

Abstract
Acid–base disorders are common in critically ill patients. Metabolic acid–base disorders are particularly common in patients who require acute renal replacement therapy. In these patients, metabolic acidosis is common and multifactorial in origin. Analysis of acid–base status using the Stewart–Figge methodology shows that these patients have greater acidemia despite the presence of hypoalbuminemic alkalosis. This acidemia is mostly secondary to hyperphosphatemia, hyperlactatemia, and the accumulation of unmeasured anions. Once continuous hemofiltration is started, profound changes in acid–base status are rapidly achieved. They result in the progressive resolution of acidemia and acidosis, with a lowering of concentrations of phosphate and unmeasured anions. However, if lactate-based dialysate or replacement fluid are used, then in some patients hyperlactatemia results, which decreases the strong ion difference and induces an iatrogenic metabolic acidosis. Such hyperlactatemic acidosis is particularly marked in lactate-intolerant patients (shock with lactic acidosis and/or liver disease) and is particularly strong if high-volume hemofiltration is performed with the associated high lactate load, which overcomes the patient's metabolic capacity for lactate. In such patients, bicarbonate dialysis seems desirable. In all patients, once hemofiltration is established, it becomes the dominant force in controlling metabolic acid–base status and, in stable patients, it typically results in a degree of metabolic alkalosis. The nature and extent of these acid–base changes is governed by the intensity of plasma water exchange/dialysis and by the 'buffer' content of the replacement fluid/dialysate, with different effects depending on whether lactate, acetate, citrate, or bicarbonate is used. These effects can be achieved in any patient irrespective of whether they have acute renal failure, because of the overwhelming effect of plasma water exchange on nonvolatile acid balance. Critical care physicians must understand the nature, origin, and magnitude of alterations in acid–base status seen with acute renal failure and during continuous hemofiltration if they wish to provide their patients with safe and effective care.
Keywords: acidosis, alkalosis, bicarbonate, hemofiltration, hemodialysis, renal replacement therapy
Introduction
Acute renal failure (ARF) in the critically ill is still associated with a poor prognosis [1,2]. Metabolic acid–base disorders are particularly common in these patients, especially acidosis. The pathogenesis of such acidosis remains poorly understood because its main cause in ARF patients is not fully understood. However, the nature of this metabolic acidosis is likely multifactorial and probably includes the effect of chloride-rich fluid resuscitation [3] and the accumulation of lactate, phosphate, and unexcreted metabolic acids such as sulfate [4]. This multifactorial metabolic acidosis associated with ARF often leads to acidemia. Furthermore, persistent acidosis has been demonstrated to be an indicator of poor prognosis [5]. The rationale behind the perceived need to correct severe acidosis lies in the potential adverse cellular effects of such metabolic disturbance on myocardial function, likelihood of arrhythmias, and pulmonary vascular tone. However, very few studies [6] have in fact established that clinically significant benefits might arise from the correction of such acidosis.
Nonetheless, renal replacement therapy (RRT) such as intermittent hemodialysis (IHD), continuous venovenous hemofiltration (CVVH), continous venovenous hemodailysis, and continuous venovenous hemodiafiltration (CVVHDF) has been applied to the treatment of critically ill patients with ARF to improve fluid overload, uremia, and acid–base disorders. The use of RRT and adjustments in the replacement solutions administered to acidotic critically ill patients with ARF can have a substantial effect on acid–base homeostasis. Furthermore, high-volume hemofiltration (HVHF) may have an even stronger effect on acid–base disorders. Therefore, improving our understanding of the impact of RRT on acid–base disorders and gaining insights into the nature of such disorders and the mechanisms of action of RRT are important.
In the present review we explore the acid–base disorders seen in ARF, the effect of RRT and its modalities on acid–base disorders, the effect of replacement fluid on acid–base balance, and the effect of HVHF on acid–base balance. A strong focus is given to the clinical implications of these interventions, with the aim of helping clinicians better understand and manage the acid–base disorders in ARF and critically ill patients in general.
