

Abstract
Lung injury caused by inhalation of dust from swine-concentrated animal-feeding operations (CAFO) involves the release of inflammatory cytokine interleukin 8 (IL-8), which is mediated by protein kinase C-ε (PKC-ε) in airway epithelial cells. Once activated by CAFO dust, PKC-ε is responsible for slowing cilia beating and reducing cell migration for wound repair. Conversely, the cAMP-dependent protein kinase (PKA) stimulates contrasting effects, such as increased cilia beating and an acceleration of cell migration for wound repair. We hypothesized that a bidirectional mechanism involving PKA and PKC regulates epithelial airway inflammatory responses. To test this hypothesis, primary human bronchial epithelial cells and BEAS-2B cells were treated with hog dust extract (HDE) in the presence or absence of cAMP. PKC-ε activity was significantly reduced in cells that were pretreated for 1 h with 8-bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP) before exposure to HDE (P < 0.05). HDE-induced IL-6, and IL-8 release was significantly lower in cells that were pretreated with 8-Br-cAMP (P < 0.05). To exclude exchange protein activated by cAMP (EPAC) involvement, cells were pretreated with either 8-Br-cAMP or 8-(4-chlorophenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate (8-CPT-2Me-cAMP) (EPAC agonist). 8-CPT-2Me-cAMP did not activate PKA and did not reduce HDE-stimulated IL-6 release. In contrast, 8-Br-cAMP decreased HDE-stimulated tumor necrosis factor (TNF)-α-converting enzyme (TACE; ADAM-17) activity and subsequent TNF-α release (P < 0.001). 8-Br-cAMP also blocked HDE-stimulated IL-6 and keratinocyte-derived chemokine release in precision-cut mouse lung slices (P < 0.05). These data show bidirectional regulation of PKC-ε via a PKA-mediated inhibition of TACE activity resulting in reduced PKC-ε-mediated release of IL-6 and IL-8.
Keywords: lung, cytokine, PKC-ε, PKA, organic dust
acute and long-term exposure to organic dusts present in the air of concentrated animal-feeding operations (CAFO) causes inflammation of the airways and lung function to progressively decline over time (22, 29, 30). A high prevalence of airway disease, such as chronic bronchitis, chronic obstructive pulmonary disease, and asthma, has been observed in agricultural workers who farm animals in such facilities (13). Exposure to dusts from swine CAFO results in the release of tumor necrosis factor (TNF-α), interleukin-6 (IL-6), and interleukin-8 (IL-8) from airway epithelial cells (19, 20). IL-6 initiates proinflammatory processes and is elevated in the blood and airway secretions of dust-exposed individuals, whereas IL-8 release promotes recruitment of neutrophils to the airways. Compared with other organic dusts, swine CAFO dust stimulates the largest amount of TNF-α from lung monocytes and epithelial cells (17).
Whereas swine CAFO dust-induced TNF-α release in monocytes is impacted by endotoxins present in the dust (31), airway epithelial cells lack lipopolysaccharide signaling proteins such as CD14 and MD-2 (3, 11) and are stimulated to release TNF-α by the peptidoglycan contained in CAFO dust (17). In vitro, we have found that small concentrations (200 pg/ml) of swine CAFO dust-stimulated TNF-α are capable of inducing the autocrine/paracrine stimulation of bronchial epithelial protein kinase C-ε (PKC-ε)-dependent IL-8 release (36). Also in an autocrine/paracrine manner, dust-induced TNF-α stimulates the PKC-ε-independent release of IL-6, as both dust-stimulated PKC activation and cytokine release can be blocked by TNF-neutralizing antibodies or fusion proteins (36). Soluble TNF-α release from cells requires the posttranslational cleavage of a pro-TNF-α form by the TNF-α-converting enzyme (TACE; ADAM-17) (21). Evidence exists that cAMP-PKA activation blocks TNF-α expression (9) and that cAMP-elevating agents inhibit TACE activity in alveolar epithelial cells (25). Moreover, swine CAFO dust-stimulated TNF-α gene expression was inhibited by cAMP in alveolar epithelial cells (7). These findings have not been extended to swine CAFO dust-mediated airway inflammation with regard to TNF-α-mediated IL-6 and IL-8 release.
Both IL-6 and IL-8 release from airway epithelial cells is mediated by the dust-stimulated sequential activation of PKC isoforms, namely, PKC-α and PKC-ε (36). PKC has profound effects on the regulation of airway epithelial cell function. Agents that activate PKC slow cell migration during wound repair (34), and hog dust extract (HDE) specifically activates PKC and retards bronchial epithelial wound repair (24). Conversely, agents that activate the cAMP-dependent protein kinase (PKA) demonstrate accelerated wound closure in airway epithelium (26). Similar to wound repair, the ciliary motility of airway epithelium is stimulated by agents that activate PKA (37) but slowed by agents that activate PKC (32). Thus a bidirectional control mechanism likely exists involving a PKA-PKC axis that regulates certain epithelial airway innate immune responses.
We hypothesized that swine barn dust-induced cytokine release would be minimized in airway epithelial cells pretreated with cAMP. In the present study, we used a specific, cell-permeable agonist analog to PKA, 8-Bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP), to elevate cAMP levels. This pretreatment minimizes swine barn dust-stimulated release of IL-8 by preventing the activation of PKC-ε. Furthermore, we show that the action of cAMP on its major cellular receptor, PKA, results in the inhibition of dust-stimulated TACE activity governing TNF-α release upstream of both IL-6 release and PKC-ε activation in the airway epithelium. Thus our findings provide evidence that cAMP-PKA activating agents, including adenosine (1, 2) or β-adrenergic agonists, may be beneficial preventative and therapeutic agents in mitigating the inflammatory response induced by swine CAFO dusts.
MATERIALS AND METHODS
Cell isolation and culture.
Primary normal human bronchial epithelial cells (HBECs) were isolated from deidentified human lungs that were rejected for transplantation using a method previously described (4). The lungs were obtained from the International Institute for the Advancement of Medicine (IIAM). The protocol was approved by the IIAM ethics committee and the University of Nebraska Medical Center Institutional Review Board, and the care and use of animals was approved by The University of Nebraska Medical Center Institutional Animal Care and Use Committee. Cells were used in experiments from passages 1 to 4. Each experiment included cells derived from five donors. BEAS-2B cells were commercially obtained (ATCC, Manassas, VA).
HDE.
Aqueous extracts of HDE were prepared using settled dusts collected from swine confinement feeding operations and prepared as previously described (18). Dust (0.1 g/ml) was solubilized in sterile Hank's Balanced Salt Solution (pH 7.4) for 1 h at room temperature, centrifuged for 20 min at 2,000 g, and filter-sterilized (0.22 μm) to eliminate large particles and any microorganisms. Diluted (5%) extracts contained protein (1–2 mg/ml), endotoxin (22.5 to 48.75 EU/ml), and muramic acid (∼400 pmol/mg; marker for peptidoglycan) as previously determined (17).
Human IL-6, IL-8, and TNF-α detection by ELISA.
