

Summary
Once upon a time immunology was a black box, inflammatory and autoimmune diseases were a mystery, and relatively blunt tools were used to treat these diseases. In the last forty years, advances in molecular biology, DNA recombination technology and genome sequencing allowed immunologists to open the box. As the complexity and diversity of the immune response is unveiled, targeted cellular and molecular therapies now offer rational approaches to treat immune-mediated diseases. Here, we discuss how the tried and true bench-to-bedside strategies resulted in some spectacular successes, along with some puzzling failures. Conversely, the advent of targeted therapies in the clinic has led to a wealth of information that changes how we think about the pathogenesis of immune-mediated diseases and how we categorize disease. In turn, these insights can inform next generation drug discovery and refine targeted therapies for the appropriate patient subsets.
Introduction – A glimpse into the not distant past
Therapeutic intervention using products of immune cells dates back to the late 19th century with the development of anti-diphtheria toxin by Emil von Behring and Shibasaburo Kitasato. However, in the 1970’s, immunology was regarded by many molecular biologists and biochemists as a soft science – heavily phenomenological with limited molecular understanding of the immune response. Erythropoietin, prolactin and interferon had been isolated in the 1960’s, but the ‘gemisch’ of cytokines studied by immunologists in this era was derisively referred to as “lymphodreck” (Oppenheim and Gery, 1993). Only in 1974, Zinkernagel and Doherty would report that the ability of T cells to mount an immune response requires foreign and, surprisingly, self-encoded antigens. That is, virus-infected fibroblasts were killed only if the T cells were derived from a genetically identical strain of mice. The molecular basis of this phenomenon are gene products known as major histocompatibility antigens, a concept that explains “self” and “non-self” recognition, and was awarded the Nobel Prize in Physiology or Medicine in 1996. Although this breakthrough had profound implications for understanding diseases that disrupted self-tolerance, in 1974, most diseases were still characterized just as a collection of symptoms, with no mechanistic understanding of their pathophysiology. Psoriasis, today recognized as an IL-23-mediated autoimmune skin disease, for instance used to be described as a “scaling dermatosis of unknown etiology”.
The molecular cloning of interleukins, and hundreds of other factors, dramatically changed the landscape of immunological research. Further fueling the revolution were advances in fluorescence based flow cytometry, recombinant DNA technology and development of monoclonal antibody (mAb) technology. These tools enabled dissection of what once was thought to be a homogeneous CD4+ T cell population to what actually represents a large family of different lineages/subsets from Th1 to Th22 cells, and various regulatory T cells. Discovery of receptors and co-receptors and adhesion molecules and downstream signaling pathways provided a more precise understanding of immunity and how immune deregulation can result in disease.
The opportunity and challenges provided by these basic discoveries were how basic knowledge about the functioning of the immune system can be used to treat immune-mediated disease. The spectrum of molecules and cells identified facilitated the development of what would become known as “targeted therapies” (Figure 1). In contrast to drugs identified empirically found to be “immunosuppressive” in cell-based assays, molecular techniques identified key nodes, both extracellular and intracellular, against which therapies could be designed and deployed. Just as the complexity of immunology has evolved over the past 40 years, our understanding of human diseases likewise advanced. Nowadays, with a greater in-depth cellular and molecular understanding of immunological disease, the heterogeneous nature of autoimmune disorders has become more apparent. Greater delineation of the underlying pathogenic mechanisms of autoimmune diseases began to enable the identification of patient subsets whose diseases are driven by different biological mechanisms, thus improving our ability to match new and old therapies for each of these subsets.
Figure 1. Timeline of targeted therapies.
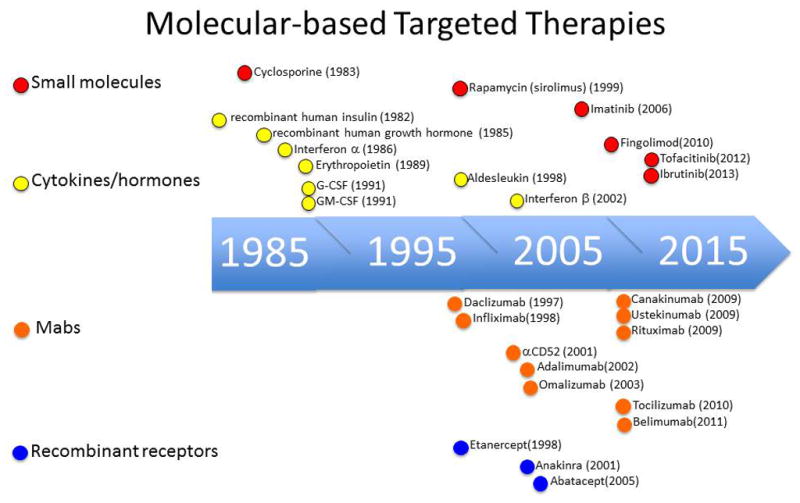
Selected examples of recombinant cytokines and cytokine receptors, monoclonal antibodies (mAbs) and small molecules illustrate the evolution of targeted therapies.
In this article, we will highlight some of the triumphs and disappointments in the translation of basic immunologic discoveries into impactful therapeutics, and how these have shaped our current therapeutic paradigms. We will focus particularly on a sampling of therapeutic targets that have revealed new insights into basic biology and the pathogenesis of human immune-mediated disease, illustrating themes that relate to success versus failure. We will touch on the challenges in designing targeted therapies, ranging from blocking secreted cytokines to deleting immune cells and how the intricacy of the immune system impacts these strategies. Given the pleiotropic effects of cytokines, complexity of cytokine receptors and human disease heterogeneity, we discuss what was anticipated of targeted therapies and substantiated in the clinic as well as the surprises - both good and bad. We will look for lessons that can be gleaned and what might these lessons mean for the development of next-generation targeted therapies. First though, a brief historical perspective is in order.
Immune therapies – the oldies but goodies
In considering the lessons provided by the new generation of targeted therapies, it is worth revisiting some classic drugs and considering the implications of their use. Many of these drugs arose from natural compounds and their immunosuppressive activities were found empirically and not by design, acetylsalicylic acid, being such example (Dinarello, 2010). Corticosteroids, endogenous immunosuppressive molecules, were first isolated in the 1940s. Though revolutionary, their toxicities limited their long-term use. Nonetheless, clinicians devised dosing regimens that provided efficacy while limiting, albeit incompletely, adverse effects. Cyclosporine, a fungal product, was first employed as an anti-fungal antibiotic and later recognized to have immunosuppressive properties. Its target too is ubiquitously expressed and therefore, it presents many non-immunologic side effects. The first synthetic class of small molecules used to treat autoimmune diseases and transplantation were anti-metabolites co-opted from Oncology, including azathioprine, methotrexate, and cyclophosphamide. In the era of targeted therapies, it may be easy to dismiss the importance of these agents, but it is worth emphasizing that these drugs remain useful therapeutic options in many autoimmune diseases nonetheless. In many cases, even “blunt” drugs like cyclophosphamide (derived from mustard gas!) can be the right drug for the right patient at the right time.
Cytokines and cytokine antibodies: the beginnings of targeted therapies
The ground-breaking efforts of Cohen and Boyer in recombinant DNA expression technology in the 1970’s provide the foundation for commercial biotherapeutics. Human insulin was the first commercialized product based on recombinant DNA technology and was approved in 1982 for the treatment of Type 1 diabetes mellitus (DM) (Figure 1). Within the next decade, ten additional cytokines, growth factors, or enzymes were approved for human use. The pioneering work of Kohler and Milstein in 1975 with the discovery of mAb technology led to the development of therapeutic antibodies. Development of antibody humanization technologies and the ability to generate human antibodies have overcome immunogenicity problems of mouse antibodies and are now the standards for human use (Figure 1 and Table 1). In the next section, we will discuss the advantages and pitfalls of the prominent classes of cytokines and antibodies that have emerged as candidates for targeted therapy.
Table I.