Acid–base analysis using the Stewart–Figge methodology
As described above, the pathogenesis of acid–base disorders of ARF remains unknown and the cause of acidosis in ARF patients is probably multifactorial. It is hard to quantitatively approciate such multifactorial metabolic disorders by means of the classical Henderson–Hasselbach method. Recently, however, quantitative acid–base analysis using the Stewart–Figge approach [7,8] was introduced. This method first involves calculating the apparent strong ion difference (SID; all concentrations in mEq/l):
Apparent SID = [Na+] + [K+] + [Mg2+] + [Ca2+] - [Cl-] - [lactate]
The calculation then takes into account the role of weak acids (carbon dioxide, albumin, and phosphate) in the balance of electrical charges in plasma water, as expressed through calculation of the effective SID (partial carbon dioxide tension [PCO2] in mmHg, albumin in g/l, and phosphate in mmol/l):
Effective SID = 1000 × 2.46 × 10-11 × PCO2/(10-pH) + [albumin] × (0.12 × [pH - 0.631]) + [phosphate] × (0.309 × [pH - 0.469])
Once weak acids are quantitatively taken into account, the difference between apparent and effective SID should be zero, unless there are unmeasured charges (anions). Such charges are then described by the strong ion gap (SIG):
SIG = apparent SID – effective SID.
The component of albumin and phosphate is defined as the total concentration of nonvolatile weak acid (Atot). [Atot], along with SID and PCO2, is an independent determinant of [H+] or pH. According to the Stewart–Figge approach, metabolic acidosis can then result from a reduction in the SID or from an increase in Atot, and respiratory acidosis can result from a gain in PCO2. The changes in each of these variables can be quantified to express how much each one is responsible (in mEq/l) for the findings on blood analysis.
Acid–base balance in acute renal failure
Classically, metabolic acidosis in renal failure is described as a high anion gap metabolic acidosis. However, in the clinical setting, the anion gap is not always elevated. These findings might lead clinicians to diagnostic and therapeutic confusion. In these situations, quantitative analysis using the Stewart–Figge approach can be helpful. In this regard, Rocktaeschel and coworkers [9] recently examined the acid–base status of ARF patients using the Stewart–Figge methodology and demonstrated several features. First, critically ill patients with ARF were typically acidemic compared with control patients (Fig. 1). Second, this acidemia appeared secondary to metabolic acidosis with a mean base excess of approximately -7 mEq/l, which appeared secondary to the accumulation of lactate, phosphate, and unmeasured anions (possible candidates for these unmeasured anions include sulfate, urate, hydroxypropionate, oxalate, and furanpropionate [10]; Fig. 2). Third, in these patients there was also a marked failure to alter the apparent SID to achieve a degree of metabolic compensation (Fig. 3). Despite this finding, half of the ARF patients had an anion gap within the normal range. Furthermore, these acidifying disorders were attenuated by a concomitant metabolic alkalosis, which was essentially secondary to hypoalbuminemia. Hypoalbuminemia lowered the anion gap and masked the presence of acidifying anions to those clinicians using conventional acid–base analysis.
Figure 1.
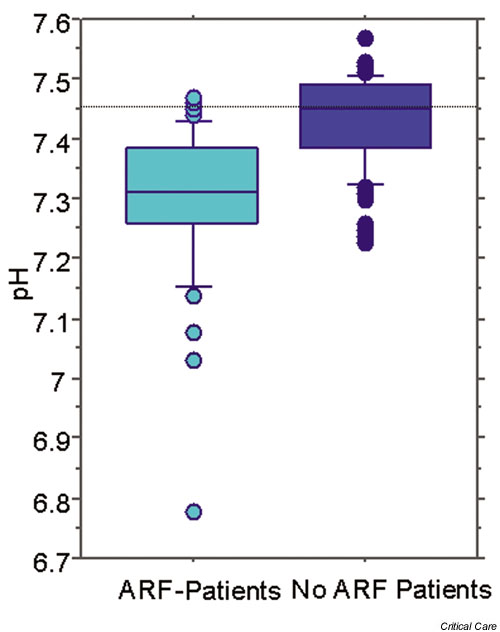
Difference in pH between patients with acute renal failure (ARF) in an intensive care unit (ICU) and a control population of ICU patients.
Figure 2.
