, Nyhan D, Shoukas AA, Berkowitz DE. Impaired shear stress-induced nitric oxide production through decreased NOS phosphorylation contributes to age-related vascular stiffness. J Appl Physiol 101: 1751–1759, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Taddei S, Virdis A, Ghiadoni L, Salvetti G, Bernini G, Magagna A, Salvetti A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 38: 247–279, 2001 [DOI] [PubMed] [Google Scholar]
- 43.Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res 5: 264–273, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev 89: 957–989, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, Lob HE, Santhanam L, Mitchell GF, Cohen RA, Seta F. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension 62: 1105–1110, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation 113: 664–670, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Wolinsky H, Glagov S. Structural basis for the static mechanical properties of the aortic media. Circ Res 14: 400–413, 1964 [DOI] [PubMed] [Google Scholar]
- 48.Wu J, Thabet SR, Kirabo A, Trott DW, Saleh MA, Xiao L, Madhur MS, Chen W, Harrison DG. Inflammation and mechanical stretch promote aortic stiffening in hypertension through activation of p38 mitogen-activated protein kinase. Circ Res 114: 616–625, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamin R, Morgan KG. Deciphering actin cytoskeletal function in the vascular smooth muscle cell. J Physiol 590: 4145–4154, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix. Proc Am Thorac Soc 5: 32–39, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]