A Summary of Selected Targeted Therapies 1986-present
| Target | Mechanism | Molecule Type | Brand Name (generic name) | Approval/Clinical Phase | Clinical application |
|---|---|---|---|---|---|
| Targeting Cytokines and Cytokine Receptors | |||||
| Type 1 IFNs | Anti-viral | cytokine | Intron A® (interferon alpha 2b) | Approved 1986 | Chronic HBV & HCV Hairy Cell Leukemia Malignant Melanoma AIDS associated KS |
| cytokine | Roferon® (interferon alpha 2a) | Approved 1986 | Chronic HCV | ||
| cytokine | Infergen® (interferon alfacon 1) | Approved 1997 | Chronic HCV | ||
| cytokine | Pegintron® (peginterferon alpha 2b) | Approved 2001 | Chronic HCV | ||
| cytokine | Pegasys® (peginterferon alpha 2a) | Approved 2002 | Chronic HBV & HCV | ||
| Anti-IFNα | humanized IgG1 mAb | rontalizumab | Phase II (completed) | SLE – benefit observed only in IFN-low subgroup (NCT00962832) | |
| Immunosuppression | cytokine | Rebif® (interferon beta 1a) | Approved 2002 | RRMS | |
| cytokine | Betaseron® (interferon beta 1b) | Approved 2003 | RRMS | ||
| cytokine | Avonex® (interferon beta 1a) | Approved 1995 | RRMS | ||
| Interleukin-2 | Anti-tumor | cytokine | Proleukin® (aldesleukin) | Approved 1998 | Metastatic renal cell cancer |
| Treg cell expansion | cytokine | Low dose Proleukin® (aldesleukin) | Phase II (completed) | Benefit in GVHD (NCT00529035) Benefit in HCV-associated cryoglobulinemia (NCT00574652) Worsening in Type 1 DM (NCT00525889) |
|
| Treg cell expansion | Immunocomplex | IL-2/anti-IL2 mAb (JES6.1) | Preclinical stage | Autoimmune diseases | |
| Expansion of NK and CD8 T cells | Immunecomplex | IL-2/anti-IL2 mAb (S4B6, MAB602) | Preclinical stage | Anti-tumor therapy | |
| superkine | IL-2 superkine | Preclinical stage | Anti-tumor therapy | ||
| Anti-IL-2Rα mAb | chimeric IgG1 mAb | Simulect® (basiliximab) | Approved 1998 | Acute organ rejection | |
| humanized IgG1 mAb | Zenapax® (daclizumab) | Approved 1997 Withdrawn 2009 | Acute organ rejection Benefit observed in RRMS | ||
| TNF | TNFRI-Fc | fusion receptor | lenercept | Terminated | Worsens RRMS |
| TNFRII-Fc for TNFα and LTα | fusion receptor | Enbrel® (etanercept) | Approved 1998 | RA, JIA, PsA, AS, Psoriasis No benefit in CD |
|
| Anti-TNFα mAb | chimeric IgG1 mAb | Remicade® (infliximab) | Approved 1998 | RA, CD, UC, AS, PsA | |
| Anti-TNFα mAb | human IgG1 mAb | Humira® (adalimumab) Simponi® (golimumab) |
Approved 2002 Approved 2009 |
RA, CD, UC, AS, PsA, JIA, Psoriasis RA, UC, AS, PsA |
|
| Anti-TNFα Fab | humanized Fab | Cimzia® (certolizumab pegol) | Approved 2008 | RA, CD, AS, PsA | |
| IL-1 | IL-1Ra | recombinant protein | Kineret® (anakinra) | Approved 2001 | RA, CAPS |
| IL-1R1/IL-RAcP | fusion receptor | Arcalyst® (rilonacept) | Approved 2008 | CAPS | |
| Anti-IL-1β mAb | human IgG1 mAb | Ilaris® (canakinumab) | Approved 2009 | CAPS | |
| IL-6 | Anti-IL-6R mAb | humanized IgG1 mAb | Actemra® (tocilizumab) | Approved 2005 (Japan) Approved 2010 |
Castleman’s Disease RA, Polyarticular JIA, Systemic JIA |
| Anti-IL6R mAb | human IgG1 mAb | sarilumab | Phase III | RA | |
| Anti-IL6 mAb | human IgG1 mAb | sirukumab | Phase III | RA | |
| Targeting T Cells | |||||
| T cell depletion/moduration | Anti-CD3 mAb | mouse IgG2a mAb | OKT3® (muronomab-CD3) | Approved 1986 (withdrawn by sponsor in 2010) | Transplant rejection |
| Anti-T cell antisera | rabbit anti- thymoglobulin | Thymoglobulin® (anti-thymocyte globulin) | Approved 1998 | Transplant rejection | |
| Anti-CD52 mAb | humanized IgG1 mAb | Campath® (alemtuzumab) | Approved 2001 | CLL, cutaneous T cell lymphoma | |
| CTLA-Fc Costimulatory blockade | Receptor fusion | Orencia® (abatacept) | Approved 2005 | RA | |
| Anti-CD4 mAb | mouse IgG1 mAb | 16H5 BB14 BF5 |
Terminated | ||
| mouse IgG2a mAb | 19THY5D7 BL4 MT151 |
||||
| rat IgG1 mAb | YNB46.1.8 | ||||
| chimeric IgG1 mAb | keliximab priliximab |
||||
| chimeric primatized IgG4 mAb | clenoliximab | ||||
| humanized IgG | cedelizumab MDX-CD4 | ||||
| humanized IgG1 mAb | 4162W94 Campath 9H tregalizumab |
||||
| humanized IgG1 mAb (effectoress & FcRn enhanced) | MTRX1011A | ||||
| human IgG1 mAb | zanolimumab | ||||
| Th2 cells | anti-IgE mAb | humanized IgG1 mAb | Xolair® (omalizumab) | Approved 2003 | Moderate to severe asthma |
| Anti-IL-13 mAb | humanized IgG4 mAb | lebrikizumab | Phase III | Asthma | |
| Anti-IL-5 mAb | humanized IgG1 mAb | mepolizumab | Phase III | Asthma | |
| Anti-IL-4Rα mAb | human IgG4 mAb | dupilumab | Phase II | Asthma | |
| Th17 cells | Anti-p40 mAb | human IgG1 mAb | Stelara® (ustekinumab) | Approved 2009 Phase II (completed) |
Psoriasis CD |
| Anti-IL-17A/A mAb | human IgG1 mAb | secukinumab | Phase III Phase II |
Psoriasis, RA, AS RRMS |
|
| Anti-IL17RA mAb | human IgG2 mAb | brodalumab | Phase III | Psoriasis | |
| Anti-IL-17A/A & A/F mAb | humanized IgG4 mAb | ixekizumab | Phase III | Psoriasis | |
| Targeting B Cells | |||||
| B cell depletion/modulation | Anti-CD20 mAb | chimeric IgG1 mAb | Rituxan® (rituximab) | Approved 2006 | RA, AAV, MPA |
| Anti-BAFF/BLys mAb | human IgG1 mAb | Benlysta® (belimumab) | Approved 2011 | SLE | |
| Anti-CD20 mAb | humanized IgG1 mAb | ocrelizumab | Phase III | RRMS, PPMS | |
| Anti-CD20 mAb (enhanced complement activation) | human IgG1 mAb | Arzerra® (ofatumumab) | Approved 2009 Phase III |
B-CLL Pemphigus vulgaris |
|
| Targeting Intracellular Signaling Components by Small Molecules | |||||
| mTOR/FKBP12 | inhibitor | Bacterial macrolide | Rapamune® (sirolimus) | Approved 1999 | Renal transplantation rejection |
| S1P1R | agonist | Fungal metabolite | Gilenya® (fingolimod) | Approved 2010 | RRMS |
| JAK | inhibitor | Specificity JAK1/2 | Jakafi® (ruxolitinib) | Approved 2011 | Myelofibrosis |
| inhibitor | Specificity JAK1/2/3 | Xeljanz® (tofacitinib) | Approved 2012 | RA | |
| BTK inhibitor | inhibitor | Imbruvica® (Ibrutinib) | Approved 2013 | Mantle cell lymphoma B-CLL |
|
| unknown; modulates nuclear factor-2 pathway | Tecfidera® (dimethyl-fumarate) | Approved 2013 | RRMS | ||
| PI3K-δ | inhibitor | idelalisib | Phase III | Refractory indolent NHL, B-CLL | |
| HDAC | inhibitor | Zolinza® (vorinostat) | Approved 2006 | Cutaneous T cell lymphoma | |
| inhibitor | Istodax® (romidepsin) | Approved 2009 | Cutaneous T cell lymphoma | ||
| inhibitor | panobinostat | Phase III | lymphoma | ||
| inhibitor | CI-994 | Phase III | Non-small cell lung carcinoma | ||
Abbreviations - AAV - ANCA-associated vasculitis; AS - ankylosing spondylitis; CAPS - cyropyrin-associated periodic syndromes; CD - Crohn’s disease; CLL - chronic-lymphocytic leukemia, HBV - Hepatitis B virus; HCV - Hepatitis C virus; JIA - juvenile idiopathic arthritis; KS - Kaposi sarcoma, MPA – microscopic polyarteritis; NHL - non-Hodgkins lymphoma; PPMS - primary progressive multiple sclerosis; PsA - psoriatic arthritis; RA - rheumatoid arthritis; RRMS - relapsing remitting multiple sclerosis; SLE - systemic lupus erythematosus
Type 1 IFNs
Interferons (IFNs), as indicated by their name, interfere with viral infections (Pestka, 2007). The type I IFNs comprise 12 different α subtypes, β, ε, κ and ω IFNs, are ubiquitously induced by viral and bacterial infections, although plasmacytoid dendritic cells (pDCs) are the major producers. They bind a common IFNα/β (IFNAR) heterodimeric receptor, inducing hundreds of genes termed interferon stimulated genes (ISGs) that mediate a wide array of biological functions, including both activating and inhibitory immunomodulatory effects (Gonzalez-Navajas et al., 2012).