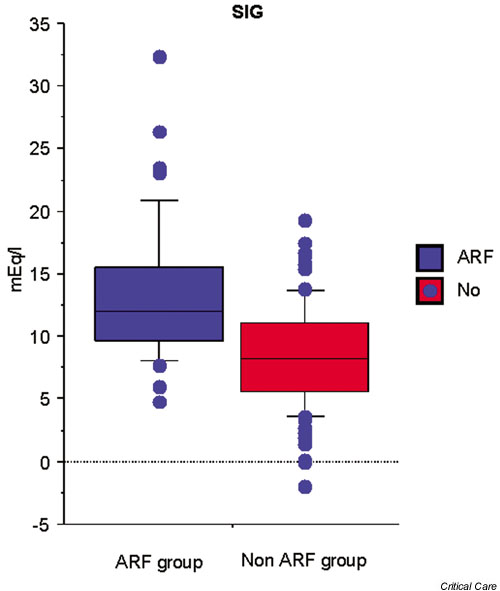
Differences in strong ion gap (SIG) between (ARF) patients and controls in an intensive care unit.
Figure 3.
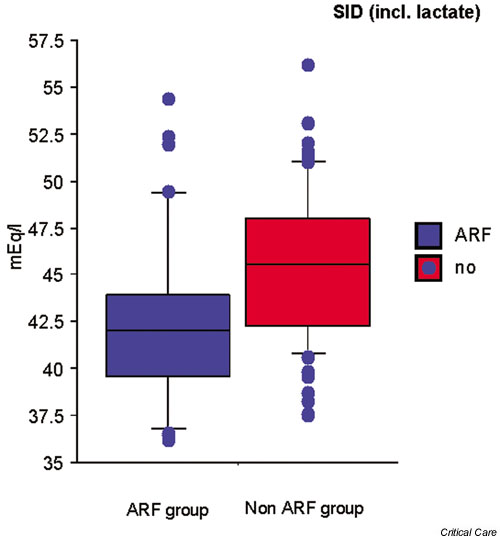
Differences in apparent strong ion difference (SIDa) between acute renal failure (ARF) patients and control individuals in an intensive care unit.
Effect of renal replacement therapy on acid–base balance
There are two major modalities of RRT. One is intermittent and the other continuous. Few studies have been done to detect which modality is better in terms of acid–base control. Uchino and coworkers [11] compared the effect on acid–base balance of IHD and CVVHDF. Before treatment, metabolic acidosis was common in both groups (63.2% for IHD and 54.3% for CVVHDF). Both IHD and CVVHDF corrected metabolic acidosis. However, the rate and degree of correction differed significantly. CVVHDF normalized metabolic acidosis more rapidly and more effectively during the first 24 hours than did IHD (P < 0.01). IHD was also associated with a higher incidence of metabolic acidosis than was CVVHDF during the subsequent 2 week treatment period (P < 0.005; Fig. 4). Accordingly, CVVHDF can be considered physiologically superior to IHD in the correction of metabolic acidosis. The overwhelming superiority of continuous RRT in terms of control of acidosis was also recently established in comparison with peritoneal dialysis, with all patients randomized to CVVH achieving correction of acidosis by 50 hours of treatment, compared with only 15% of those treated by peritoneal dialysis (P < 0.001) [12]. How does continuous RRT correct acidosis?
Figure 4.
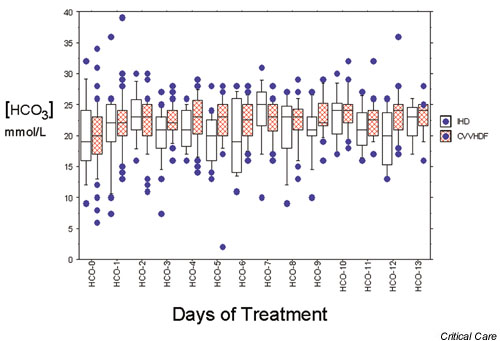
Box plot illustrating bicarbonate control with intermittent dialysis (IHD) and continuous therapy (continuous venovenous hemodiafiltration [CVVHDF]).
To gain insights into the mechanisms by which continuous RRT corrects metabolic acidosis in ARF, Rocktaschel and coworkers [13] studied the effect of CVVH on acid–base balance using the Stewart–Figge methodology. Before commencing CVVH, patients had mild acidemia secondary to metabolic acidosis. This acidosis was due to increased unmeasured anions (SIG 12.3 mEq/l), hyperphosphatemia, and hyperlactatemia. It was attenuated by the alkalizing effect of hypoalbuminemia. Once CVVH was commenced, acidemia was corrected within 24 hours. This change was associated with a decreased SIG, and decreased phosphate and chloride concentrations. This correction was so powerful and dominant that, after 3 days of CVVH, patients developed alkalemia secondary to metabolic alkalosis (bicarbonate 29.8 mmol/l, base excess 6.7 mmol/l; Fig. 1). This alkalemia appeared due to a further decrease in SIG and a further decrease in serum phosphate concentration in the setting of persistent hypoalbuminemia. Hence, CVVH appears to correct metabolic acidosis in ARF through its effects on unmeasured anions, phosphate, and chloride. Once hemofiltration is established, it becomes the dominant force in controlling metabolic acid–base status, and in stable patients it typically results in a degree of metabolic alkalosis.
