

Abstract
Myhre syndrome is characterized by short stature, brachydactyly, facial features, pseudomuscular hypertrophy, joint limitation and hearing loss. We identified SMAD4 mutations as the cause of Myhre syndrome. SMAD4 mutations have also been identified in laryngotracheal stenosis, arthropathy, prognathism and short stature syndrome (LAPS). This study aimed to review the features of Myhre and LAPS patients to define the clinical spectrum of SMAD4 mutations. We included 17 females and 15 males ranging in age from 8 to 48 years. Thirty were diagnosed with Myhre syndrome and two with LAPS. SMAD4 coding sequence was analyzed by Sanger sequencing. Clinical and radiological features were collected from a questionnaire completed by the referring physicians. All patients displayed a typical facial gestalt, thickened skin, joint limitation and muscular pseudohypertrophy. Growth retardation was common (68.7%) and was variable in severity (from −5.5 to −2 SD), as was mild-to-moderate intellectual deficiency (87.5%) with additional behavioral problems in 56.2% of the patients. Significant health concerns like obesity, arterial hypertension, bronchopulmonary insufficiency, laryngotracheal stenosis, pericarditis and early death occurred in four. Twenty-nine patients had a de novo heterozygous SMAD4 mutation, including both patients with LAPS. In 27 cases mutation affected Ile500 and in two cases Arg496. The three patients without SMAD4 mutations had typical findings of Myhre syndrome. Myhre–LAPS syndrome is a clinically homogenous condition with life threatening complications in the course of the disease. Our identification of SMAD4 mutations in 29/32 cases confirms that SMAD4 is the major gene responsible for Myhre syndrome.
Keywords: Myhre syndrome, SMAD4, LAPS, long-term follow-up
Introduction
Myhre syndrome was first described in 1981 by Myhre et al.1 The canonical description reported two unrelated males with growth deficiency (adult height between −5.5 and −4.5 SD), mental retardation and unusual facies (short palpebral fissures, mid-face hypoplasia, narrow mouth and prognathism) associated with joint limitations, generalized muscular hypertrophy and skeletal findings (thickened calvarium, prominent broad mandible, broad ribs, shortened tubular bones and large pedicles of the vertebrae). Other features noted included hearing loss, hypermetropia, congenital heart defect, cryptorchidism and pilonidal dimple. A total of 19 cases have subsequently been reported (6 females and 13 males)2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12 highlighting variability in (i) the severity of the growth deficiency ranging from −6 to −2 SD and (ii) the degree of intellectual disability in which intelligence ranges from low to normal.
Laryngotracheal stenosis, Arthropathy, Prognathism and Short stature syndrome (LAPS)13 were described as a separate entity based on the presence of a recurrent laryngotracheal stenosis.14 However, the similarity between both the syndromes, particularly the short stature, progressive joint limitation and the facial anomalies, made Lindor15 postulate that they represent variable expressivity of the same entity. Recently, Le Goff et al16 identified missense SMAD4 heterozygous mutations, affecting the conserved Ile500 residue as a cause of Myhre syndrome. Shortly after, Caputo et al17 reported eight other Myhre syndrome cases with SMAD4 mutations altering the Ile500 residue in all their patients. The same mutation was also identified in two patients with LAPS syndrome18 supporting the concept that Myhre and LAPS syndromes are phenotypic variants of a single entity.
The aim of the present study was to review a large cohort of patients with Myhre and LAPS syndrome and to assess their clinical and molecular variability.
Methods
Patients
Thirty-two unrelated patients, followed up in several international genetics departments, were included in this study. Inclusion criteria were as follows: (i) the association of short stature, brachydactyly, joint limitation and muscular hypertrophy or thickened skin and a typical facial appearance; (ii) Myhre or LAPS syndrome diagnosis made by a clinical geneticist. Considering the rarity of these syndromes, a previously published description of a specific patient was not considered as an exclusion criteria.
Informed consent for participation, sample collection and publication of clinical photographs was obtained using protocols approved by the Necker Hospital ethics board.