Type 1 IFNαs are approved for cancer treatment based on their anti-proliferative and immune adjuvant effects (Table 1). They are also approved for treatment of hepatitis B and C (HBV and HCV, respectively), based on their direct inhibitory effect on viral replication, through the induction of anti-viral genes like ISG15, Mx GTPases, RNase L, and PKR (Sadler and Williams, 2008) and immunostimulatory activities. While IFNα has served as the backbone for HCV therapy, the advent of viral protease, polymerase and replication complex inhibitors in IFNα-free treatment regimens have significantly changed the HCV treatment landscape. In studying patients that spontaneously resolve HCV infection as well as those successfully treated, a robust multi-epitope CD4+ and CD8+ memory T cell response to HCV appears to be a requisite for viral control (Schmidt et al., 2013). Patients unable to clear HCV infections are more likely to have transient and weak T cell responses that react to a limited spectrum of HCV epitopes. In addition, HCV reactive T cells are intrinsically dysfunctional- possibly due to expression of inhibitory receptors (e.g., PD1 or CD279) on T cells or ligands for inhibitory receptors (e.g, PDL1) on other cell types. In vitro blockade of PD1 or the addition of IL-2 restores T cell functions (Urbani et al., 2006). The recent discovery of a diagnostic marker, SNPs linked to the IL28B (λ3) gene, which predicts patient response to IFNα therapy (Ge et al., 2009) should also provide mechanistic insights into host-pathogen interactions to improve curative paradigms for the treatment of HCV and other chronic viral illnesses (e.g., HBV and HIV) as well as for cancer immunotherapy.
While exogenous IFNα is of therapeutic benefit in chronic viral diseases, overproduction of type I IFNs can tip immune balance and cause autoimmune disorders (Lichtman et al., 2012). Indeed, a small fraction of patients treated with IFNα develops systemic lupus erythematosus (SLE). In two-thirds of patients with SLE, a prototype autoimmune disorder characterized by the production of autoantibodies, high levels of ISGs are detected in peripheral blood cells of patients (designated as “IFN-high” SLE) (Baechler et al., 2003). Interestingly, treatment of SLE patients with an anti-IFNα mAb (rontalizumab- NCT00962832) provided clinical benefit only in the IFN-low subset of patients and reiterates the need for diagnostic markers (e.g., IFN-signature) to guide therapeutic choices.
Paradoxically, IFNs are approved for another disease, a relapsing and remitting form of multiple sclerosis (RRMS), based on the immunosuppressive actions of IFN β-1 (Dhib-Jalbut and Marks, 2010). What is especially mystifying is that all Type I IFNs bind the same IFNAR complex. How binding of different ligands to the same receptor complex triggers a broad range of different biological responses remains a puzzling question for which there is yet no definitive answer. The recent description of the unique ability of IFN-β to bind IFNAR1 and transduce signals in a Jak/STAT-independent manner in the absence of IFNAR2 compared to the requirement for IFNAR1/2 for IFN-α may provide a mechanistic basis for this paradoxical phenomenon (de Weerd et al., 2013). As will further discuss, paradoxical actions of a cytokine are not an exclusivity of interferons. Rather, understanding how a single cytokine produces different biological effects and how different molecules use a limited set of receptor to produce a broad range of responses is one of the main challenges in designing targeted therapies (Figure 2).
Figure 2.

Lessons learnt and Challenges ahead.
Interleukin 2
IL-2 and the IL-2 receptor (IL2R) were the first cytokine and cytokine receptors to be cloned (Boyman and Sprent, 2012; Malek and Castro, 2010). Produced predominantly by activated CD4+ T cells, IL-2 is the prototypic, autocrine T-cell growth factor. Based on these immunostimulatory actions, IL-2 was approved for metastatic renal cell carcinoma in 1992. A limiting toxicity of IL-2 is vascular leak syndrome (VLS) and relates in part to the fact that endothelial cells express IL2Rs and release pro-inflammatory cytokines and vasoactive mediators. By virtue of their ability to block the immunostimulatory effects of IL-2, daclizumab and basiliximab, anti-IL-2R mAbs, are approved for the prevention of acute organ rejection.
The biology of IL-2 turns out to be more complex than just its immunostimulatory role and thus provides some interesting lessons for targeted therapies. A major surprise that reshaped our view of IL-2 function was that Il2−/−, Il2ra−/− and Il2rg−/− mice develop systemic autoimmunity. This counterintuitive phenotype is due to the requisite role of IL-2 in Foxp3+ regulatory T cells (Tregs), a cellular subset that was recognized long after the discovery of IL-2. Like IFNs, IL-2 has both pro- and anti-inflammatory actions. The latter effect can limit anti-tumor efficacy but may also provide new therapeutic opportunities.
Aside from actions on diverse cell populations, other factors help to explain the complexity of action of IL-2. The IL-2R consists of the IL2Rα (CD25), IL2Rβ (CD122) and the common cytokine receptor γc (CD132) subunits. IL2Rα is not involved in signaling, but rather influences affinity, and differential expression of these three receptor subunits on distinct immune cell types accounts for varying activities of IL-2. To improve efficacy and to diminish toxicities, efforts have been made to selectively target IL-2 to the IL2Rβ/γc complex and thereby preferentially activating CD8+ and NK cells and promoting tumoricidal activity. An immunocomplex of IL-2 coupled with an anti-IL-2 mAb (e.g., S4B6 and MAB602) favors IL-2Rβ/γc binding and provides superior anti-tumor activity in mice. Alternatively, selective IL-2 targeting can also be achieved by generating IL-2 ‘superkines’ with improved binding to IL2Rβ (Levin et al., 2012). Hence, selective activation of different cell types by engineered ‘superkines’ or immunocomplexes may provide greater therapeutic benefit with less toxicity.
Conversely, IL-2 immunocomplexes (e.g., JES6.1) that favor binding to IL2-Rα can drive Treg cell expansion and function, and limit autoimmune disease (Boyman and Sprent, 2012). The concept of expanding Treg cells to tame autoimmunity has gained recent clinical validation with efficacy observed in the treatment of graft-versus-host disease (GVHD) and HCV-associated cryoglobulinemia – a type of vasculitis, though the same low-dose IL-2 regimen in combination with rapamycin resulted in clinical worsening of Type 1 Diabetes Mellitus (NCT00529035, NCT0574652, NCT00525889). New insights have also come from the use of daclizumab (a monoclonal antibody specific for IL-2-Rα) in RRMS patients (Bielekova, 2013; Martin, 2012). As expected, daclizumab decreases Treg cells, but it also increases CD56bright CD122hi NK cells that have novel immunosuppressive functions.
Despite being the first cytokine cloned 31 years ago, new aspects of the biology of IL-2 continue to be revealed. Coupled to the identification of “new” cellular subsets, such as Foxp3+ Treg and CD56bright CD122hi NK cells, the capacity to manipulate distinct receptor complexes provides the ability to capitalize upon the seemingly paradoxical functions of this prototypic cytokine. Understanding its complex actions and dissecting of its role in immune homeostasis should allow more effective therapeutic use of this fascinating cytokine and its antagonists.
Tumor Necrosis Factor (TNF)
Tumor necrosis factor (TNF) is the archetype cytokine of a 19-member TNF superfamily and a 32-member TNF superfamily receptors (Aggarwal et al., 2012). Its discovery as a serum substance found in mice infected with bacillus Calmette-Guerin that selectively induced necrosis of tumor cells provides the basis for its coining “tumor necrosis factor”. Gene cloning of TNFα and identification of its receptors- TNFR1 and TNFR2 enabled the field to rapidly explore potential therapeutics.
TNFα is synthesized and presented on the cell surface as a type 2 transmembrane protein. Cleavage by membrane TNFα (mTNFα) by TNFα-converting enzyme gives rise to its secreted (sTNFα) form. Both forms bind and activate TNFRs. Conversely, binding of TNFRs to mTNFα can confer “reverse signaling” to the mTNFα-bearing cell. Biological consequences of this “reverse signaling” include secretion of IL-2 and IFNγ by T cells and increased cytotoxicity by NK cells.
Based on the discovery of TNFα as a tumor necrosis-inducing agent, initial drug discovery efforts were focused in cancer treatment (Palucka et al., 2005), but results showed minimal efficacy associated with significant toxicity. Since TNFα is also induced in bacterial sepsis, early drug discovery efforts also tested TNF antagonists in this setting. Eleven randomized placebo controlled studies involving >7000 patients testing three different anti-TNFα mAbs, TNFR1-Fc and TNFR2-Fc failed to demonstrated benefit of anti-TNF therapy for sepsis.
The subsequent testing of TNFα antagonists in inflammatory diseases was supported by the presence of TNFα as part of the “lymphodrek” in the synovial fluid of RA patients (Feldmann, 2002). Treatment of synovial cultures with anti-TNFα mAbs decreased IL-1, GM-CSF and IL-6 levels suggesting that TNFα may be at the top of a cytokine cascade. Overexpression of a TNFα transgene in mice, in which a 3’-regulatory element was deleted that resulted in TNFα overproduction, caused erosive polyarthritis and inflammatory bowel disease (IBD). These pre-clinical studies provided the basis for broad clinical experimentation in humans over the past 22 years. To date, there are five FDA-approved TNFα antagonists. As a therapeutic class, TNFα antagonists represent the largest commercial class of therapeutics with over $25B of annual sales.
TNF antagonists offer interesting lessons again pointing to unanticipated immunosuppressive roles of prototypic pro-inflammatory cytokines. Although Tnfα−/− mice or mice treated with TNFα antagonists have delayed onset of experimental autoimmune encephalitis (EAE), a phase II study of patients with MS with the TNF blocker, lenercept, demonstrated worsening of disease (Group, 1999). Pre-clinical work indicates that TNFR2 is required for oligodendrocyte regeneration, whereas TNFR1 promotes autoimmunity (Arnett et al., 2001), raising the possibility that selective inhibition of TNFR1 might be clinically useful. Additionally, TNFα inhibits plasmacytoid DC IFNα production and patients treated with TNFα antagonists demonstrate increased ISGs (Palucka et al., 2005). These and other mechanisms likely account for some of the autoimmune phenomenon (e.g., increased autoantibodies) and toxicities (e.g., worsening of RRMS) observed with anti-TNF treatment.