Effect of replacement fluid composition (lactate, acetate, bicarbonate, and citrate)
The exchange of approximately 30 l plasma water per day is necessary to achieve adequate control of uremia and acid–base disorders in ARF [14]. During continuous RRT, according to conventional acid–base thinking, there is a substantial loss of endogenous bicarbonate, which must be substituted by the addition of 'buffer' substances. (According to the Stewart–Figge approach, the explanation for this is that there is loss of a fluid with an SID of approximately 40 mEq/l, which must be replaced by a fluid with a similar SID.)
Lactate, acetate, and bicarbonate have been used as 'buffers' (or SID generators according to Stewart [7]) during RRT. Citrate has been used as a 'buffer' and for anticoagulation. These 'buffers' affect acid–base balance, and therefore we must understand their physiologic characteristics.
Bicarbonate has the major advantage of being the most physiologic anion equivalent. However, the production of a commercially available bicarbonate-based solution is not easy because of the formation of calcium and magnesium salts during long-term storage. Furthermore, the cost of this solution is approximately three times greater than that of other 'buffer' solutions. Accordingly, acetate and lactate have been used widely for RRT. Under normal conditions, acetate is rapidly converted on a 1:1 basis to carbon dioxide and then bicarbonate by both liver and skeletal muscle. Lactate is also rapidly converted in the liver on a 1:1 basis [15].
Studies of acetate-based solutions appear to exert a negative influence on the mean arterial blood pressure and cardiac function in the critically ill [16-18]. Morgera and coworkers [19] compared acid–base balance between acetate-buffered and lactate-buffered replacement fluids, and reported that the acetate-buffered solution was associated with a significant lower pH and bicarbonate levels than was the lactate-buffered solution. However, the acetate-buffered solution had 9.5 mmol/l less 'buffer' than the lactate-buffered one. Therefore, the difference is probably simply a matter of dose rather than choice of 'buffer'. From the Stewart–Figge perspective, the acetate-buffered solution contained 8 mmol/l chloride more than the lactate-buffered solution to achieve electrical equilibrium. This reduces the SID of the replacement fluid and acidifies blood more.
Thomas and coworkers [20] compared the effects of lactate-buffered versus bicarbonate-buffered fluids. Hemofiltration fluids contained either 44.5 mmol/l sodium lactate or 40.0 mmol/l sodium bicarbonate with 3 mmol/l lactate (43 mmol/l). Lactate-buffered fluids contained 142 mmol/l sodium and 103 mmol/l chloride (SID 39 mEq/l), and bicarbonate-buffered fluids contained 155 mmol/l sodium and 120 mmol/l chloride (SID 35 mEq/l). Lactate rose from approximately 2 mmol/l to 4 mmol/l when lactate-based fluids were given but not with bicarbonate. Both therapies resulted in a similar improvement in metabolic acidosis. Potentially, the lactate-buffered fluid could have had a more alkalinizing effect. However, the accumulation of lactate in blood might have offset this effect and attenuated the trend toward a higher base excess with the lactate-buffered fluids.
Tan and coworkers [21] studied the acid–base effect of CVVH with lactate-buffered and bicarbonate-buffered solutions. The lactate-buffered solution had an SID of 46 mEq/l, as compared with 35 mEq/l for the bicarbonate fluid. From the Stewart–Figge point of view, the lactate-buffered solution should have led to a greater amount of alkalosis. However, that study found a significant increase in plasma lactate levels and a decrease in base excess with the lactate-buffered solution (Figs 5 and 6). Lactate, if not metabolized and still present in blood, acts as a strong anion, which would have the same acidifying effect of chloride. Accordingly, iatrogenic hyperlactatemia can cause a metabolic acidosis (Fig. 7). The controversy can, of course, also be resolved by failure to convert exogenous lactate into bicarbonate.