SMAD4 Sequencing
Genomic DNA was obtained from peripheral blood leukocytes using standard procedures. The exons and exon–intron boundaries of the SMAD4 gene (GenBank NM_005359.5) were amplified using specific primers (available upon request). Amplification products were purified by ExoSapIT (Amersham, Buckinghamshire, UK) and directly sequenced using the Big Dye Terminator Cycle Sequencing Ready Reaction kit v.1.1 on an automatic sequencer (ABI3130xl; PE Applied Biosystems, Foster City, CA, USA).
Clinical data collection
After a review of the literature, a questionaire was designed to collect details of the clinical and radiological symptoms of patients diagnosed with Myhre and LAPS syndromes, including their differential diagnoses. This was submitted to each referring physician. Completed forms, photographs and radiographs (when authorized by the patients or their legal representatives) were collected.
Results
Patients
Thirty-two unrelated patients, 17 of whom had been previously described (11 patients in Le Goff et al;16 3 in McGowan et al;7 1 in Rosser et al19 1 in Burglen et al3 and 1 in Al Ageeli et al20) were recruited. The cohort consisted of 17 females and 15 males, ranging in age from 8 to 48 years. All were sporadic cases. Ethnic origin included Caucasian (n=24), North African (n=2), African (n=1), Pakistani (n=1), Ashkenaze Jew (n=2), mixed Caucasian-North African (n=1) and mixed Caucasian-AfroCaribbean (n=1). Among the 32 patients, 30 had been diagnosed as having Myhre syndrome and 2 as having LAPS.
The clinical and radiological details are summarized in Table 1. The facial appearance of some patients are displayed in Figure 1.
Table 1. Clinical and radiological details of MSS patients investigated in this study.
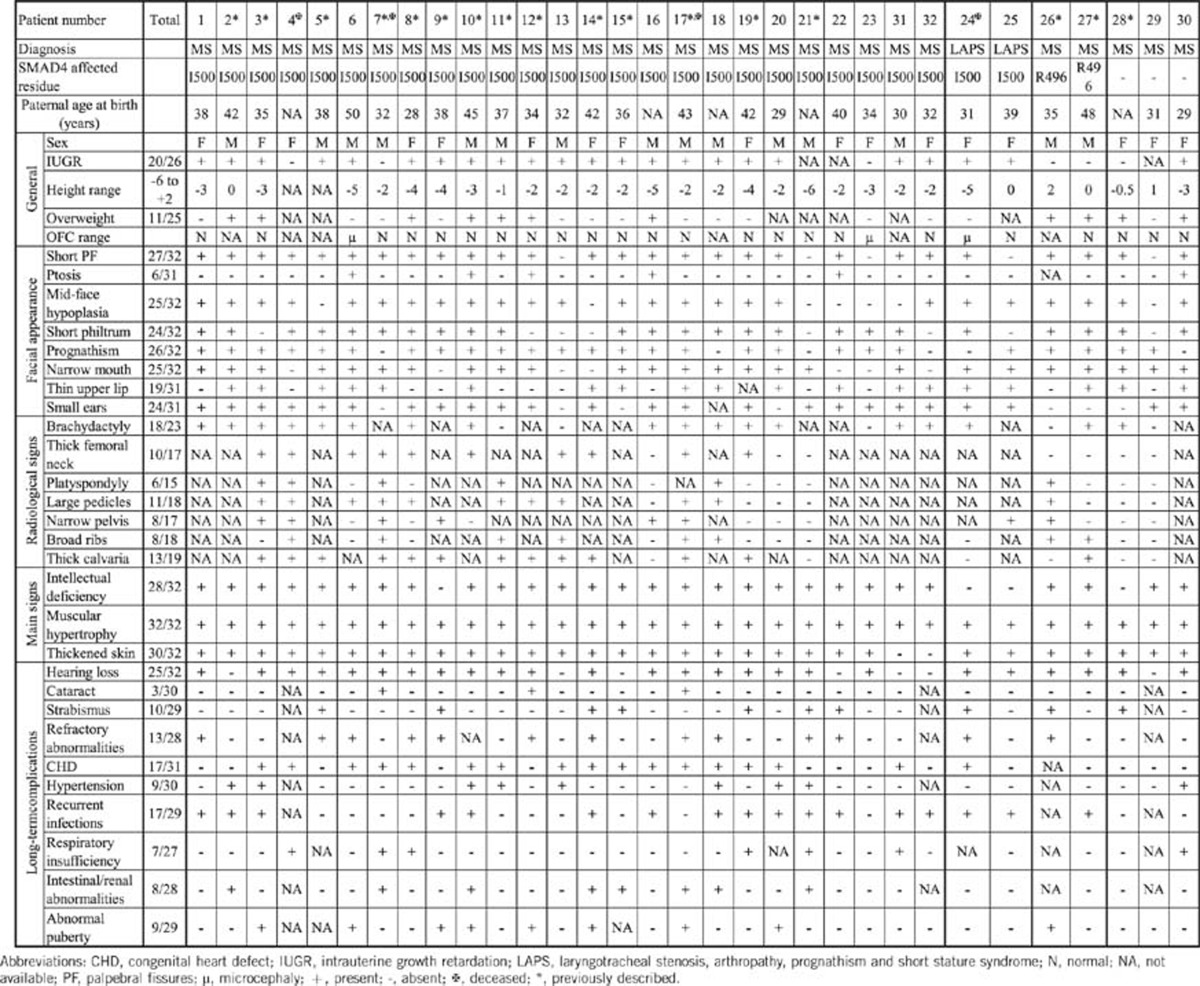
Figure 1.

Pictures of some patients with Myhre syndrome, with mutations altering the Ile500 (a–i: patients 3, 7, 9, 11, 13, 14, 16, 17 and 18), with the p.Arg496Cys mutation (j: patient 27) and without mutation (k: patient 28).