Another aspect to consider is the effectiveness of infliximab, an anti-TNFα monoclonal antibody, but not of etanercept, a TNFRII-Fc fusion receptor, in the treatment of Crohn’s Disease, a form of inflammatory bowel disease. This differential effect led to important questions whether the efficacy of different biologicals that target TNFα relate to their ability to affect the function of membrane-bound versus soluble TNFα. Membrane-bound TNFα is important for protective immunity and lymphoid organization, except for primary B cell follicle formation which is reliant on sTNFα (Nielsen and Ainsworth, 2013; Ruuls, 2001). Conversely, sTNFα plays a dominant role in several inflammatory conditions except for inflammatory bowel disease. One plausible explanation for the differential efficacy of infliximab and etanercept in Crohn’s Disease was that infliximab would have the unique ability to induce apoptotic cell death. However, this model has been put into question because another TNFα-targeting drug, certolizumab pegol (CZP), also binds mTNFα, has shown clinical efficacy in Crohn’s Disease, but does not induce apoptosis. This conundrum may be partly enlightened by the ability of infliximab and CZP to induce mTNFα signaling that downregulates pro-inflammatory signals without inducing apoptosis (Derer et al., 2013).
Interleukin-1
IL-1, one of the first cytokines identified, was described in the 1940’s as a product of leukocytes termed “endogenous pyrogen” and is now recognized as a prototypic cytokine that induces inflammation. Its actions are attributable to two related gene products IL-1α and IL-1β (Gabay et al., 2010). Inactive pro-IL-1β is processed by the inflammasome complex to generate the active cytokine. There are many products that activate the inflammasome ranging from crystals to microbial products. Like TNF, IL-1β blockade was efficacious in many models of inflammatory arthritis. Because IL-1 has an endogenous inhibitor, IL-1 receptor antagonist (IL-1Ra), a recombinant version (kineret) was developed and approved in 2001 for the treatment of RA.
A collection of genetic autoinflammatory disorders due to mutations of NLRP3 (designated the cryopyrin-associated periodic syndromes or CAPS) that result in constitutive inflammasome activation and IL-1β secretion has returned IL-1 therapies to the spotlight (Federici et al., 2013; Gabay et al., 2010; Goldbach-Mansky and Kastner, 2009). Three IL-1β antagonistis (kineret, rilonacept, and canukinumab) are now approved for these disorders and underscores the power of human genetics in identifying diseases where targeted therapeutics may provide their greatest clinical impact.
Aside from IL-1, the IL-1 family of ligands consists of ten additional family members. Many of these additional family members (e.g., IL-18 and IL-33) are genetically linked to autoimmune diseases and much will be learned as these novel cytokines are targeted in the clinic.
Interleukin-6
Produced by many immune and non-immune cells, IL-6 is the third member of inflammatory cytokine triumvirate: TNF, IL-1 and IL-6 (Tanaka et al., 2012). It has widespread effects in B cell Ig production, induction of acute phase reactants, Th17 cell differentiation, megakaryocyte maturation and osteoclast function. Blocking IL-6 is effective in many models of inflammation and an anti-IL6R mAb (toclilizumab) is approved for Castleman’s disease (a rare benign lymphoproliferative disorder of germinal center B cells), RA and juvenile idiopathic arthritis (JIA) (Nishimoto and Kishimoto, 2006).
Given the multitude of therapeutic options for patients with RA, how then might physicians choose the appropriate therapy for patients? In fact, there are few biomarkers that help guide therapy (Daien and Morel, 2014). A recent analysis of rheumatoid arthritis synovial transcriptome phenotypes has identified four major phenotypes (Dennis et al., 2014). Each phenotype has a distinct underlying gene expression signature reflecting different cellular compositions and correlate with differential clinical responses to adalimumab and tocilizumab treatment. This is clearly an important area to pursue.
Targeting Cytokine Signaling – the Jakinibs
In addition to directly blocking of cytokines and cytokine receptors, an alternative strategy is to interfere with receptor downstream signaling. The possibility that cytokine signaling could be targeted by small molecules stemmed from several developments - some derived from basic research and some from clinical studies of a cohort of patients with a rare disease. The importance of reversible protein phosphorylation in signal transduction along with the elucidation of the human kinome facilitated the development of therapeutically useful kinase inhibitors (Manning et al., 2002). With their prominent role in cancer, proteins tyrosine kinases (PTKs) became obvious targets, although developing specific PTK inhibitors was challenging. Some of the first PTK inhibitors, like genistein and herbimycin A, were natural products isolated from fungi and their poor target specificity limited their therapeutic usefulness. Aided by new insights into the protein structure of kinases and increasing sophistication of medicinal chemistry, many successes emerged stemming from structure-enabled rational drug design and high-throughput screening approaches. Imatinib, for example, revolutionized the treatment of chronic myeloid leukemia (Druker et al., 2001) and now kinases are among the most attractive targets in cancer therapeutics, with 20 FDA-approved inhibitors.
Many key immune receptors initiate signaling via PTKs linked to downstream serine-threonine kinases. Cytokines that bind type I and type II cytokine receptors constitute an excellent example. They comprise a range of factors that include: interleukins, interferons, colony stimulating factors, hormone-like cytokines and all exert their effects through Janus kinases (JAKs) (Leonard and O’Shea, 1998). Specifically, the intracellular domains of cytokine receptors selectively bind to different JAKs (TYK2, JAK1, JAK2 and JAK3) in various combinations. Mutant cell lines initially revealed the criticality of TYK2, JAK1 and JAK2, but the first in vivo proof of the importance of JAKs came from the discovery of children with severe combined immunodeficiency and JAK3 mutations. Gene knock-out mice subsequently confirmed the essential, non-redundant functions of the JAKs.
The first JAK inhibitor (jakinib) to be tested in humans was tofacitinib (Changelian et al., 2003). Tofacitinib was approved for moderate-to-severe RA and has shown efficacy in ulcerative colitis (UC) and psoriasis (Sandborn et al., 2012; Strober et al., 2013). The initial strategy underlying the development of tofacitinib was JAK3’s critical role in immunity. Although tofacitinib was intended to be a JAK3 inhibitor, in reality it also inhibits JAK1 and, to a lesser extent, JAK2. One might assume that this could be a drawback, given lethal phenotypes associated with Jak1−/− or Jak2−/− mice. In fact, use of tofacitinib is associated with mild to moderate anemia. Moreover, the discovery that gain-of-function JAK2 mutations that underlie myeloproliferative disorders provided a rationale for purposefully targeting JAK2. Ruxolitinib, a JAK1 and JAK2 inhibitor, was approved for myelofibrosis (Verstovsek et al., 2010) and was the first JAK inhibitor approved for human use.
There are useful lessons offered by first generation jakinibs (O’Shea et al., 2013). First, because they affect multiple JAKs, they interfere with multiple cytokines that contribute to RA immunopathogenesis. As a result, these drugs inhibit both adaptive and innate immunity. Thus, jakinibs provide a good example of how genetics can both inform and mislead drug development. A priori, a reasonable expectation was that selectivity would be critical for the development of a successful drug and inhibition of JAK2 would be problematic. However, this may be less detrimental than expected and interfering with multiple cytokines has the benefit of modulating both innate and adaptive immunity.
Another point to consider is that although jakinibs interfere with cytokines, unlike the other biologicals previously discussed, they do not provide long-term inhibition; thus, a potential advantage of small molecule inhibitors is their short half-lives. Drug discontinuation typically results in rapid reversibility of the immunomodulatory effects when compared to biotherapeutics that have longer half-lives. Much is still to be learned from the next-generation Jakinibs with different ratios of JAK1:2:3:Tyk2 inhibition and how these differences may translate into efficacy and toxicities.
Targeting T cells and their products
Polyclonal preparations of rabbit or equine anti-human thymocyte globulin have been used for allograft rejection and aplastic anemia since the late 1990s. Unfortunately, their use was complicated by cytokine release syndrome and immunogenicity to rabbit/equine antibodies resulting in serum sickness. Given the importance of T cells in autoimmune disease and organ rejection, significant efforts were focused on generating monoclonal antibodies to target specifically T cells. The identity of these antibody targets, however, was often not known until much later. A mAb against the T cell receptor (TCR) complex (anti-CD3 Ab or OKT3) was the first to be approved and commercialized for use in solid organ allograft rejection (Table 1). Another such anti-T-cell mAb was anti-CD52 mAbs (CAMPATH-1M, G, H or alemtuzumab) that demonstrate clinical efficacy in GVHD, non-Hodgkin’s lymphoma, vasculitis, and RRMS (Waldmann and Hale, 2005). The target antigen was subsequently discovered to be CD52, a glycosylphosphatidylinositol linked protein expressed on T cells, but also on other immune cells. The breadth of expression contributes not only to its clinical effectiveness, but also its toxicities including prolonged cytopenias, paradoxical autoimmunity and opportunistic infections. The recent description of a suppressive CD52+CD4+ T cell provides additional mechanisms for alemtuzumab action either through depletion of CD52+ suppressor cells and/or binding and neutralization of the immunosuppressive activities of soluble CD52 released from CD52+ T cells (Bandala-Sanchez et al., 2013).