Figure 5.
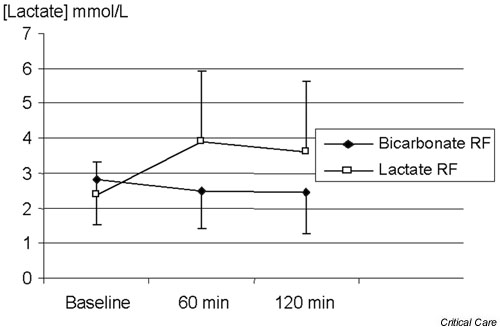
Effect of bicarbonate-based replacement fluids (bicarbonate RF) and lactate-based replacement fluids (lactate RF) on blood lactate levels.
Figure 6.
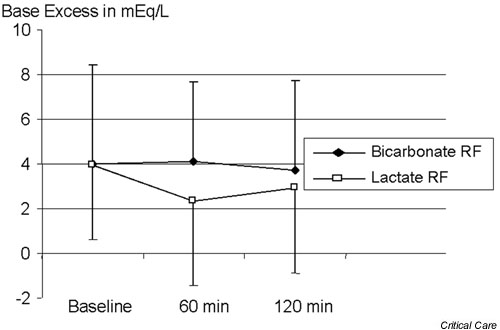
Effect of bicarbonate-based replacement fluids (bicarbonate RF) and lactate-based replacement fluids (lactate RF) on base excess.
Figure 7.
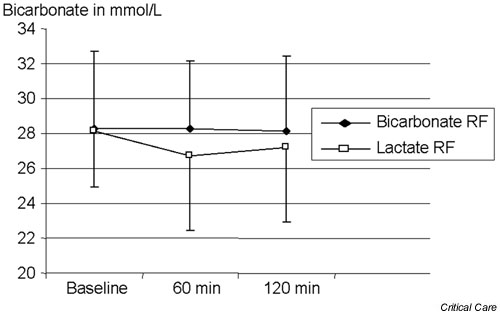
Effect of bicarbonate-based replacement fluids (bicarbonate RF) and lactate-based replacement fluids (lactate RF) on serum bicarbonate levels.
Most commercially available replacement fluids are buffered with approximately 40–46 mmol/l lactate. In the vast majority of patients, the administration of such replacement fluid maintains a normal serum bicarbonate level without any significant increase in blood lactate concentration. Because the ability of the liver to metabolize lactate is in the region of 100 mmol/hour [22], even aggressive CVVH at 2 l/hour exchange would still deliver less than the normal liver can handle.
However, if lactate-based dialysate or replacement fluids are used in some patients with liver dysfunction or shock, then the administration of lactate-buffered fluids can induce significant hyperlactatemia and acidosis because the metabolic rate is insufficient to meet the additional lactate load. Although lactate normally acts as a 'buffer' by being removed from the circulation and thereby lowering the SID, if lactate is only partly metabolized and accumulates in plasma water then it acts like a strong anion. Thus, hyperlactatemia decreases the apparent SID, which results in increased dissociation of plasma water and thereby lowers the pH.
Citrate has been used for regional anticoagulation. During this procedure, citrate is administered to the circuit before the filter and chelates calcium, thus impeding coagulation. Once citrate enters the circulation, it is metabolized to carbon dioxide and then bicarbonate on a 1: 3 basis; thus, 1 mmol citrate yields 3 mmol carbon dioxide and then bicarbonate.
Under these circumstances, citrate acts as the 'buffer' as well as the anticoagulant. If the method described by Mehta and coworkers [23] is applied, then approximately 48 mmol/hour 'bicarbonate equivalent' is given as citrate. This rate of alkali administration may result in metabolic alkalosis (in up to 25% of cases). Caution is warranted in patients with liver disease, who may not be able to metabolize citrate. In these patients, citrate may accumulate and result in severe ionized hypocalcemia and metabolic acidosis because the citrate anion (C6H5O73-) acts as an unmeasured anion and increases the SIG, which has acidifying effects.