BEAS-2B cells or HBECs were cultured on 12-well plates and grown to 95% confluence using LHC-9:RPMI media (1:1) in 37°C incubators containing 5% CO2. Cells were pretreated with or without 100 μM 8-Br-cAMP (Sigma, St. Louis, MO) for 1 h. HDE (5%) was then added and incubated for 2, 6, and 18 h, and the supernatant was collected. Concentrations of human TNF-α, IL-6, and IL-8 were determined by antibody detection using ELISA (R&D Systems, Minneapolis, MN) as previously described (36). Values for cytokines measured in culture supernatants were normalized for total protein in the cell pellet for each condition using the Bradford protein assay (6).
Assay for PKC activity.
BEAS-2B cells were cultured on 60-mm tissue culture dishes (B-D Falcon, Franklin Lakes, NJ) to 95% confluence and pretreated for 1 h with or without 100 μM 8-Br-cAMP. Cells were then stimulated with either 1% or 5% HDE for 1 or 6 h in a total volume of 2 ml per dish. Previous data demonstrated the sequential activation of PKC-α at 1 h and PKC-ε at 6 h (36). As a positive control, 100 ng/ml of the direct classical and novel PKC isoform activator, phorbol-12-myristate-13-acetate (PMA; Sigma), was added to some dishes for 5–15 min. Medium was removed, and 250 μl of cell lysis buffer (35 mM Tris·HCl, 0.4 mM EGTA, and 10 mM MgCl) containing a 1:10 dilution of protease inhibitor cocktail (Sigma), 0.2 mM PMSF, and 0.1% Triton X-100 was added; dishes were then snap frozen as previously described (35). Thawed dishes were then scraped, the buffer containing the cells was collected and sonicated for 5 s, crude cell protein was fractionated by centrifugation at 10,000 g for 30 min at 4°C, and the cell fractions containing either PKC-α or PKC-ε were measured for activity as previously described (32). Experiments were performed a minimum of three separate times.
PKA activity assay.
BEAS-2B cells were cultured on 60-mm dishes, treated with 0.1 μM, 1.0 μM, and 10 μM concentrations of either 8-Br-cAMP, the analog specific for cAMP-dependent protein kinase (PKA) activation, or 8-(4-chlorophenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate (8-CPT-2Me-cAMP) (R&D Systems-Tocris), the analog specific for exchange protein activated by cAMP (EPAC) activation. After 1 h, the supernatant was removed, 250 μl of cell lysis buffer was added, and cells were flash frozen. Dishes were thawed and scraped into centrifuge tubes and kept on ice. The supernatant containing the cells was sonicated, and cells were centrifuged at 10,000 g at 4°C for 30 min. PKA activity was measured in the cytosolic fraction of the bronchial epithelial cells as previously described (37). Data were standardized to the total amount of cell protein assayed and expressed as picomoles of radiolabeled phosphate transferred onto a standard amount of heptapeptide substrate (Leu-Arg-Arg-Ala-Ser-Leu-Gly; Sigma) per minute of reaction time. Each unique condition was measured in a minimum of three separate experiments.
TACE assay.
TACE activity was measured using a commercially available fluorometric assay (SensoLyte 520 TACE activity assay kit; AnaSpec, Fremont, CA). BEAS-2B were grown to ∼90% confluence on collagen-coated six-well plates in serum-free (LHC-9:RPMI) medium. After treatment with 8-Br-cAMP and/or HDE, cells were removed from plates with prewarmed trypsin/EDTA and collected in microfuge tubes. Cells were pelleted at 500 g and counted, and 2.5 × 105 cells were aliquoted per condition. Aliquoted cells were pelleted and resuspended in the kit lysis buffer and vortexed 10 min at 4°C. Lysed cells were centrifuged at 10,000 g for 10 min. Supernatant fractions were assayed using the prediluted TACE substrate solution per the manufacturer's instructions. Reactions were halted with 2 M H2SO4, and fluorescence was read at 490/520 nm. Results were interpolated from a standard curve of rhTACE (ADAM-17) and expressed as micrograms per milliliter.
NF-κB binding.
NF-κB binding activities were assessed using the BEAS-2B cell line transfected with the NF-κB Cignal Vector Reporters (SABiosciences, Valencia, CA), as previously described (16). Briefly, BEAS-2B cells were reverse-transfected onto 96-well plates with a vector reporter construct containing an NF-κB-inducible firefly luciferase reporter as well as a constitutively expressed Renilla luciferase reporter. Following transfection, cells were preincubated for 1 h with 0, 10, or 100 μM 8-Br-cAMP and then stimulated with 5% HDE for 12 h. Cells were harvested using Promega Dual-GLo-Luciferase Reagents (Promega, Madison, WI) and read on a Victor 3 V plate reader (Perkin Elmer, Waltham, MA). Data were normalized for analyses by determining the ratio of induced (firefly luciferase) vs. constitutive (Renilla luciferase) luminescence for each treatment condition.
Ex vivo mouse lung slice model.
Precision-cut lung (PCL) slices were made as previously described (14). Briefly, C57BL/6 mice were killed, and the lungs were inflated with low-melting-point agarose (Invitrogen, Carlsbad, CA). Chilled lungs were then sliced into 150-μm precision-cut slices (OTS 4500 Tissue slicer; Electron Microscopy Sciences, Hatfield, PA) followed by incubation at 37°C to remove the agarose. Slices (3–5 per well) were placed in 12-well tissue culture plates. After 5 days of incubation with daily change of media, slices were exposed overnight to 5% HDE in the presence or absence of 100 μM 8-Br-cAMP or isoproterenol, and media supernatants were collected. Media were assayed for murine IL-6 and keratinocyte-derived chemokine (KC) by ELISA. Data were normalized to the total amount of PCL protein contained in each well per milliliter of media.
Cell viability assay.
An aliquot (50 μl) of supernatant media from BEAS-2B monolayers or PCL slices treated with HDE, cAMP analogs, or media alone were assayed for cell viability using a TOX-7 kit (Sigma) to measure lactate dehydrogenase (LDH) release, according to the manufacturer's instructions. As a positive control, confluent 60-mm dishes of BEAS-2B cells were lysed with 0.1% Triton-X 100, and LDH was measured.
Statistical analysis.
Replicate data from at least three separate experiments are presented as the means ± SE. One-way ANOVA with Tukey's multicomparison posttest was employed to compare responses between three or more groups. Differences between groups were accepted as significant using a 95% confidence interval (P < 0.05). In all analyses, GraphPad Prism (San Diego, CA; version 5.01) software was utilized to determine statistical significance.
RESULTS
cAMP decreases hog barn dust-stimulated interleukin release.