Anti-CD4 mAbs offered the first opportunity to selectively target CD4+ T cells at a time when we did not fully appreciate Th cell heterogeneity (Isaacs et al., 1997). Pre-clinical experimentation had demonstrated robust efficacy of anti-CD4 mAbs in SLE, EAE, inflammatory arthritis, GVHD and tolerance induction. Over a dozen different anti-CD4 antibodies have entered clinical testing and can be segregated into four generations reflecting the many obstacles encountered (Table 1). Those include immunogenicity, long-lasting depletion of CD4+ T cells, short drug half-life due to internalization, clearance of the bound antibody and, surprisingly, development of dose-limiting rash. Overall, targeting CD4 has not been a clinically or commercially attractive venture. Based on these experiences, one might assume that wholesale blocking of T cells would be problematic, but contrasting with the relative poor track record of anti-CD4 mAbs, is abatacept, a CD152-Fc fusion protein that inhibits CD28/CD80 (B7.1) and CD86 (B5.2) costimulatory pathways that are important for T cell activation and acquisition of effector functions. Abatacept is approved for the treatment of rheumatoid arthritis and a form of arthritis that develops in children and adolescents, called juvenile idiopathic arthritis, (Keating, 2013). Admittedly, targeting co-stimulation is much less draconian than depleting T cells, but it is still not entirely obvious why the outcomes of these two strategies are so different.
Helper T cells
With the realization of the heterogeneity of CD4+ T cell subsets, more recently drug discovery efforts have focused on targeting specific cytokines associated with different CD4+ T cell subpopulations. In fact, recognition in the late 1970’s that CD4+ T cells could present two different flavors, Th1 and Th2, was just the beginning of understanding the dynamic nature and heterogeneity of Th cell differentiation (Zhu and Paul, 2008). Four decades of investigation has led to detailed characterization of receptors, signaling events and transcriptional control that regulate Th1 cells to secrete IFNγ and respond to intracellular bacteria and protozoa, Th2 cells to secrete IL-4, IL-5 and IL-13 and respond to helminths, Th17 cells to secrete IL-17 to mediate immunity against extracellular bacteria and fungi, and Treg cells to secrete IL-10 or TGF-β and dampen immune responses and inflammation. Dissection of each T helper subset has permitted more precise targeting of pathogenic T cell responses without disarming all CD4+ T cell functions.
Th2 cells
Th2 immunity is associated not only with Th2 cells, but accumulation of eosinophils, alternatively activated macrophages, basophils and mast cells, elevated IgE, mucus production, and smooth muscle hyperplasia (Van Dyken and Locksley, 2013) and aberrant activation of Th2 pathways is associated with the development of atopic/allergic diseases. Both IL-4 and IL-13 bind a heterodimeric IL13R1/IL4Rα receptor that signals through a Jak1/Tyk2 pathway. IL-4 has an additional private IL-4Rα/γc receptor expressed on lymphocytes that promotes class-switching and increases in IgE. IL-13 also has a private receptor- IL13R2 whose function is not fully understood. In addition to Th2 cells, many other cell types including basophils, mast cells, eosinophils, innate-like lymphocyte type 2 cells (ILC2) and NKT cells secrete IL-4 and IL-13.
The impetus to target these pathways in allergic diseases [atopic dermatitis (AD), allergic rhinitis and allergic asthma] is supported, in part, by demonstration that IL-4, IL-5 and IL-13 play important roles in models of allergic lung inflammation and because of the identification of TSLP, IL13 and IL4R single-nucleotide polymorphisms (SNPs) associated with AD and asthma susceptibility.
There have been several interesting insights gained from targeting Th2 cytokines. Just as CD4+ T cells are heterogeneous, so are asthma patients. Significant efforts have been put on defining asthma phenotypes based on clinical and laboratorial parameters. Age of onset, history of atopy, body mass index, smoking, lung function, serum IgE levels, peripheral eosinophilia and sputum eosinophilia, are examples, just to name a few (Ingram and Kraft, 2012). Eosinophilia and serum IgE have been utilized to stratify patients for clinical trials. Anti-IgE (omalizumab) therapy is approved for patients with moderate to severe asthma and its use and dose are determined, in part, on serum IgE levels. A significant breakthrough was made recently with the discovery of a new biomarker, serum periostin- a bronchial epithelial cell gene induced by IL4/13, which identifies asthma patients who respond to an anti-IL13 mAb (lebrikizumab) (Arron et al., 2013). Similarly, treatment with an anti-IL4Rα mAb (dupilumab) or an anti-IL-5 mAb (mepolizumab) is efficacious in a selected subset of asthma patients with elevated eosinophil levels (NCT01312961, ISRCTN75169762, NCT00292877). These experiences further underscore the need to develop companion diagnostic tests that reflect a patient’s underlying disease to enable the best therapeutic decision.
Additionally, the clinical experiences with targeted Th2 therapies have revealed not only the heterogeneity of human allergic diseases, but also the different contributions within the Th2-associated cytokines with IL4/13 demonstrating broader inflammatory roles and IL-5 having a more restricted role through eosinophil recruitment. As these Th2 targeting agents progress in the clinic and because Th2 cytokines are known now to contribute to other biological processes beyond immunity, we will learn much more about their contributions not only in allergic, but also fibrotic and metabolic diseases
Interleukin-17
The biological understanding of the IL-17 family members in immunity and disease has rapidly emerged and with some quite unexpected results (Miossec and Kolls, 2012). IL-17 exists in three forms- IL-17A/A, IL-17A/F and IL-17F/F. All three forms bind IL-17RA/IL-17RC and through ACT1/TRAF6 signaling and activates NF-κB. IL-17 operates on a variety of cell types and synergizes with TNF to promote inflammatory responses. IL-17 family members are important in controlling extracellular bacterial and fungal infections. Development of Th17 cells requires IL-23 and IL-1β and transcription factors RORα and RORC (RORγt in mice). In addition to IL-17A and F, Th17 cells also secrete IL-21, IL-22 and GM-CSF.
Multiple agents are in clinical development to target the IL-17 family members and the clinical experience has revealed both concordant and discordant clinical results from IL-12/23 versus IL-17 neutralization. The most impressive clinical efficacy with IL-17 and IL12/23 neutralization, to date, is in psoriasis and ankylosing spondylitis (AS) (NCT00809159, NCT01107457, NCT00267969 and NCT01330901). Disappointingly, while elevated IL-17 levels were first described in synovial fluid of RA patients, IL-17 antagonists, with the exception of one study, have not demonstrated efficacy.
In contrast, discordant results have been demonstrated in RRMS and CD. Secukinumab (anti-IL17A/A mAb) is efficacious in RRMS (NCT01874340), while ustekinumab (anti-IL12/23 mAb) demonstrated no benefit (NCT00207727). This discordance may be explained, in part, by pre-clinical experiments that demonstrate a requirement of IL-23 in the priming, but not effector, phase of EAE (Thakker et al., 2007) as well as the ability of IL-12 to induce IFN-γ which is protective in EAE (Gran et al., 2004). Conversely, while ustekinumab is efficacious in patients with moderate to severe CD (NCT00771667), treatment of patients with secukinumab resulted in worsened disease (NCT01009281). Why targeting different points of the Th17 axis leads to differences in therapeutic outcome for different diseases remains to be understood. These results nonetheless exemplify not only how much remains to be learned but also that results of pre-clinical trials can also be informative and offer guidance to basic research (Figures 2 and 3).
Figure 3. Flow of information in development of targeted therapies.
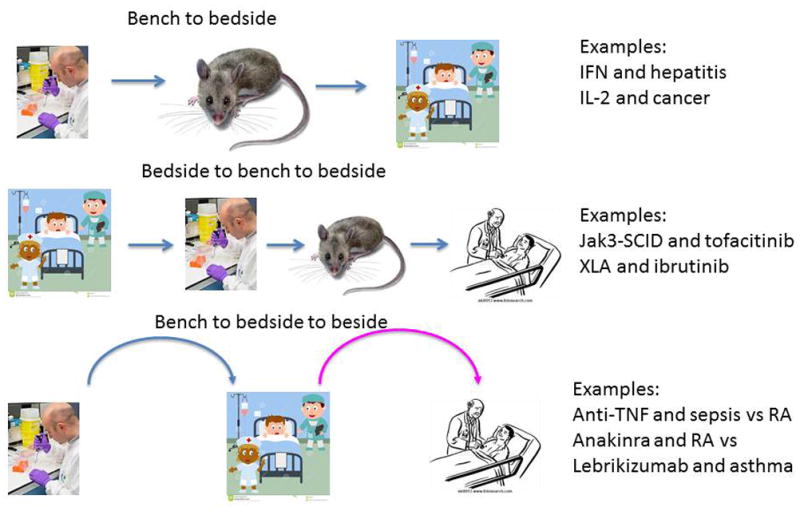
Advances in molecular biology revealed a number of targets that were identified at the bench and led to successful drugs at the bedside (‘bench to bedside’) Conversely though, advances in sequencing technology led to the discovery of various genetic disorders that also provided convincing targets for intervention. Such “experiments of nature” facilitated the understanding of what the consequence of interfering with a target might be and thus provided impetus to go back to the bench (‘bedside to bench to bedside’). In other circumstances, development of a targeted therapy did not have the expected result. Fortunately, in a number of cases this led investigators to go back to the bedside and rethink disease mechanisms, and find the right disease for the therapy (‘bench to bedside to bedside’).