When oxidizable anions are used in the replacement fluids, the anion (acetate, lactate, and citrate) must be completely oxidized to carbon dioxide and water in order to generate bicarbonate. If the metabolic conversion of nonbicarbonate anions proceeds without accumulation, then their buffering capacity is equal to that of bicarbonate. Thus, the effect on acid–base status depends on the 'buffer' concentration rather than on the kind of 'buffer' used [15]. When the metabolic conversion is impaired, the increased blood concentration of the anions leads to an increased strong anion in lactate or unmeasured anions for acetate and citrate. All lower the apparent SID and acidify blood. The nature and extent of these acid–base changes is governed by the intensity of plasma water exchange/dialysis, by the 'buffer' content of the replacement fluid/dialysate, and by the metabolic rate for these anions.
Effect of high volume hemofiltration on acid–base balance
Recently, HVHF was applied to the treatment of septic shock patients, with favorable hemodynamic results [24]. However, if commercial lactate-buffered replacement fluid is used during HVHF, then patients might receive more than 270 mmol/hour exogenous lactate. This lactate load could overcome endogenous lactate metabolism, even in healthy subjects [25], and result in progressive hyperlactatemia. Hyperlactatemia has been reported with lactate-buffered fluids in critically ill ARF patients treated with intermittent hemofiltration and a lactate load of 190–210 mmol/hour [16]. Such hyperlactatemia might induce a metabolic acidosis. Cole and coworkers [26] studied the effect of HVHF on acid–base balance. HVHF with lactate-buffered replacement fluids (6 l/hour of lactate-buffered fluids) induced iatrogenic hyperlactatemia. Plasma lactate levels increased from a median of 2.51 mmol/l to a median of 7.3 mmol/l at 2 hours (Fig. 8). This change was accompanied by a significant decrease in bicarbonate and base excess. However, such hyperlactatemia had only a mild and transient acidifying effect. A decrease in chloride and effective SID and the removal of unmeasured anions (decrease in SIG) all rapidly compensated for this effect (Fig. 9). Thus, the final effect was that HVHF induced only a minor change in pH from 7.42 to 7.39 at 2 hours. In the period from 2 to 8 hours, the blood lactate concentration remained stable at around 7–8 mmol/l, whereas compensatory effects continued, which restored bicarbonate levels to 27.2 mmol/l and pH to 7.44 by 8 hours of treatment.
Figure 8.
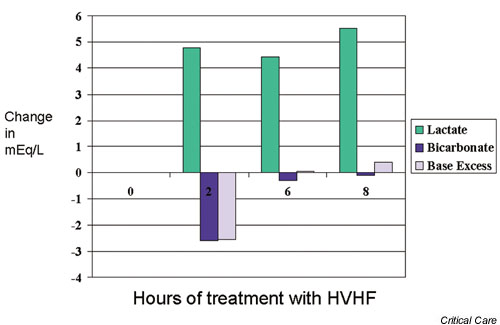
Effect of high-volume hemofiltration (HVHF) on lactate, bicarbonate, and base excess.
Figure 9.
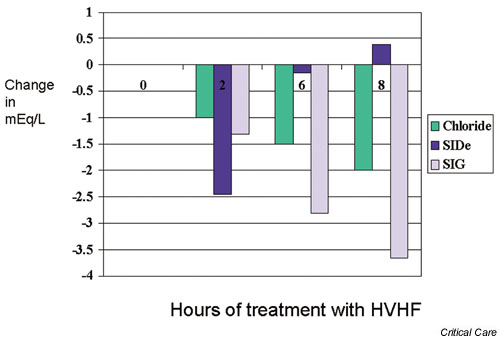
Effect of high-volume hemofiltration (HVHF) on chloride, effective strong ion difference (SIDe), and strong ion gap (SIG).
Although the chloride concentration in the replacement fluid was high compared with the serum chloride level, a progressive decrease in chloride was observed. This might be due to chloride losses in excess of gains. Uchino and coworkers [27] examined the sieving coefficient for chloride during HVHF and found a sieving coefficient for chloride in excess of 1. Another possible explanation for hypochloremia would be the intracellular movement of chloride in response to metabolic acidosis (chloride shift). A decrease in effective SID was explained by the aggregate minor changes in arterial PCO2, albumin, and phosphate. The changes in SIG appeared most likely to be due to simple filtration of unmeasured anion.
Consequently, HVHF with lactate-buffered fluids induced a marked hyperlactatemia but did not induce a progressive acidosis. However, caution is warranted in particular patients who have marked pretreatment hyperlactatemia (>5 mmol/l) or liver dysfunction, or where the intensity of HVHF exceeds 6 l/hour plasma water exchange. Bicarbonate use is warranted in such patients.