SMAD4 Sequencing
SMAD4 sequencing revealed a mutation in 29 patients (Table 1), including the two patients with LAPS syndrome. All mutations found were missense heterozygous changes, occurring de novo in all 29 cases. In 27 cases, mutations affected the Ile500 residue. Three different missense mutations were found to change this residue: c.1498A>G (p.(Ile500Val); (16/25)), c.1499T>C (p.(Ile500Thr); (10/25)) and c.1500A>G (p.(Ile500Met); (1/25)). The c.1486C>T (p.(Arg496Cys)) change was identified in two patients and this mutation was considered as probably damaging by the Polyphen software. The three remaining cases had no mutation of the whole SMAD4 coding sequence. Identified missense mutations were submitted to the ClinVar (NCBI) public database.
Clinical data analysis
The average paternal age at birth was 36.7 years. No relevant familial medical history was found in any case. Intrauterine growth retardation was observed in 23/29 (79.3%) cases (with data available). In three cases, additional features were detected antenatally including unilateral renal agenesis (1), common umbilical artery (1) and aortic coarctation (1). Postnatally, a growth retardation, ranging from −2SD to −5.5SD was present in 22/32 (68,7%) cases. All patients, except one (31/32, 96.8%), had small hands and feet. Five (15.6%) patients were treated by growth hormone, with an increase of the growth velocity in four cases. Obesity was observed in 11 (34.3%) patients, the youngest one being 6 years old. Abnormal onset of puberty was also frequently reported (seven precocious, three males and four females), and two delayed (two females) and early menopause was reported in 1/6 adult females. A total of 6/15 male patients had cryptorchidism, associated with micropenis in one case and abnormal onset of puberty in two (one advanced and one delayed). Ten patients were only aged 8–11 years at the time of the study and did not display abnormalities in the development of secondary sexual characteristics.