Of note, with respect to the biology of IL-17, it is important to emphasize that Th17 cells are just one of many types of cells that produce IL-17, and are likely not its most prominent source. Other producers of IL-17 include γδ T cells, innate lymphoid cells (ILCs) and neutrophils. The importance of Rorγt as a key transcription factor that controls IL-17 production in all these cells suggests that it might be a reasonable target for generation of small molecule inhibitors. Rorγ is part of the 48 member nuclear receptor superfamily, which includes glucocorticoid and steroid hormone receptors. Remarkably, an old drug, digoxin, can bind to Rorγt and inhibit IL-17 production (Huh et al., 2011). Generation of selective Rorγ inhibitors is an active area of research and holds great promise in targeting Th17 biology.
Targeting B cells
The contribution of B cells to humoral immunity was already suggested in 1952 by Colonel Ogden Bruton with the description of the case of a boy with history of pneumonia and bacterial respiratory infections who had no serum immunoglobulins (Ig). Bruton type or X-linked agammaglobulinemia (XLA) would become the first primary immunodeficiency syndrome to be described. Patients with XLA suffer a developmental abnormality caused by mutations in Bruton’s tyrosine kinase (BTK) and lack mature B cells.
The mechanisms by which B cells contribute to autoimmunity have exponentially increased since the description of XLA (Pillai et al., 2011). The pathogenic roles of autoantibodies and immune complexes were the first to be established. However, B cells also secrete cytokines and express co-stimulatory receptors that contribute to T cell function. In addition, activated B cells are potent antigen presenting cells and are found in ectopic lymphoid aggregates in non-lymphoid organs in autoimmune diseases, where they promote germinal center (GC) reactions. Plasmablasts or plasma cells resident within these GC-like structures secrete autoantibodies that propagate inflammation. The identification of regulatory B cells that attenuate T cell responses through IL-10 or TGF-β secretion or through intercellular interactions provides an additional dimension by which B cell deregulation can contribute to autoimmunity (Mauri and Bosma, 2012).
The approval of two B cell modulating therapies- rituximab and belimumab has enabled to begin understanding the effects of B cell modulation in humans (Isaacs et al., 1997; Jacobi and Dorner, 2010) (Table 1). Rituximab is a chimeric anti-CD20 mAb that depletes most CD20+ B cells and is approved for the treatment of autoimmune disorders including patients with RA who have failed TNF antagonist therapies, ANCA-associated vasculitis (AAV) and microscopic polyarteritis (MPA). Belimumab is an anti-BAFF/BLys mAb that neutralizes a growth factor critical for B cell survival and is approved for the treatment of SLE, a disease where rituximab did not demonstrate benefit (Merrill et al., 2010; Rovin et al., 2012).
Treatment of patients with rituximab and belimumab has provided support for many of the proposed B-cell mediated mechanisms in autoimmunity. Treatment with rituximab decreases a subset of autoantibodies that are casually linked to autoimmune diseases as well as decreasing IL-6 secreting B cells, IL-17 production and skewing cytokine profiles that favor immunosuppression (Barr et al., 2012; Gran et al., 2004). Treatment of SLE patients with belimumab decreases naïve and activated B cell numbers, anti-dsDNA Abs and normalizes complement levels (Stohl et al., 2012).
A surprise in B-cell drug discovery was the efficacy of rituximab therapy observed in patients with RRMS (Hauser et al., 2008). At the time, the major paradigm was that RRMS was primarily a T-cell mediated disease. T cell-directed therapies were extremely efficacious in EAE when administered before or after disease onset, whereas B cell modulation was only effective before disease onset. Another puzzle yet to be solved is that while rituximab is efficacious, treatment of RRMS patients with a TACI-Fc fusion protein (atacicept), that neutralizes both BAFF/BLys and APRIL (a related family member), results in disease worsening (NCT00642902).
An additional aspect of B cell modulation therapy is the need to better understand the status of the immune lymphocyte repertoire following therapy. In the case of rituximab causes B cell depletion, but B cells begin to repopulate the circulation 6 months following the end of therapy and return to baseline levels ~12 months following treatment (Roll et al., 2006). These repleted B cells, however, are mostly naïve CD27-IgD+ B cells and not memory CD27+IgD+ B cells, the latter subset requiring >2 years to return to baseline levels. B cells undergo both central and peripheral selection processes to delete autoreactive B cells (Meffre, 2011). Patients with SLE, type 1 diabetes and rheumatoid arthritis demonstrate loss of both central and peripheral tolerance while patients with MS only demonstrate defects in peripheral tolerance. Might this difference in selection checkpoints contribute to the more sustained clinical effects observed with B cell depletion therapy in MS? Further analysis of immune repertoires following B cell modulatory therapies will provide greater insights into both basic immunology and the mechanistic basis of efficacy or lack of efficacy in the treatment of autoimmune diseases.
Targeting Immunoreceptor signaling
The TCR, B-cell receptor (BCR) and Fc receptors (FcRs), are structurally related and their shared modes of signal transduction were elucidated during the late 1980s. Phosphorylation of receptor subunits on specific tyrosine residues termed immunoreceptor tyrosine based activation motifs (ITAMs) by SRC family PTKs recruits spleen tyrosine kinase (SYK) family of PTKs by virtue of their tandem SRC homology 2 domains (Chan et al., 1994a). There are two SYK PTK family members- ZAP-70 and SYK. While patients with ZAP70 mutations have a severe combined immunodeficiency making this kinase a logical therapeutic target, a successful inhibitor of this kinase has yet to be developed (Chan et al., 1994b). A candidate clinical SYK inhibitor, fostamatinib (R788), was developed and demonstrated utility in preclinical models of allergy, RA and SLE. It showed promised in initial trials, but was terminated in pivotal trials in RA (NCT00665925, NCT01242514, NCT01197755). New clinical trials are evaluating more specific and potent SYK inhibitors in the setting of chronic lymphocytic leukemia (CLL) and autoimmunity. As SYK has important functions outside of the immune system including thrombosis and vascular biology, additional clinical investigation of SYK inhibitors will inform their suitability in immune and non-immune mediated diseases and potential toxicity liabilities.
Besides SRC- and SYK- family of PTKs, a third family, the TEC PTKs, is important in immunoreceptor signaling. One member of this family, BTK, is especially notable, since mutations of BTK are often the underlying cause of X-linked agammaglobulinemia (XLA). Since XLA patients can live normal and healthy lives with as long as they receive immunoglobulin replacement, BTK makes a logical target for diseases of the B cell compartment. Indeed, Ibrutinib (PCI-32765)a BTK inhibitor has been approved for patients with mantle cell lymphoma and chronic lymphocytic lymphoma (Byrd et al., 2013; Wang et al., 2013), and testing of BTK inhibitors in autoimmune disorders is currently ongoing.
In addition to these immunoreceptor activated PTKs, leukocytes activate a variety of additional signaling pathways required for function. These include phosphoinositide 3-kinase (PI3K), protein kinase B, mammalian target of rapamycin, protein kinase C family, and mitogen-activated protein kinases (MAPK). Many of these kinases are active areas of cancer drug discovery. Given their broad functions, targeting this kinase can be associated with toxicities that are tolerable for cancer therapy, but may be problematic for long-term treatment of chronic diseases. We will limit our discussion on two targets- PI3K-δ and p38. Phosphoinositide 3’ kinases (PI3K) consist of a p85 regulatory subunit that associates with one of four- α, β, γ or δ catalytic subunits. Specifically, PI3K-δ is required for B and T cell immunity, whereas PI3K-γ is critical for chemokine receptor signalling in neutrophils and macrophages and for optimal T cell responses (Rommel et al., 2007; Winkler et al., 2013). A gain-of-function PI3KCD mutations underlie an immunologic disorder characterized by T cell senescence and immunodeficiency and may benefit from PI3K-δ inhibitors (Angulo et al., 2013). Mice deficient in PI3K-δ or PI3K–γ, or mice treated with selective PI3K-δ and PI3K-γ inhibitors are protected from development of autoimmunity and inflammatory diseases (Banham-Hall et al., 2012).
Idelalisib, a PI3k-δ inhibitor, has demonstrated clinical efficacy in refractory indolent non-Hodgkins lymphoma and in combination with rituximab in relapsed CLL (NCT01539512 and NCT01282424). While acceptable for cancer patients, the incidence of fatigue, diarrhea, fever and rash observed in idelalisib treated patients is unlikely to be suitable for chronic use in autoimmune disorders. In addition, PI3K-δ has been demonstrated to be required for Treg function and also plays an inhibitory role in TLR signalling in macrophages (Patton et al., 2006; Uno et al., 2010). These mechanisms may contribute to the spontaneous colitis observed in mice expressing PI3K-δ(D910A). Whether some of the toxicities observed with idelalisib are on- or off-target will require additional evaluation with other PI3Kδ and dual PI3Kδ/γ inhibitors.