We have previously established that HDE stimulates proinflammatory cytokine release, including IL-6 and IL-8, from bronchial epithelial cells (20). Intracellular elevations in cAMP have been associated with anti-inflammatory mechanisms in some cell types (15). To determine the effect of cAMP on HDE-stimulated interleukin release, BEAS-2B cells, a human bronchial epithelial cell line, were pretreated for 1 h with 100 μM of a cell-permeable analog of cAMP, 8-Br-cAMP, before 24-h treatment with 5% HDE in the continued presence of 100 μM 8-Br-cAMP. A significant (P < 0.05 vs. HDE only) decrease in IL-6 release was observed in those cells pretreated with 8-Br-cAMP before HDE treatment (Fig. 1A). Whereas both HDE and the positive control secretagogue, PMA (100 ng/mL), significantly (P < 0.05) stimulated IL-6 release over 24 h compared with the media control, 8-Br-cAMP itself had no effect on baseline IL-6 release. In addition, 8-Br-cAMP did not decrease PMA-stimulated IL-6 release (data not shown). Likewise, 8-Br-cAMP pretreatment significantly (P < 0.05) decreased HDE-stimulated IL-8 release (Fig. 1B). These data reveal that cAMP blocks HDE-stimulated IL-6 and IL-8 release from bronchial epithelial cells.
Fig. 1.
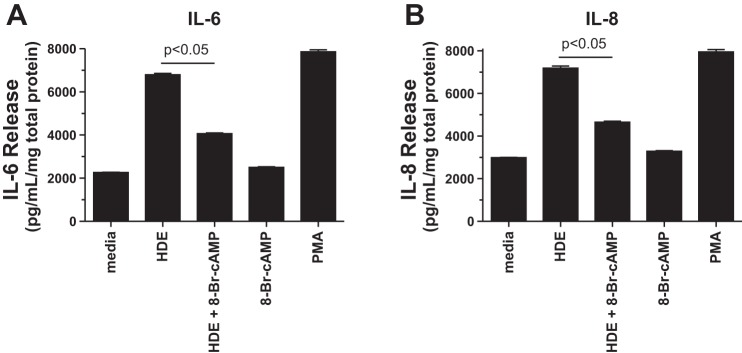
Pretreatment of BEAS-2B cells with 8-bromoadenosine 3′,5′-cyclic monophosphate (8-Br-cAMP) decreases hog dust extract (HDE)-stimulated IL-6 and IL-8 release. BEAS-2B cells were pretreated with 100 μM 8-Br-cAMP for 1 h and then stimulated with 5% HDE for 24 h. Phorbol-12-myristate-13-acetate (PMA; 100 ng/ml for 10 min) was used as a positive control for stimulated interleukin release. The supernatant was collected and assayed for either IL-6 (A) or IL-8 (B) by ELISA. Cells pretreated with 8-Br-cAMP showed significantly less cytokine concentrations compared with controls (P < 0.05). Bars represent SE of experiments performed in triplicate.
Effects of cAMP on swine barn dust-stimulated PKC activity.
Previous studies have demonstrated that HDE stimulates proinflammatory cytokine release from bronchial epithelial cells through a PKC-dependent mechanism (36). To determine whether cAMP-mediated inhibition of interleukin release occurs at the level of PKC regulation, BEAS-2B cells were pretreated with 8-Br-cAMP before stimulation with HDE, and PKC-α and PKC-ε activation was determined. HDE (5%) treatment significantly (P < 0.001) increased maximal PKC-ε activity at 6 h, but 1-h pretreatment with 100 μM 8-Br-cAMP blocked HDE-stimulated PKC-ε (Fig. 2A). Even though 1% HDE failed to optimally increase PKC-ϵ activity, a statistically significant reduction in 1% HDE-stimulated PKC-ε activity was still observed in response to 100 μM 8-Br-cAMP. Whereas 10–100 μM of 8-Br-cAMP blocked 5% HDE-stimulated PKC-ε activity, 1 μM 8-Br-cAMP failed to have any effect (Fig. 2B). Higher concentrations of 8-Br-cAMP were avoided, as we have previously demonstrated that 8-Br-cAMP concentrations above 100 μM can cross-activate the cGMP-dependent protein kinase in bronchial epithelium (33). 8-Br-cAMP (100 μM) blocked HDE-stimulated increases in PKC-ε at all time points tested and did not differ from baseline media control PKC-ε activity up to 24 h (Fig. 2C). Direct activation of PKC-ε by PMA was unaffected by pretreatment with any concentration of 8-Br-cAMP (Fig. 2D), similar to what was observed with cytokine release and suggesting that 8-Br-cAMP does not directly inhibit PKC-ε. Whereas 5% HDE optimally stimulates PKC-α at 1-h treatment, 8-Br-cAMP-pretreatment did not inhibit HDE-stimulated PKC-α activity (Fig. 3). In summary, these data show that cAMP exerts its effect downstream of PKC-α yet upstream of both IL-6 and IL-8 production. These data also demonstrate that cAMP reduces HDE-induced PKC-ε activity. Moreover, this effect by cAMP is not a result of directly inhibiting PKC-ε although the action of cAMP is downstream of PKC-α.
Fig. 2.
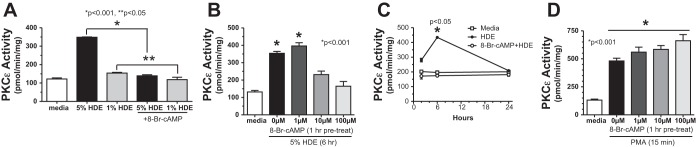
Pretreatment of BEAS-2B cells with 8-Br-cAMP inhibits HDE-stimulated protein kinase C-ε (PKC-ε) activity. A: BEAS-2B cells were pretreated for 1 h with 100 μM 8-Br-cAMP and stimulated for 6 h with either 1% or 5% HDE, and PKC-ε activity was assayed. 8-Br-cAMP pretreated cells showed significantly less activity than 5% (*P < 0.001) or 1% (**P < 0.05) HDE alone. B: BEAS-2B cells pretreated with 0–100 μM 8-Br-cAMP before 6-h stimulation with 5% HDE, and PKC-ε activity was assayed. HDE-stimulated PKC-ε was blocked by 10–100 μM 8-Br-cAMP. C: BEAS-2B cells were pretreated with 100 μM 8-Br-cAMP before 2-, 6-, and 24-h stimulation with 5% HDE, and PKC-ε activity was assayed. HDE-stimulated PKC-ε was significantly (*P < 0.05) blocked by 8-Br-cAMP at 6 h vs. HDE only. D: BEAS-2B cells were pretreated with 100 μM 8-Br-cAMP before 15-min stimulation with 100 ng/ml PMA, and PKC-ε activity was assayed. PMA significantly (*P < 0.001) stimulated PKC-ε activity. 8-Br-cAMP did not affect PMA-stimulated PKC-ε activity. Bars represent SE of experiments performed in triplicate.
Fig. 3.
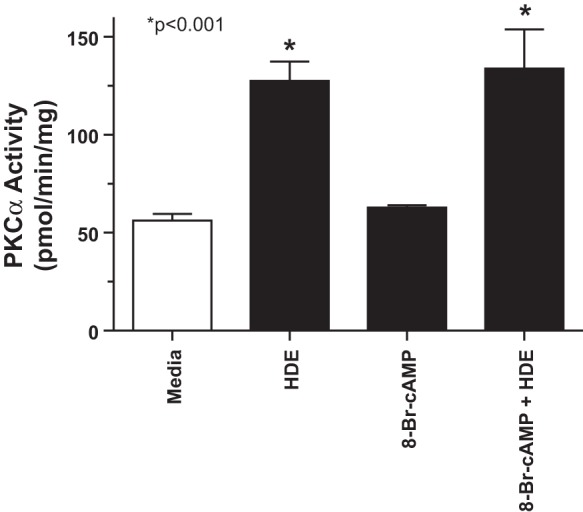
Pretreatment of BEAS-2B cells with 8-Br-cAMP does not affect HDE-stimulated PKC-α activity. BEAS-2B cells were pretreated for 1 h with 100 μM 8-Br-cAMP, stimulated with 5% HDE for 1 h, and assayed for PKC-α activity. HDE significantly (*P < 0.001 vs. media control) stimulates PKC-α activity, but 8-Br-cAMP pretreatment does not block this effect. Bars represent SE of experiments performed in triplicate.