Microcephaly was noted in 3/25 (12%) (data not available for seven patients). Idiopathic intracranial hypertension occured in two females and their brain MRI scans were normal. Four patients had mild brain abnormalities including: mild dilatation of lateral ventricules (n=1), focal white matter hypersignal (n=2) and short corpus callosum (n=1). Intellectual disability was reported in 28/32 patients ranging from mild to moderate, except for one case who had severe disability. Impairment of speech development was present in 22/32 patients. Behavioral disorders were frequently reported (18/32) and consisted of a wide range of problems, including hyperactivity, stubbornness, aggressiveness, frustration, intolerance, poor communication skills, autistic features and polyphagia. Only six cases received normal school education, whereas the other 26 patients received special education.
As far as the facial dysmorphism is concerned, no single feature appeared to be present in all patients, but the previously well-described features were frequently seen (each feature being observed in 24–27/32 cases, Table 1). These consisted of short palpebral fissures, mid-face hypoplasia, a short philtrum, prognathism, a narrow mouth and small ears. Only the thin upper lip (19/31) and ptosis (6/31) were reported less often. Taken together, the features resulted in a typical and recognizable facial gestalt. In older patients, the prognathism and small palpebral fissures appeared more obvious, and there was thickening of the soft tissues of the face.
Besides the dysmorphic facial features, all patients had joint limitation, either of small joints (26/32) or of large joints (27/32), leading to tiptoe walking in 14 cases. All but two also had thickened skin. These features often became more obvious with time. Some patients displayed other dermatological features, such as keratosis pilaris (n=2), palmoplantar hyperkeratosis (n=1) and abnormal skin healing with keloid formation (n=2). Muscular pseudohypertrophy was found in all but one of the patients. Muscle biopsies were performed in five patients and showed either no or minor nonspecific abnormalities, except in one case, where there was moderate irregularity of myogenic fibers and decreased immunostaining for dystrophin. For this latter patient, the whole-coding sequence of DMD gene was analyzed by Sanger sequencing and was normal. Electromyography was also undertaken in three patients, revealing low amplitudes and turns/amplitude ratios in the myopathic range in one case. Nevertheless, all patients had a normal neurologic examination.
The spectrum of the other abnormalities was quite wide. Hearing loss was frequently reported, affecting 25/32 patients. The type of hearing loss was predominantly conductive or mixed (nine cases and four cases, respectively), with only four cases of pure sensorineural deafness. The deafness was unilateral or bilateral and of any degree of severity. At least in 4/15 adult patients, the deafness was described as slowly progressive. Recurrent respiratory or ear infections were also frequently observed (17/32 cases). This infection susceptibility was associated with a humoral immune deficiency in two cases, but systematic screens for specific immunoglobin levels were performed in very few cases. In two other cases, the recurrent infections were complicated by choanal atresia, and the frequency of infections improved after surgical treatment of the atresia. Velopharyngeal insufficiency was observed in eight cases, giving rise to nasal voice and velar dysfunction; cleft palate was present in only one patient. Laryngotracheal stenosis was reported in five cases, including the two cases diagnosed as LAPS syndrome. The stenosis was not congenital and was of variable degree, but required invasive surgery with tracheal resection in one case. Bronchopulmonary insufficiency was reported in seven cases, including obstructive disorders (chronic obstructive pulmonary disease (1) and obstructive syndrome associated with sleep apnea (1)), restrictive interstitial disorder (1), post infectious (1, pleural empyema) and pulmonary hypertension due to stenoses of pulmonary arteries either isolated (1) or associated with a restrictive disorder (1).
Congenital heart malformations were found in 17 patients, mainly patent ductus arteriosus (8), aortic coarctation (4), mild-to-moderate valvular aortic stenosis (4) and small membranous ventricular septal defect with mildly dilated aortic root (3.5 cm, patient 24). Arterial hypertension was also found in nine patients, mainly the older ones, but one patient developed hypertension at 10 years of age. Three patients presented with pericarditis: one case as an isolated episode, one with a chronic form that began at 5 years of age and required immunosuppressive medications, and one with a progressive, constrictive form, which led to death. Ophthalmological anomalies were frequently reported, mostly with refractory abnormalities (13/32 including hypermetropia (5) and astigmatism (6), and more rarely myopia (3)) and strabismus (10), but 3 patients presented with congenital cataracts (1 bilateral and 2 unilateral). Of note, two patients had retinal involvement with one case of retinitis pigmentosa and one case with maculopathy. Other reported problems included intestinal abnormalities (8/32), namely duodenal atresia (1), esophageal stenosis (1), pyloric stenosis (1), gastritis (1), superior mesenteric artery syndrome (1) and hepatomegaly (1), chronic constipation (2), and urinary tract abnormalities (renal unilateral agenesis (2) and vesico-ureteral reflux (2)).