The MAPK family is an evolutionarily conserved pathway comprising three tiers of sequentially activated kinases that control broad cellular functions in response to extracellular stimuli. The p38 MAPK has been an area of intense focus of clinical development for autoimmunity (Genovese, 2009). The p38 MAPKs have four isoforms- α, β, γ and δ. At least seven small molecule inhibitors of p38 α, β and δ with different specificities have been tested in the clinic. To date, clinical efficacy has not been observed in RA, psoriasis, or CD. Some shared toxicities have included skin disorders, infection, liver abnormalities and gastrointestinal side effects making these likely to be on-target effects. A consistent finding of the various p38 inhibitor trials is that C-reactive protein, a surrogate inflammatory marker, decreases initially with therapy, but then returns to baseline levels suggesting an adaptation mechanism may occur with p38 inhibition. While p38 may not a target worth pursuing for inflammatory diseases, understanding how the underlying mechanisms of adaptation may provide insights into p38 biology as well as how inflammatory diseases circumvent therapy.
Targeting the epigenome
Various components of immune cell signaling are individually targeted by drugs in hope of rectifying aberrant immune response, and those strategies have favored molecules that are proximal to cell surface; receptors, receptor associated kinases or extracellular cytokines. Broader approaches to expand candidate drug targets now include molecules whose action is in the nucleus and on chromatin. The idea of epigenetic drugs holds great potential because the epigenome is considered plastic, responsive to perturbation, and contributes to the pathogenesis of many inflammatory diseases. Recent advance in next-generation sequencing technologies have also helped to obtain a multitude of epigenomic information as possible new biomarkers. Current spectrum of mechanism-based epigenetic drugs includes DNA methylation inhibitors, histone deacetylase inhibitors (HDACi), histone methyltransferase inhibitors and BET protein inhibitors (BETi) (Table 1).
Acetylation of histone is one of the best-characterized readouts of epigenetic information that promotes relaxed chromatin structure and active gene transcription. The organization of enzymes that constitutes “acetylome” is rather tight and includes only 18 acetyltransferases (HAT) and 18 deacetylases (HDAC). HDAC inhibitors are widely tested as cancer therapeutics and proven to be effective in slowing cell proliferation and inducing apoptosis in experimental settings. While predicting the spectrum of HDACi effect in vivo and finding appropriate regimens for clinical benefit is challenging, an oral HDACi (givinostat) has been demonstrated to provide some clinical benefit in patients with systemic JIA and with only mild reversible adverse events (Vojinovic and Damjanov, 2011). Additional placebo controlled studies will be required to assess the effectiveness of HDACi in autoimmunity.
BET proteins (Brd2-4) have been implicated in transcriptional control of multiple inflammatory genes. JQ1 is a small molecule inhibitor that binds the BET bromodomain and block interactions between Brd2-4 and acetylated histones. Treatment of macrophages with a BET inhibitor (I-BET or JQ1) or knock-down of Brd2, 3 or 4 resulted in decreased LPS-induced pro-inflammatory cytokine production (TNFα, IL-6 and MCP) (Belkina et al., 2013; Nicodeme et al., 2010). In addition, BET bromodomain inhibition with JQ1 blocks human Th17 differentiation and function (Mele et al., 2013). As many BET small molecule inhibitors are being developed, the opportunity to assess efficacy and potential toxicities of BET inhibition will be addressed in the near future.
Conclusions and speculation
The black box of immunology has given way to the discovery of numerous molecules, diverse cell populations and the identification of signaling pathways and cell biology insights. Over the past 40 years, this had led to numerous advances in dissecting molecular mechanisms underlying immune and inflammatory disease and many new therapies. “Immunology” has evolved to become “molecular immunology”, hardly a soft science by any measure. This has equipped us with the ability to identify target molecules for drug design (Figures 2 and 3). In addition to time-tested reliable compounds derived from natural sources, we have invented and compiled a wide collection of mechanism-based designer biologics as well as chemically synthesized small molecules as therapeutic options. Also our knowledge of biomarkers to predict and evaluate efficacy has expanded. Nevertheless, we should not fool ourselves to believe that our knowledge on the collection of drugs and biomarkers is sufficient to execute precision medicine in the 21st century. There will always be the “unknown unknowns”. As one example out of many, when IL-2 was approved, we did not know about Tregs, Th17s and Tfh cells as well as an essential role of IL-2 in peripheral tolerance. This will always be the case in science, medicine and drug discovery. The existence of the “unknown unknowns” always needs to be borne in mind as frontiers are approached. There will always be lessons- good (efficacy), bad (lack of efficacy), and ugly (worsening of disease) - learned from the bedside. Ultimately, we would need to decipher the rules to choose the right combination of drugs for the right person at the right time.
What might the next 40 years hold? As we further advance and refine the complexities of immunology and better understand the complexities and heterogeneity of human diseases, what might we reflect on in 2054? Where might pluripotent cells and tissue regeneration take us? Will we utilize induction and remission protocols for autoimmunity to re-establish tolerance and immune homeostasis? Can we reverse the epigenetic changes that occur in disease? How do we cure these diseases? Might we develop genetic and biomarker profiles that not only identify disease risk, but also predict when clinical disease will strike to permit treatment before end-organ damage? Might we be able to alter one’s microbiome or use vaccinations to alter immunity or delete autoreactivity for those at risk? Might 2054 reflect on the ‘cellulardrek’ of early 21st century? All said, it will undoubtedly be another breathtaking 40 years built on the integration of basic discoveries, translational insights and thoughtful, rigorous clinical studies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, Baxendale H, Coulter T, Curtis J, Wu C, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342:866–871. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett HA, Mason J, Marino M, Suzuki K, Matsushima GK, Ting JP. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nature neuroscience. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- Arron JR, Scheerens H, Matthews JG. Redefining approaches to asthma: developing targeted biologic therapies. Adv Pharmacol. 2013;66:1–49. doi: 10.1016/B978-0-12-404717-4.00001-9. [DOI] [PubMed] [Google Scholar]
- Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, Shark KB, Grande WJ, Hughes KM, Kapur V, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2610–2615. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandala-Sanchez E, Zhang Y, Reinwald S, Dromey JA, Lee BH, Qian J, Bohmer RM, Harrison LC. T cell regulation mediated by interaction of soluble CD52 with the inhibitory receptor Siglec-10. Nature immunology. 2013;14:741–748. doi: 10.1038/ni.2610. [DOI] [PubMed] [Google Scholar]
- Banham-Hall E, Clatworthy MR, Okkenhaug K. The Therapeutic Potential for PI3K Inhibitors in Autoimmune Rheumatic Diseases. The open rheumatology journal. 2012;6:245–258. doi: 10.2174/1874312901206010245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr TA, Shen P, Brown S, Lampropoulou V, Roch T, Lawrie S, Fan B, O’Connor RA, Anderton SM, Bar-Or A, et al. B cell depletion therapy ameliorates autoimmune disease through ablation of IL-6-producing B cells. The Journal of experimental medicine. 2012;209:1001–1010. doi: 10.1084/jem.20111675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina AC, Nikolajczyk BS, Denis GV. BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol. 2013;190:3670–3678. doi: 10.4049/jimmunol.1202838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielekova B. Daclizumab therapy for multiple sclerosis. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2013;10:55–67. doi: 10.1007/s13311-012-0147-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nature reviews Immunology. 2012;12:180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. The New England journal of medicine. 2013;369:32–42. doi: 10.1056/NEJMoa1215637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AC, Desai DM, Weiss A. The role of protein tyrosine kinases and protein tyrosine phosphatases in T cell antigen receptor signal transduction. Annual review of immunology. 1994a;12:555–592. doi: 10.1146/annurev.iy.12.040194.003011. [DOI] [PubMed] [Google Scholar]
- Chan AC, Kadlecek TA, Elder ME, Filipovich AH, Kuo WL, Iwashima M, Parslow TG, Weiss A. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science. 1994b;264:1599–1601. doi: 10.1126/science.8202713. [DOI] [PubMed] [Google Scholar]
- Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, Rizzuti BJ, Sawyer PS, Perry BD, Brissette WH, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003;302:875–878. doi: 10.1126/science.1087061. [DOI] [PubMed] [Google Scholar]
- Daien CI, Morel J. Predictive Factors of Response to Biological Disease Modifying Antirheumatic Drugs: Towards Personalized Medicine. Mediators of inflammation. 2014;2014:386148. doi: 10.1155/2014/386148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, Zaker-Tabrizi L, Fung KY, Forster SC, Beddoe T, et al. Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nature immunology. 2013;14:901–907. doi: 10.1038/ni.2667. [DOI] [PubMed] [Google Scholar]
- Dennis G, Holweg CTJ, Kummerfeld SK, DFC, Setiadi AF, Hackney JA, Haverty PM, Gilbert H, Lin WY, Diehl L, et al. Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Research & Therapy. 2014 doi: 10.1186/ar4555. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derer S, Till A, Haesler R, Sina C, Grabe N, Jung S, Nikolaus S, Kuehbacher T, Groetzinger J, Rose-John S, et al. mTNF reverse signalling induced by TNFalpha antagonists involves a GDF-1 dependent pathway: implications for Crohn’s disease. Gut. 2013;62:376–386. doi: 10.1136/gutjnl-2011-300384. [DOI] [PubMed] [Google Scholar]