PKA, but not EPAC, agonist blocks HDE-stimulated interleukin release.
PKA is the major cellular target for the action of cAMP, but EPAC are present in airway epithelium that can also be directly activated by cAMP. To determine whether the cAMP-mediated inhibition of HDE-stimulated interleukin release signals through PKA or EPAC, we pretreated BEAS-2B with agonist analogs of cAMP that were selective for EPAC (8-CPT-2Me-cAMP) or PKA (8-Br-cAMP). Using direct PKA activity assays of BEAS-2B cells treated with 0.1–10 μM of each analog, we found that 1-h treatment with 8-Br-cAMP significantly (P < 0.05) increased PKA activity in a dose-dependent manner, but 8-CPT-2Me-cAMP did not activate PKA (Fig. 4A). Correspondingly, 1-h pretreatment with 10 μM 8-CPT-2Me-cAMP had no effect on 5% HDE-stimulated IL-6 release, whereas 10 μM 8-Br-cAMP inhibited IL-6 release (Fig. 4B). The same results were also observed for HDE-stimulated IL-8 release (data not shown). Neither analog alone had an effect on baseline levels of the interleukins. To establish that the inhibitory action of cAMP on HDE-induced PKC-ε activation was signaling through PKA, we pretreated BEAS-2B cells with an antagonist analog of cAMP [Rp-adenosine-3',5'-cyclic monophosphorothioate (Rp-cAMPS)] before subsequent exposure of the cells to 8-Br-cAMP and HDE. Preloading the cells for 30 min with 10 μM Rp-cAMPS blocked the ability of 10 μM 8-Br-cAMP to reduce 5% HDE stimulation of PKC-ε activity (Fig. 4C). This blockade of action was due to inhibition of PKA activation, as pretreatment with 10 μM Rp-cAMPS negated the ability of 8-Br-cAMP to activate PKA in the presence or absence of HDE (Fig. 4D). These data support the notion that the action of cAMP on HDE-stimulated interleukin release is dependent on PKA and not EPAC.
Fig. 4.

PKA activation, but not exchange protein activated by cAMP (EPAC) activation, results in reduced HDE-stimulated IL-6 release. A: dose-response curve for PKA activity in BEAS-2B cells. Cells were treated with 0.1 μM, 1 μM, and 10 μM concentrations of 8-Br-cAMP (PKA agonist), or 8-(4-chlorophenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate (8-CPT-2Me-cAMP), the stimulatory analog for EPAC for 1 h. 8-Br-cAMP (*P < 0.05), but not 8-CPT-2Me-cAMP, stimulated PKA activity. B: effect of cAMP analogs on HDE-stimulated IL-6 release in BEAS-2B cells. Cells were stimulated with 5% HDE 24 h in the presence or absence of 10 μM 8-Br-cAMP or 8-CPT-2Me-cAMP, and media supernatants were assayed for IL-6 release. Cells pretreated with 8-CPT-2Me-cAMP did not significantly alter HDE-stimulated IL-6 release compared with HDE-only treated cells (*P < 0.05 vs. media control). C: effect of PKA antagonist analog [Rp-adenosine-3',5'-cyclic monophosphorothioate (Rp-cAMPS)] on HDE-stimulated PKC-ε activity in BEAS-2B. Cells pretreated with 10 μM Rp-cAMPS (30 min) before 10 μM 8-Br-cAMP pretreatment for 1 h restored the ability of 5% HDE to activate PKC-ε (**P < 0.001 vs. 8-Br-cAMP + HDE). D: effect of PKA antagonist analog (Rp-cAMPS) on HDE-stimulated PKA activity in BEAS-2B. Pretreatment with Rp-cAMPS (10 μM for 30 min) competitively antagonized the ability of 8-Br-cAMP (*P < 0.001 vs. no treatment control) to activate PKA. Bars represent SE of experiments performed in triplicate.
Pretreatment of BEAS-2B and primary bronchial epithelial cells with 8-Br-cAMP decreases HDE-stimulated TNF-α release.
PKC-α stimulation of TNF-α is an intermediary in the stimulated release of IL-6 and the sequential activation of PKC-ε, leading to IL-8 release after HDE treatment, as determined by TNF-neutralizing antibodies and fusion protein (36). Because our data revealed that cAMP blocks HDE-stimulated PKC-ε, but not PKC-α activity (Fig. 3), we examined the effect of cAMP on HDE-stimulated TNF-α levels in BEAS-2B. A 1-h pretreatment with 100 μM 8-Br-cAMP significantly (P < 0.001) decreased HDE-stimulated TNF-α release at 2 h, 6 h, and 18 h compared with HDE-treated cells that were not pretreated with 8-Br-cAMP (Fig. 5). Similar responses were observed when primary HBECs were examined compared with BEAS-2B. Under the same treatment conditions as the BEAS-2B assays, 8-Br-cAMP significantly (P < 0.05) decreased HDE-stimulated TNF-α, IL-6, and IL-8 release (Fig. 6). To control for any direct effect of 8-Br-cAMP on TNF-α, we conducted “add back” studies in which TNF-α, an activator of bronchial epithelial cell PKC-ε (34), was used to stimulate BEAS-2B cells that had been pretreated with 8-Br-cAMP. Whereas 1-h pretreatment with 100 μM 8-Br-cAMP significantly (P < 0.001) inhibited 6 h HDE-stimulated PKC-ε activity, pretreatment with 100 μM 8-Br-cAMP did not block PKC-ε activation when BEAS-2B were subsequently stimulated for 1 h with 250 pg/ml TNF-α (Fig. 7). Likewise, 8-Br-cAMP had no direct effect on inhibiting NF-κB activity, as 5% HDE significantly activated NF-κB vs. control media whether or not BEAS-2B cells were pretreated with 10 or 100 μM 8-Br-cAMP (Fig. 8). These data demonstrate that the action of cAMP-PKA diminishes HDE-stimulated TNF-α production, leading to decreased PKC-ε activation. These data also show that the action of cAMP is not the result of direct NF-κB inhibition or any interference on the ability of TNF-α to directly activate PKC-ε.
Fig. 5.