In our cohort, four patients died at an early age (at 9, 19, 22 and 28 years), including one of the two patients diagnosed with LAPS. The cause of death was restrictive respiratory insufficiency (1), restrictive pericarditis (1), sudden death with cardiogenic shock (1) and sudden death due to massive mesenteric ischemia of unexplained cause (1).
Radiological data were available for 23/32 patients – no sign was pathognomic (Table 1). The most frequently reported features were brachy-metacarpia/tarsia (18/23), enlarged vertebral pedicules (11/18) and thick calvarium (13/19). No patient presented with all radiological features, suggesting that these should not be considered as mandatory for the diagnosis.
The two males with the Arginine 496 mutation had both the typical facial gestalt and developed joint limitations, thickened skin, muscular build, mild hearing loss and intellectual deficiency. No intrauterine, or postnatal growth retardation was observed, but they developed obesity early. One of them had a maculopathy with abnormal electroretinogram.
We specifically analyzed the three Myhre syndrome patients with no SMAD4 mutation. All of them had typical muscular, skin and joint abnormalities and facial appearance. Only one of them presented with postnatal growth retardation; 1/3 had normal intellectual development and attended normal school. Other features included early hypertension, velopharyngeal insufficiency and progressive chronic obstructive pulmonary disease in 1/3 patients; early menopause (1/3); hearing loss (2/3); myopia (1/3) and pigmentary retinal alterations (1/3).
Discussion
We report here a series of 32 patients with Myhre (30) or LAPS (2) syndrome with molecular data on the SMAD4 status. Twenty-nine patients were found to have SMAD4 mutations, including the two patients with LAPS, but three Myhre patients did not have any SMAD4 mutation.
Among all patients, we consistently observed the canonical signs of Myhre syndrome (progressive thickness of the skin, muscular pseudohypertrophy and joint limitations) and the facial features – short palpebral fissures, mid-face hypoplasia, a small philtrum, prognathism, a narrow mouth and small ears – supporting clinical homogeneity. Interestingly, the two LAPS patients also present with the same gestalt as all other patients. They also shared the same set of malformations and complications. The laryngotracheal stenosis, which led to the description of the LAPS entity, was also observed in three Myhre cases. These findings combined with the previous results of Lindor et al18 clearly support the view that Myhre and LAPS syndromes are phenotypic variants of the same syndrome.
Apart from those characteristic features, other associated signs were less frequently observed and were variable in their severity, including growth retardation, cognitive skills or behavioral disorders. A wide range of other anomalies were also observed mainly: (i) hearing loss (78%), (ii) congenital heart malformations (54%) and (iii) ophthalmological anomalies (46%). The presence of multiple congenital malformations correlates with the expression profile of SMAD4, which is ubiquitously expressed throughout the embryonic development and in most adult tissue and cell types.21 Notably conditional knockout of Smad4 in mouse chondrocytes leads to dwarfism, with histological anomalies of the growth plates,22 associated with a sensorineural hearing loss due to abnormalities of the cochlea in size and histology.23 However, skeletal features were not consistently observed in our cohort, although brachydactyly, thick calvarium and large pedicles were frequent. As skeletal surveys were not done in all patients, future clinical reports will require more consistent radiographic assessment.