Conclusion
RRT can strongly affect acid–base disorders and can be used to correct severe metabolic acidosis. If the dose of treatment is titrated to achieve such a goal, essentially even the most dramatic metabolic acidosis can be corrected. Replacement fluid solutions containing 'buffers' such as lactate, acetate, bicarbonate, and citrate can have a variable effect on acid–base balance, depending on the dose and rate of metabolic disposition, as clearly seen in the setting of HVHF. Critical care physicians must understand the nature, origin, and magnitude of the alterations in acid–base status seen with ARF and associated disorders, and the powerful effects of continuous hemofiltration if they wish to provide their patients with safe and effective care.
Competing interests
None declared.
Abbreviations
ARF = acute renal failure; Atot = total concentration of nonvolatile weak acid; CVVH = continuous venovenous hemofiltration; CVVHDF = continuous venovenous hemodiafiltration; HVHF = high-volume hemofiltration; IHD = intermittent hemodialysis; PCO2 = partial carbon dioxide tension; RRT = renal replacement therapy; SID = strong ion difference; SIG = strong ion gap.
References
- Spiegel DM, Ullian ME, Zerbe GO, Berl T. Determinant of survival and recovery in acute renal failure patients dialysed in the intensive care unit. Am J Nephrol. 1991;11:44–47. doi: 10.1159/000168271. [DOI] [PubMed] [Google Scholar]
- Brivet F, Kleinknecht D, Loirat P. Acute renal failure in intensive care units – causes, outcome, and prognostic factors: a prospective, multicenter study. Crit Care Med. 1996;24:192–198. doi: 10.1097/00003246-199602000-00003. [DOI] [PubMed] [Google Scholar]
- Kellum JA, Bellomo R, Kramer DJ, Pinsky MR. Etiology of metabolic acidosis during saline resuscitation in endotoxemia. Shock. 1998;9:1–5. doi: 10.1097/00024382-199805000-00009. [DOI] [PubMed] [Google Scholar]
- Prough DS, Bidani A. Hyperchloremic metabolic acidosis is a predictable consequence of intraoperative infusion of 0.9% saline. Anesthesiology. 1999;90:1247–1249. doi: 10.1097/00000542-199905000-00003. [DOI] [PubMed] [Google Scholar]
- Wendon J, Smithies M, Sheppard M, Bullen K, Tinker J, Bihari DJ. Continuous high volume venous-venous hemofiltration in acute renal failure. Intensive Care Med. 1989;15:358–363. doi: 10.1007/BF00261493. [DOI] [PubMed] [Google Scholar]
- Kirshbaum B. Effect of hemodialysis on the hypersalphatemia of chronic renal failure. ASAIO J. 1998;44:314–318. doi: 10.1097/00002480-199807000-00014. [DOI] [PubMed] [Google Scholar]
- Stewart PA. Modern quantitative acid-base chemistry. Can J Physiol Pharmacol. 1983;61:1444–1461. doi: 10.1139/y83-207. [DOI] [PubMed] [Google Scholar]
- Figge J, Mydosh T, Fencl V. Serum proteins and acid-base equilibria: a follow up. J Lab Clin Med. 1992;120:713–719. [PubMed] [Google Scholar]
- Rocktaeschel J, Morimatsu H, Uchino S, Goldsmith D, Pousie S, Story DA, Gutteridge G, Bellomo R. Acid-base status of critically ill patients with acute renal failure: analysis based on Stewart-Figge methodology. Crit Care. 2003;7:60–66. doi: 10.1186/cc2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa T. Organic acids and the uremic syndrome: protein metabolite hypothesis in the progression of chronic renal failure. Semin Nephrol. 1996;16:167–182. [PubMed] [Google Scholar]
- Uchino S, Bellomo R, Ronco C. Intermittent versus continous renal replacement therapy in the ICU: impact on electrolyte and acid-base balance. Intensive Care Med. 2001;27:1037–1043. doi: 10.1007/s001340100953. [DOI] [PubMed] [Google Scholar]