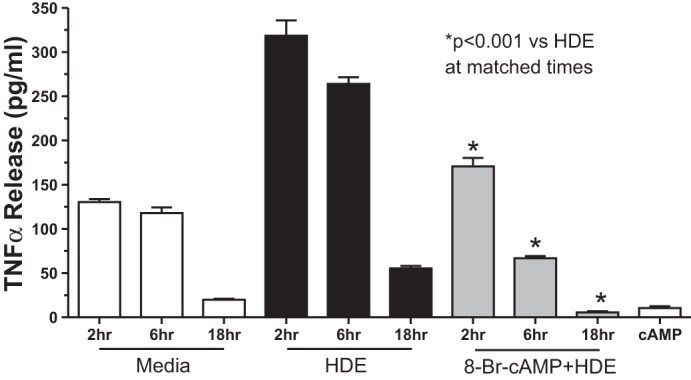
Pretreatment of BEAS-2B cells with 8-Br-cAMP decreases HDE-stimulated TNF-α release. BEAS-2B cells were pretreated with 100 μM 8-Br-cAMP for 1 h and then stimulated with 5% HDE for 2–18 h, and supernatant media were collected and assayed for TNF-α release. Cells pretreated with 8-Br-cAMP and stimulated with HDE showed significantly lower concentrations of TNF-α at all time points compared with those cells treated only with HDE (*P < 0.001). Bars represent SE of experiments performed in triplicate.
Fig. 6.
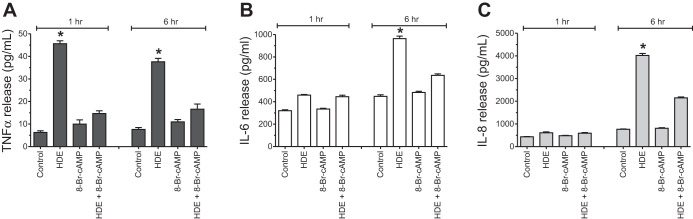
Pretreatment of primary human bronchial epithelial cells (HBECs) with 8-Br-cAMP decreases HDE-stimulated TNF-α, IL-6, and IL-8 release. HBECs were pretreated with 100 μM 8-Br-cAMP for 1 h and then stimulated with 5% HDE for 2–6 h, and supernatant media were collected and assayed for TNF-α, IL-6, and IL-8 release. Cells pretreated with 8-Br-cAMP and stimulated with HDE showed significantly lower concentrations of TNF-α (A), IL-6 (B), and IL-8 (C) by 6 h compared with those cells treated only with HDE (*P < 0.001). Bars represent SE of experiments performed in triplicate.
Fig. 7.
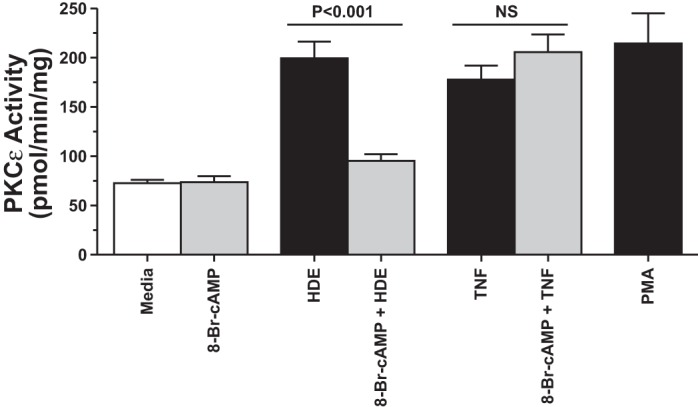
Pretreatment of BEAS-2B cells with 8-Br-cAMP does not affect TNF-α-stimulated PKC-ε activation. BEAS-2B cells were pretreated for 1 h with or without 100 μM 8-Br-cAMP, followed by stimulation with either 5% HDE for 6 h or 250 pg/ml TNF-α for 1 h, and PKC-ε activity was assayed. PMA (100 ng/ml for 5 min) was used as a positive control. 8-Br-cAMP blocked HDE-stimulated PKC-ε (P < 0.001), but not TNF-α-stimulated PKC-ε (NS; no significance) activity. Bars represent SE of experiments performed in triplicate.
Fig. 8.
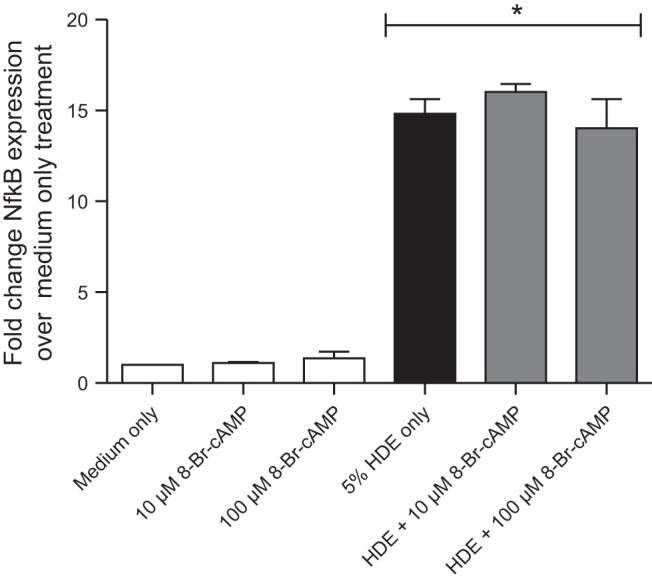
Pretreatment of BEAS-2B cells with 8-Br-cAMP does not alter HDE-stimulated NF-κB activation. BEAS-2B were preincubated for 1 h with 0, 10, or 100 μM 8-Br-cAMP and then stimulated with 5% HDE for 12 h. NF-κB transcriptional activity was assayed as described and expressed as fold change activation over medium-only-treated cells. HDE significantly (*P < 0.001) stimulated NF-κB activity in the presence or absence of 8-Br-cAMP. Bars represent SE of experiments performed in triplicate.
Pretreatment of BEAS-2B cells with 8-Br-cAMP blocks HDE-stimulated TACE activation.
BEAS-2B cells were pretreated in the presence or absence of 10 μM 8-Br-cAMP for 1 h before stimulation with 5–10% HDE for 2 h, and TACE activity was measured. Both 5 and 10% HDE significantly elevated TACE activity (P < 0.05), but HDE-stimulated TACE activity increases were blocked by pretreatment with 8-Br-cAMP (Fig. 9). Collectively, these data (Figs. 5, 6, and 9) demonstrate that cAMP blocks HDE-stimulated TACE activity and TNF-α conversion in bronchial epithelial cells and contributes to decreased HDE-induced interleukin release (IL-6 and IL-8).
Fig. 9.
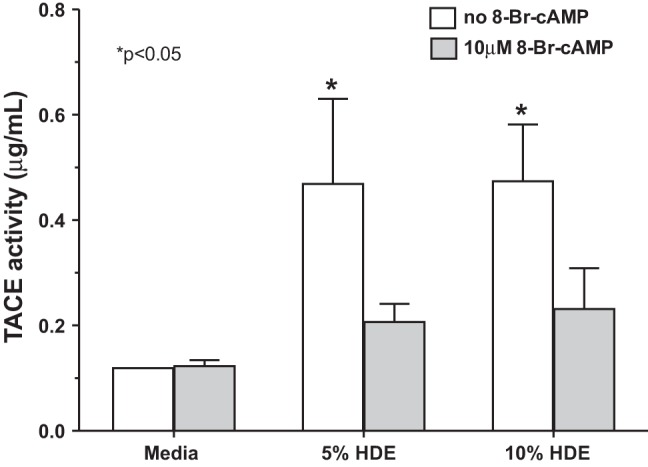
Pretreatment of BEAS-2B cells with 8-Br-cAMP blocks HDE-stimulated TNF-α-converting enzyme (TACE) activation. BEAS-2B cells were pretreated with 10 μM 8-Br-cAMP for 1 h, and then cells were stimulated with 5% and 10% HDE and incubated for 2 h. Cell lysates were assayed for TACE activity. HDE significantly (*P < 0.05) elevated TACE activity compared with media controls, and pretreatment with 8-Br-cAMP inhibited HDE-stimulated TACE. Bars represent SE of experiments performed in triplicate.