- Dhib-Jalbut S, Marks S. Interferon-beta mechanisms of action in multiple sclerosis. Neurology. 2010;74(Suppl 1):S17–24. doi: 10.1212/WNL.0b013e3181c97d99. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Anti-inflammatory Agents: Present and Future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. The New England journal of medicine. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Federici S, Martini A, Gattorno M. The Central Role of Anti-IL-1 Blockade in the Treatment of Monogenic and Multi-Factorial Autoinflammatory Diseases. Frontiers in immunology. 2013;4:351. doi: 10.3389/fimmu.2013.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann M. Development of anti-TNF therapy for rheumatoid arthritis. Nature reviews Immunology. 2002;2:364–371. doi: 10.1038/nri802. [DOI] [PubMed] [Google Scholar]
- Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nature reviews Rheumatology. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis and rheumatism. 2009;60:317–320. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. The Journal of allergy and clinical immunology. 2009;124:1141–1149. doi: 10.1016/j.jaci.2009.11.016. quiz 1150-1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nature reviews Immunology. 2012;12:125–135. doi: 10.1038/nri3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gran B, Chu N, Zhang GX, Yu S, Li Y, Chen XH, Kamoun M, Rostami A. Early administration of IL-12 suppresses EAE through induction of interferon-gamma. J Neuroimmunol. 2004;156:123–131. doi: 10.1016/j.jneuroim.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. The New England journal of medicine. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- Huh JR, Leung MW, Huang P, Ryan DA, Krout MR, Malapaka RR, Chow J, Manel N, Ciofani M, Kim SV, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature. 2011;472:486–490. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram JL, Kraft M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies. The Journal of allergy and clinical immunology. 2012;130:829–842. doi: 10.1016/j.jaci.2012.06.034. quiz 843-824. [DOI] [PubMed] [Google Scholar]
- Isaacs JD, Burrows N, Wing M, Keogan MT, Rebello PR, Watts RA, Pye RJ, Norris P, Hazelman BL, Hale G, et al. Humanized anti-CD4 monoclonal antibody therapy of autoimmune and inflammatory disease. Clin Exp Immunol. 1997;110:158–166. doi: 10.1111/j.1365-2249.1997.tb08312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobi AM, Dorner T. Current aspects of anti-CD20 therapy in rheumatoid arthritis. Current opinion in pharmacology. 2010;10:316–321. doi: 10.1016/j.coph.2010.02.002. [DOI] [PubMed] [Google Scholar]
- Keating GM. Abatacept: a review of its use in the management of rheumatoid arthritis. Drugs. 2013;73:1095–1119. doi: 10.1007/s40265-013-0080-9. [DOI] [PubMed] [Google Scholar]
- Leonard WJ, O’Shea JJ. Jaks and STATs: biological implications. Annual review of immunology. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484:529–533. doi: 10.1038/nature10975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman EI, Helfgott SM, Kriegel MA. Emerging therapies for systemic lupus erythematosus--focus on targeting interferon-alpha. Clin Immunol. 2012;143:210–221. doi: 10.1016/j.clim.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek TR, Castro I. Interleukin-2 receptor signaling: at the interface between tolerance and immunity. Immunity. 2010;33:153–165. doi: 10.1016/j.immuni.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Martin R. Anti-CD25 (daclizumab) monoclonal antibody therapy in relapsing-remitting multiple sclerosis. Clin Immunol. 2012;142:9–14. doi: 10.1016/j.clim.2011.10.008. [DOI] [PubMed] [Google Scholar]
- Mauri C, Bosma A. Immune regulatory function of B cells. Annual review of immunology. 2012;30:221–241. doi: 10.1146/annurev-immunol-020711-074934. [DOI] [PubMed] [Google Scholar]
- Meffre E. The establishment of early B cell tolerance in humans: lessons from primary immunodeficiency diseases. Annals of the New York Academy of Sciences. 2011;1246:1–10. doi: 10.1111/j.1749-6632.2011.06347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mele DA, Salmeron A, Ghosh S, Huang HR, Bryant BM, Lora JM. BET bromodomain inhibition suppresses TH17-mediated pathology. The Journal of experimental medicine. 2013;210:2181–2190. doi: 10.1084/jem.20130376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill JT, Neuwelt CM, Wallace DJ, Shanahan JC, Latinis KM, Oates JC, Utset TO, Gordon C, Isenberg DA, Hsieh HJ, et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: the randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis and rheumatism. 2010;62:222–233. doi: 10.1002/art.27233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nature reviews Drug discovery. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto N, Kishimoto T. Interleukin 6: from bench to bedside. Nature clinical practice Rheumatology. 2006;2:619–626. doi: 10.1038/ncprheum0338. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Laurence A, McInnes IB. Back to the future: oral targeted therapy for RA and other autoimmune diseases. Nature reviews Rheumatology. 2013;9:173–182. doi: 10.1038/nrrheum.2013.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim JJ, Gery I. From lymphodrek to interleukin 1 (IL-1) Immunology today. 1993;14:232–234. doi: 10.1016/0167-5699(93)90169-l. [DOI] [PubMed] [Google Scholar]
- Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J. Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3372–3377. doi: 10.1073/pnas.0408506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, Rowan WC, Sancho S, Walker LS, Vanhaesebroeck B, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177:6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- Pestka S. The interferons: 50 years after their discovery, there is much more to learn. The Journal of biological chemistry. 2007;282:20047–20051. doi: 10.1074/jbc.R700004200. [DOI] [PubMed] [Google Scholar]
- Pillai S, Mattoo H, Cariappa A. B cells and autoimmunity. Current opinion in immunology. 2011;23:721–731. doi: 10.1016/j.coi.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP. Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis and rheumatism. 2006;54:2377–2386. doi: 10.1002/art.22019. [DOI] [PubMed] [Google Scholar]
- Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nature reviews Immunology. 2007;7:191–201. doi: 10.1038/nri2036. [DOI] [PubMed] [Google Scholar]
- Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, Maciuca R, Zhang D, Garg JP, Brunetta P, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis and rheumatism. 2012;64:1215–1226. doi: 10.1002/art.34359. [DOI] [PubMed] [Google Scholar]
- Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nature reviews Immunology. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandborn WJ, Ghosh S, Panes J, Vranic I, Su C, Rousell S, Niezychowski W, Study AI. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. The New England journal of medicine. 2012;367:616–624. doi: 10.1056/NEJMoa1112168. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Blum HE, Thimme R. T-cell responses in hepatitis B and C virus infection: similarities and differences. Emerging Microbes and Infections. 2013;2:e15. doi: 10.1038/emi.2013.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stohl W, Hiepe F, Latinis KM, Thomas M, Scheinberg MA, Clarke A, Aranow C, Wellborne FR, Abud-Mendoza C, Hough DR, et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis and rheumatism. 2012;64:2328–2337. doi: 10.1002/art.34400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strober B, Buonanno M, Clark JD, Kawabata T, Tan H, Wolk R, Valdez H, Langley RG, Harness J, Menter A, et al. Effect of tofacitinib, a Janus kinase inhibitor, on haematological parameters during 12 weeks of psoriasis treatment. The British journal of dermatology. 2013;169:992–999. doi: 10.1111/bjd.12517. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Narazaki M, Kishimoto T. Therapeutic targeting of the interleukin-6 receptor. Annual review of pharmacology and toxicology. 2012;52:199–219. doi: 10.1146/annurev-pharmtox-010611-134715. [DOI] [PubMed] [Google Scholar]
- Thakker P, Leach MW, Kuang W, Benoit SE, Leonard JP, Marusic S. IL-23 is critical in the induction but not in the effector phase of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:2589–2598. doi: 10.4049/jimmunol.178.4.2589. [DOI] [PubMed] [Google Scholar]
- Uno JK, Rao KN, Matsuoka K, Sheikh SZ, Kobayashi T, Li F, Steinbach EC, Sepulveda AR, Vanhaesebroeck B, Sartor RB, et al. Altered macrophage function contributes to colitis in mice defective in the phosphoinositide-3 kinase subunit p110delta. Gastroenterology. 2010;139:1642–1653. 1653 e1641–1646. doi: 10.1053/j.gastro.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbani S, Amadei B, Tola D, Massari M, Schivazappa S, Missale G, Ferrari C. PD-1 expression in acute hepatitis C virus (HCV) infection is associated with HCV-specific CD8 exhaustion. Journal of virology. 2006;80:11398–11403. doi: 10.1128/JVI.01177-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annual review of immunology. 2013;31:317–343. doi: 10.1146/annurev-immunol-032712-095906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. The New England journal of medicine. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vojinovic J, Damjanov N. HDAC inhibition in rheumatoid arthritis and juvenile idiopathic arthritis. Mol Med. 2011;17:397–403. doi: 10.2119/molmed.2011.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann H, Hale G. CAMPATH: from concept to clinic. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2005;360:1707–1711. doi: 10.1098/rstb.2005.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. The New England journal of medicine. 2013;369:507–516. doi: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DG, Faia KL, DiNitto JP, Ali JA, White KF, Brophy EE, Pink MM, Proctor JL, Lussier J, Martin CM, et al. PI3K-delta and PI3K-gamma inhibition by IPI-145 abrogates immune responses and suppresses activity in autoimmune and inflammatory disease models. Chemistry & biology. 2013;20:1364–1374. doi: 10.1016/j.chembiol.2013.09.017. [DOI] [PubMed] [Google Scholar]
- Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. 2008;112:1557–1569. doi: 10.1182/blood-2008-05-078154. [DOI] [PMC free article] [PubMed] [Google Scholar]