- Phu NH, Hien TT, Mai NT, Chau TT, Chuong LV, Loc PP, Winearls C, Farrar J, White N, Day N. Hemofitlration and peritoneal dialysis in infection-associated acute renal failure in Vietnam. N Engl J Med. 2002;347:895–902. doi: 10.1056/NEJMoa020074. [DOI] [PubMed] [Google Scholar]
- Rocktaschel J, Morimatsu H, Uchino S, Ronco C, Bellomo R. Impact of continuous veno-venous hemofiltration on acid-base balance. Int J Artif Organs. 2003;26:19–25. doi: 10.1177/039139880302600104. [DOI] [PubMed] [Google Scholar]
- Kierdorf H, Sieberth HG. Continuous treatment modalities in acute renal failure. Nephrol Dial Transplant. 1995;10:2001–2008. [PubMed] [Google Scholar]
- Heering P, Ivens K, Thumer O, Brause M, Grabensee B. Acid-base balance and substitution fluid during continuous hemofiltration. Kidney Int. 1999;56:s37–s40. [PubMed] [Google Scholar]
- Davenport A, Will E, Davison AM. The effect of lactate-buffered solutions on the acid-base status of patients with renal failure. Nephrol Dial Transplant. 1989;4:800–804. [PubMed] [Google Scholar]
- Mansell MA, Morgan SH, Moore L, Kong CH, Laker MF, Wing AJ. Cardiovascular and acid-base effects of acetate and bicarbonate haemodialysis. Nephrol Dial Transplant. 1987;1:229–232. [PubMed] [Google Scholar]
- Saman S, Opie LH. Mechanism of reduction of action potential duration of ventricular myocardium by exogenous lactate. J Mol Cell Cardiol. 1984;10:659–662. doi: 10.1016/s0022-2828(84)80629-x. [DOI] [PubMed] [Google Scholar]
- Morgera S, Heering P, Szentandrasi T, Manassa E, Heintzen M, Willers R, Passlick Deetjen J, Grabensee B. Comparison of a lactate-versus acetate-based hemofiltration replacement fluid in patients with acute renal failure. Renal Fail. 1997;19:155–164. doi: 10.3109/08860229709026270. [DOI] [PubMed] [Google Scholar]
- Thomas AN, Guy JM, Kishen R, Geraghty IF, Bowles BJM, Vadgama P. Comparison of lactate and bicarbonate buffered haemofiltration fluids: use in critically ill patients. Nephrol Dial Transplant. 1997;12:1212–1217. doi: 10.1093/ndt/12.6.1212. [DOI] [PubMed] [Google Scholar]
- Tan HK, Uchino S, Bellomo R. The acid-base effects of continuous hemofiltration with lactate or bicarbonate buffered replacement fluids. Int J Artif Organs. 2003;26:477–483. doi: 10.1177/039139880302600605. [DOI] [PubMed] [Google Scholar]
- Cohen RD, Iles RA. Lactic acidosis. Clin Endocrinol Metab. 1980;9:513–527. [PubMed] [Google Scholar]
- Mehta RL, McDonald B, Aguilar M, Ward DM. Regional citrate anticoagulation for continuous arteriovenous hemodialysis in critically ill patients. Kidney Int. 1990;38:976–981. doi: 10.1038/ki.1990.300. [DOI] [PubMed] [Google Scholar]
- Cole L, Bellomo R, Journois D, Davenport P, Baldwin I, Tipping P. High volume hemofiltration in human septic shock. Intensive Care Med. 2001;27:978–986. doi: 10.1007/s001340100963. [DOI] [PubMed] [Google Scholar]
- Levraut J, Ciebera JP, Jambou P, Ichiai C, Labib Y, Grimaud D. Effect of continuous veno-venous hemofiltration with dialysis on lactate clearance in critically ill patients. Crit Care Med. 1997;25:58–62. doi: 10.1097/00003246-199701000-00013. [DOI] [PubMed] [Google Scholar]
- Cole L, Bellomo R, Baldwin I, Hayhoe M, Ronco C. The impact of lactate-buffered high volume hemofiltration on acid-base balance. Intensive Care Med. 2003;29:1113–1120. doi: 10.1007/s00134-003-1812-1. [DOI] [PubMed] [Google Scholar]
- Uchino S, Cole L, Morimatsu H, Goldsmith D, Ronco C, Bellomo R. Solute mass balance during isovolaemic high volume haemofiltration. Intensive Care Med. 2003;29:1541–1546. doi: 10.1007/s00134-003-1857-1. [DOI] [PubMed] [Google Scholar]