Pretreatment of mouse lung slices with 8-Br-cAMP blocks HDE-stimulated IL-6 and KC release.
To extend our observations beyond cultured human cells, we examined the effect of cAMP on HDE-stimulated cytokine release in mouse PCL slices. PCL slices were pretreated with either media alone, the β-agonist isoproterenol (100 μM), or 100 μM 8-Br-cAMP for 1 h before stimulation with 5% HDE for 24 h. Media supernatants were collected and measured for IL-6 and KC (CXCL1, an IL-8 homolog) by ELISA. 8-Br-cAMP pretreatment significantly (P < 0.05) inhibited 5% HDE-stimulated release of IL-6 (Fig. 10A) and KC (Fig. 10B) in mouse PCL slices. In addition, 1-h pretreatment with 100 μM isoproterenol, another cAMP agonist utilized in clinical settings, also inhibited HDE-stimulated cytokine/chemokine production in PCL slices. Similar to the BEAS-2B data, 1-h pretreatment with 100 μM 8-Br-cAMP effectively (P < 0.01 vs. HDE only) blocked 5% HDE-stimulated PKC-ε activity at 6 h (Fig. 10C). In these same PCL slices, 8-Br-cAMP significantly activated PKA (Fig. 10D) and inhibited HDE-stimulated TACE activity (Fig. 10E). These data reproduce our findings in human bronchial epithelial cell cultures to a whole-tissue ex vivo mouse model of HDE-stimulated (36) cytokine release.
Fig. 10.

Pretreatment of mouse lung slices with 8-Br-cAMP blocks HDE-stimulated IL-6 and keratinocyte-derived chemokine (KC) release. A: significant reduction in cytokine release of IL-6 was seen in mouse precision-cut lung slices pretreated with 100 μM 8-Br-cAMP (cAMP) or 100 μM isoproterenol (ISO) and then stimulated with 5% HDE (*P < 0.05 vs. HDE only). B: KC release was significantly reduced in lung slices pretreated with 100 μM 8-Br-cAMP or isoproterenol and subsequently stimulated with 5% HDE (*P < 0.05 vs. HDE only). C: HDE-stimulated increases in PKC-ε activities were not observed in mouse precision-cut lung slices pretreated with 100 μM 8-Br-cAMP. D: 100 μM 8-Br-cAMP significantly (P < 0.001 vs. HDE only) stimulates PKA activity in mouse precision-cut lung slices. E: HDE-stimulated increases in TACE activity (*P < 0.05) were not observed in mouse precision-cut lung slices pretreated with 100 μM ISO or 10–100 μM 8-Br-cAMP. Bars represent SE of experiments performed in triplicate.
DISCUSSION
Our findings support a bidirectional relationship between PKA and PKC-ε in the lung epithelial cell inflammatory response to swine barn dust. Such regulation is not unlike the neutrophil bidirectional control hypothesis first postulated by Nelson Goldberg et al. (10), whereby the action of cAMP resulted in anti-inflammatory properties, but the action of cGMP was deemed to be proinflammatory. However, our findings show that cAMP pretreatment of bronchial epithelial cells reduces HDE-induced inflammatory response by stimulating PKA and subsequently inhibiting PKC-ε activity. In support of our proposed pathway (Fig. 11), we showed that cAMP does not inhibit PKC-α, an upstream activator of TNF-α, IL-6, and IL-8 following swine CAFO dust stimulation. Whereas cAMP pretreatment of cells resulted in the inhibition of dust-stimulated PKC-ε activity, cAMP did not directly affect PKC-ε, as evidenced by PMA- and rhTNF-α-stimulation of PKC-ε in the presence of exogenous, cell-permeable cAMP (8-Br-cAMP). Instead, the activation of PKA by cAMP led to the inactivation of TACE, thus preventing the dust stimulation of PKC-ε via TNF-α production and release. This was evidenced by the antagonism of PKA using Rp-cAMPS, leading to the restoration of dust-stimulated PKC-ε, even in the presence of 8-Br-cAMP (Fig. 4).
Fig. 11.
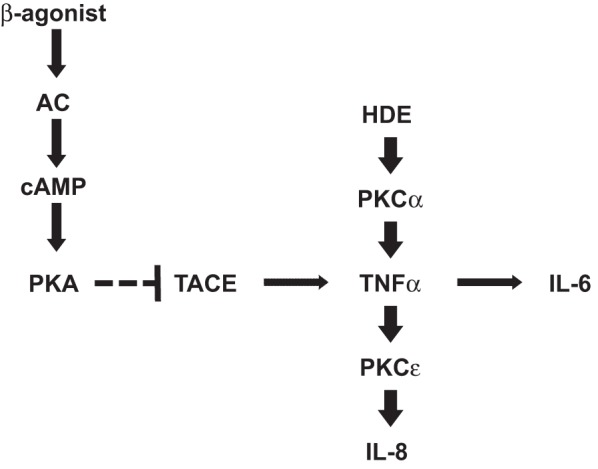
Proposed model of cAMP action on HDE-stimulated cytokine release. β-Agonists, such as isoproterenol, stimulate adenylyl cyclase (AC) production of cAMP, leading to the activation of protein kinase A (PKA). cAMP/PKA action leads to the inhibition of TACE, thus preventing the release of active TNF-α in response to HDE-stimulated PKC-α activity. This prevents the release of IL-6 (PKC-ε independent) and IL-8 (PKC-ε dependent).
Our findings support those of Burvall et al. (8), whereby 8-Br-cAMP caused a small but significant decrease in swine barn dust-stimulated IL-8 in an alveolar epithelial cell line, A549. However, our findings also differ from this study in that they observed that IL-6 was slightly elevated in response to cAMP and dust. These differences may be related to dust differences, origin of the cells (alveolar vs. bronchial), or to transformed vs. primary cell cultures. In our study, we have shown the inhibitory effect of cAMP on both human bronchial cells and mouse lung tissue exposed to HDE. Our findings also differ from those of Linden (12), in that we do not observe cAMP-stimulated release of IL-8 in BEAS-2B or primary human bronchial epithelial cells. Our observations are consistent with the general anti-inflammatory actions of cAMP reported for many cell types (15).
Although previous work has provided insight into the mechanisms for the HDE-induced inflammatory pathway in the epithelial lining, some aspects remain unknown. TACE, a metalloproteinase disintegrin, must play a role because it functions as a sheddase required for the release of soluble TNF-α, a protein that participates in immune defense, apoptosis, inflammation, and autoimmunity. TACE executes a proteolytic cleavage of pro-TNF-α imbedded in the membrane, releasing soluble extracellular TNF-α. No changes in TACE mRNA expression were found in alveolar cells treated with swine barn dust (7). Our findings support an enhancement of TACE activity as opposed to alterations in protein or gene expression in response to dust. This is supported by our observation that concentrations of 8-Br-cAMP (10 and 100 μM) that have no effect on NF-κB activity are capable of rapidly and significantly reducing TACE activity (Fig. 8). Similarly, constitutive levels of TACE expression were also described (7) in the presence or absence of dust. In that study, it was suggested that, whereas PKA phosphorylation of CREB could result in attenuation of TNF mRNA, the cAMP effect on TNF mRNA expression in response to dust in alveolar epithelium was PKA independent (7).
The mechanism for cAMP-PKA inhibition of TACE is unknown at this time. The prodomain of TACE functions as an inhibitor of the sheddase until it is cleaved by furin, a proprotein convertase, in the trans-Golgi, leading to a catalytically active mature form of TACE. In bone marrow stromal cells, elevations in cAMP have been shown to increase furin activity, resulting in alterations in target substrate processing (5). Our hypothesis presumes that PKA interacts with TACE although it is unclear whether such an interaction is a phosphorylation event (few data regarding non-PMA-mediated phosphorylation of ADAM-17 exist) or some other association in the airway epithelial cell, such as cAMP-mediated furin activation, that minimizes TACE association with TNF-α and prevents the cleavage of TNF-α from the membrane and the subsequent release of the cytokine. In addition to existing studies showing that TNF-α mRNA is reduced by cAMP in macrophages (9) and alveolar epithelium (7), TACE activity is blocked by ethanol in alveolar epithelial cells (25). An ethanol-specific adenylyl cyclase (AC7) exists (28), resulting in alcohol stimulation of cAMP and PKA in the bronchial epithelium as well (23). Our data support this hypothesis by showing reduced TACE activity when cells were pretreated with 8-Br-cAMP, but not the EPAC-activating agonist, indicating that PKA is imposing an inhibitory effect on TACE in relation to TNF-α.
The findings that a PKA activator, 8-Br-cAMP, reduced proinflammatory cytokine/chemokine release in cultured epithelial cells and PCL slices from mice suggest that pharmacological elevation of lung cAMP levels might reduce swine CAFO dust-induced lung inflammation in vivo. In proof-of-concept studies, we also demonstrated that isoproterenol, a β-adrenergic agonist that increases cAMP, reduced IL-6 and KC release from lung slices. Future lines of investigation should investigate whether various β-adrenergic agonists that stimulate adenylyl cyclase to produce cAMP will modulate swine CAFO-induced airway inflammation in an animal model via activation of PKA and subsequent inactivation of PKC-ε. There is evidence that β-adrenergic receptor agonists may be beneficial in the treatment of exposed workers because Strandberg et al. (27) found that salmeterol (a long-acting β2-agonist) reduced enhanced bronchial responsiveness following swine barn exposures (27). Such a treatment may hold the potential to reduce lung inflammation for workers in CAFO and thus reduce the risk of respiratory disease.
GRANTS
This material is the result of work supported with resources and the use of facilities at the VA Nebraska-Western Iowa Health Care System, Omaha, NE [Department of Veterans Affairs (VA I01BX000728) to T. Wyatt]. The study was supported by grants from the National Institute of Environmental Health Sciences (NIEHS R01ES019325 to J. Poole), National Institute for Occupational Safety Health (NIOSH R01OH008539 to D. Romberger), and National Institute on Alcohol Abuse and Alcoholism (NIAAA R01AA017993 to T. Wyatt). This work was also supported in part by the Central States Center for Agricultural Safety and Health (CS-CASH; U54OH010162).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
T.A.W., J.A.P., T.M.N., K.L.B., and D.J.R. conception and design of research; T.A.W., T.M.N., J.M.D., A.J.H., and K.L.B. performed experiments; T.A.W., J.A.P., T.M.N., J.M.D., and A.J.H. analyzed data; T.A.W., J.A.P., T.M.N., A.J.H., K.L.B., and D.J.R. interpreted results of experiments; T.A.W., T.M.N., J.M.D., and A.J.H. prepared figures; T.A.W. drafted manuscript; T.A.W., J.A.P., T.M.N., J.M.D., A.J.H., K.L.B., and D.J.R. edited and revised manuscript; T.A.W., J.A.P., T.M.N., J.M.D., A.J.H., K.L.B., and D.J.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors acknowledge Ms. Lisa Chudomelka for expert editorial assistance and Ms. Shannon Kline for technical support.
REFERENCES
- 1.Allen-Gipson DS, Blackburn MR, Schneider DJ, Zhang H, Bluitt DL, Jarrell JC, Yanov D, Sisson JH, Wyatt TA. Adenosine activation of A2B receptor(s) is essential for stimulated epithelial ciliary motility and clearance. Am J Physiol Lung Cell Mol Physiol 301: L171–L180, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen-Gipson DS, Spurzem K, Kolm N, Spurzem JR, Wyatt TA. Adenosine promotion of cellular migration in bronchial epithelial cells is mediated by the activation of cyclic adenosine monophosphate-dependent protein kinase A. J Investig Med 55: 378–385, 2007 [DOI] [PubMed] [Google Scholar]
- 3.Bäckhed F, Meijer L, Normark S, Richter-Dahlfors A. TLR4-dependent recognition of lipopolysaccharide by epithelial cells requires sCD14. Cell Microbiol 4: 493–501, 2002 [DOI] [PubMed] [Google Scholar]
- 4.Bailey KL, Robinson JE, Sisson JH, Wyatt TA. Alcohol decreases RhoA activity through a nitric oxide (NO)/cyclic GMP(cGMP)/protein kinase G (PKG)-dependent pathway in the airway epithelium. Alcohol Clin Exp Res 35: 1277–1281, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharyya N, Wiench M, Dumitrescu C, Connolly BM, Bugge TH, Patel HV, Gafni RI, Cherman N, Cho M, Hager GL, Collins MT. Mechanism of FGF23 processing in fibrous dysplasia. J Bone Miner Res 27: 1132–1141, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254, 1976 [DOI] [PubMed] [Google Scholar]
- 7.Burvall K, Palmberg L, Larsson K. Expression of TNFalpha and its receptors R1 and R2 in human alveolar epithelial cells exposed to organic dust and the effects of 8-bromo-cAMP and protein kinase A modulation. Inflamm Res 54: 281–288, 2005 [DOI] [PubMed] [Google Scholar]
- 8.Burvall K, Palmberg L, Larsson K. Effects by 8-bromo-cyclic AMP on basal and organic dust-induced release of interleukin-6 and interleukin-8 in A549 human airway epithelial cells. Respir Med 97: 46–50, 2003 [DOI] [PubMed] [Google Scholar]
- 9.Gobejishvili L, Avila DV, Barker DF, Ghare S, Henderson D, Brock GN, Kirpich IA, Joshi-Barve S, Mokshagundam SPL, McClain CJ. S-adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via cAMP/protein kinase A pathway. J Pharmacol Exp Ther 337: 433–443, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldberg ND, Haddox MK, Nicol SE, Glass DB, Sanford CH, Kuehle FA, Jr, Estensen R. Biologic regulation through opposing influences of cyclic GMP and cyclic AMP: the Yin Yang hypothesis. Adv Cyclic Nucleotide Res 5: 307–30, 1975 [PubMed] [Google Scholar]
- 11.Jia HP, Kline JN, Penisten A, Apicella MA, Gioannini TL, Weiss J, McCray PB. Endotoxin responsiveness of human airway epithelia is limited by low expression of MD-2. Am J Physiol Lung Cell Mol Physiol 287: L428–L437, 2004 [DOI] [PubMed] [Google Scholar]
- 12.Linden A. Increased interleukin-8 release by β-adrenoceptor activation in human transformed bronchial epithelial cells. Br J Pharmacol 119: 402–406, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.May S, Romberger DJ, Poole JA. Respiratory health effects of large animal farming environments. J Toxicol Environ Health 15: 524–541, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCaskill ML, Romberger DJ, Devasure J, Boten J, Sisson JH, Bailey KL, Poole JA, Wyatt TA. Alcohol exposure alters mouse lung inflammation in response to inhaled dust. Nutrients 4: 695–710, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moore A, Willoughby D. The role of cAMP regulation in controlling inflammation. Clin Exp Immunol 101: 387–389, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nordgren TM, Heires AJ, Wyatt TA, Poole JA, Levan TD, Cerutis DR, Romberger DJ. Maresin-1 reduces the pro-inflammatory response of bronchial epithelial cells to organic dust. Respir Res 14: 51, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poole JA, Dooley GP, Saito R, Burrell AM, Bailey KL, Romberger DJ, Mehaffy J, Reynolds SJ. Muramic acid, endotoxin, 3-hydroxy fatty acids, and ergosterol content explain monocyte and epithelial cell inflammatory responses to agricultural dusts. J Toxicol Environ Health 73: 684–700, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poole JA, Kielian T, Wyatt TA, Gleason AM, Stone J, Palm K, West WW, Romberger DJ. Organic dust augments nucleotide-binding oligomerization domain expression via an NF-κB pathway to negatively regulate inflammatory responses. Am J Physiol Lung Cell Mol Physiol 301: L296–L306, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poole JA, Wyatt TA, Oldenburg PJ, Elliott MK, West WW, Sisson JH, Von Essen SG, Romberger DJ. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol 296: L1085–L1095, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Romberger DJ, Bodlak V, Von Essen SG, Mathisen T, Wyatt TA. Hog barn dust extract stimulates IL-8 and IL-6 release in human bronchial epithelial cells via PKC activation. J Appl Physiol 93: 289–296, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Scheller J, Chalaris A, Garbers C, Rose-John S. ADAM17: a molecular switch to control inflammation and tissue regeneration. Trends Immunol 32: 380–387, 2011 [DOI] [PubMed] [Google Scholar]
- 22.Schiffman SS, Studwell CE, Landerman LR, Berman K, Sundy JS. Symptomatic effects of exposure to diluted air sampled from a swine confinement atmosphere on healthy human subjects. Environ Health Perspect 113: 567–576, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sisson JH, May K, Wyatt TA. Nitric oxide-dependent ethanol stimulation of ciliary motility is linked to cAMP-dependent protein kinase (PKA) activation in bovine bronchial epithelium. Alcohol Clin Exp Res 23: 1528–1533, 1999 [PubMed] [Google Scholar]
- 24.Slager RE, Allen-Gipson DS, Sammut A, Heires A, Devasure J, Von Essen SG, Romberger DJ, Wyatt TA. Hog barn dust slows airway epithelial cell migration in vitro through a PKCα-dependent mechanism. Am J Physiol Lung Cell Mol Physiol 293: L1469–L1474, 2007 [DOI] [PubMed] [Google Scholar]
- 25.Song K, Zhao X, Marrero L, Oliver P, Nelson S, Kolls JK. Alcohol reversibly disrupts TNF-α/TACE interactions in the cell membrane. Respir Res 6: 123, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spurzem JR, Gupta J, Veys T, Kneifl KR, Rennard SI, Wyatt TA. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am J Physiol Lung Cell Mol Physiol 282: L1108–L1116, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Strandberg K, Ek A, Palmberg L, Larsson K. Fluticasone and ibuprofen do not add to the effect of salmeterol on organic dust-induced airway inflammation and bronchial hyper-responsiveness. J Intern Med 264: 83–94, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Tabakoff B, Nelson E, Yoshimura M, Hellevuo K, Hoffman PL. Phosphorylation cascades control the actions of ethanol on cell cAMP signalling. J Biomed Sci 8: 44–51, 2001 [DOI] [PubMed] [Google Scholar]
- 29.Vogelzang PF, van der Gulden Joost WJ, Folgering H, Heederik D, Tielen MJ, van Schayck CP. Longitudinal changes in bronchial responsiveness associated with swine confinement dust exposure. Chest 117: 1488–1495, 2000 [DOI] [PubMed] [Google Scholar]
- 30.Von Essen S, Romberger D. The respiratory inflammatory response to the swine confinement building environment: the adaptation to respiratory exposures in the chronically exposed worker. J Agric Saf Health 9: 185–196, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Wilson CB, Ray M, Lutz M, Sharda D, Xu J, Hankey PA. The RON receptor tyrosine kinase regulates IFN-gamma production and responses in innate immunity. J Immunol 181: 2303–2310, 2008 [DOI] [PubMed] [Google Scholar]
- 32.Wyatt TA, Sisson JH, Allen-Gipson DS, McCaskill MK, Boten JA, DeVasure JM, Bailey KL, Poole JA. Co-exposure to cigarette smoke and alcohol decreases airway epithelial cell cilia beating in a protein kinase C epsilon-dependent manner. Am J Pathol 181: 431–440, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wyatt TA, Forget MA, Sisson JH. Ethanol stimulates ciliary beating by dual cyclic nucleotide kinase activation in bovine bronchial epithelial cells. Am J Pathol 163: 1157–1166, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wyatt TA, Ito H, Veys TJ, Spurzem JR. Stimulation of protein kinase C activity by tumor necrosis factor-α in bovine bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol 273: L1007–L1012, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Wyatt TA, Schmidt SC, Rennard SI, Sisson JH. Acetaldehyde-stimulated PKC activity in airway epithelial cells treated with smoke extract from normal and smokeless cigarettes. Proc Soc Exp Biol Med 225: 91–97, 2000 [DOI] [PubMed] [Google Scholar]
- 36.Wyatt TA, Slager RE, Heires AJ, DeVasure JM, VonEssen SG, Poole JA, Romberger DJ. Sequential activation of protein kinase C isoforms by organic dust is mediated by tumor necrosis factor. Am J Respir Cell Mol Biol 42: 706–715, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wyatt TA, Spurzem JR, May K, Sisson JH. Regulation of ciliary beat frequency by both PKA and PKG in bovine airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 275: L827–L835, 1998 [DOI] [PubMed] [Google Scholar]