Importantly, the initial evaluation and long-term follow-up of the patients from our cohort support the importance of a multidisciplinary and regular systematic clinical evaluation of Myhre or LAPS syndrome patients. In early childhood, recurrent infections of the upper and lower respiratory tracts are frequent (58%), and can be precipitated by choanal atresia or by immune deficiency. As the surgical treatment of choanal atresia improved the condition of several patients, it should be systematically sought and basic immunologic tests should be considered, specifically looking for humoral immunodeficiencies. Later in life, abnormalities of pubertal onset and possible early menopause support a requirement for a specific follow-up for females. Our study results also emphasize other medical complications, that can become life threatening including obesity, idiopathic intracranial hypertension, laryngotracheal stenosis, bronchopulmonary insufficiency (due to obstructive or restrictive disorders or to stenosis of pulmonary arteries), systemic arterial hypertension, pulmonary arterial hypertension and pericarditis. Because of early death in four patients, specific surveillance guidelines to detect these complications early should be a part of regular follow-up of these patients.
A total of 29/32 patients have SMAD4 mutations. SMAD4 encodes a protein of the SMAD (mothers-against-DPP homolog) family, which is considered as a tumor suppressor. It acts as a comediator within a complex composed of heterodimers of the receptor-regulated SMADs (SMAD1, SMAD2, SMAD3, SMAD5 and SMAD8). This complex translocates into the nucleus to induce or repress the expression of TGF-β and BMP target genes.24 Importantly, loss-of-function SMAD4 mutations are reported in juvenile polyposis syndrome,25 and somatic inactivation is found in some patients with pancreatic, gastrointestinal and skin carcinoma.26, 27 To date, the long-term follow-up of Myhre syndrome patients has not revealed any suceptibility to cancer development. The Ile500 change observed in Myhre syndrome is responsible for the increased SMAD4 stability by decreasing the SMAD4 ubiquitination.16 All Myhre mutations identified so far are located in the MH2 (MAD homolgy 2) domain and are directly involved in transcriptional activation. We have previously demonstrated in patient fibroblasts a loss of TGF-β-driven transcriptional control.16 Ongoing studies will further define the consequences on TGF-β and BMP signaling pathways.
The functional consequences of the Arg 496 mutation observed only in two cases have not been specifically studied; however, one cannot exclude similar effects on the ubiquitination process as the Arg 496 residue directly interacts with Ile500.17
All SMAD4 mutations identified occured de novo and no familial transmission has been reported so far. The high average paternal age at birth (>36 years), combined with the prevalence of Ile500 change due to mainly two nucleotides changes in SMAD4, may suggest a mechanism of protein-driven selfish selection in sperm. This phenomenon, combining hypermutability of specific nucleotides, expression of this mutant in spermatogonial stem cells and further positive selection resulting in localized clonal expansion, has been demonstrated for recurrent mutations of FGFR2 and FGFR3 in Apert syndrome and achondroplasia.28 Additional studies on mutation detection in sperm of fathers and on spatial distribution of the mutation in whole testis will be needed to test this hypothesis.28
Our findings of SMAD4 mutations in 29/32 patients may support an important genetic homogeneity of the Myhre–LAPS spectrum. Nevertheless, the three females without SMAD4 mutations also had the hallmark clinical signs of Myhre syndrome, making the clinical diagnosis of the syndrome highly probable for these patients despite the absence of mutations. As molecular investigations in our study consisted of Sanger sequencing of coding exons, it is therefore possible that these patients have a molecular abnormality of SMAD4 not detectable with this technique, such as small intragenic deletion or insertion. Supplementary tests, such as Multiple Ligation Probe Amplification, may be important to resolve the question of possible genetic heterogeneity of Myhre syndrome. Considering that this issue is the first hypothesis to explain the absence of mutations in these three patients, no gene other than SMAD4 was tested for them.
We conclude that Myhre–LAPS syndromes are a single entity consistently with a moderately wide clinical spectrum characterized by typical facial appearance, progressive thickening of the skin, muscular pseudohypertrophy, joint limitations and brachydactyly. The natural history of the disorder we observed should lead to specific management recommendations. The finding of the Ile500 change in SMAD4 in the majority of cases and of a significant increase in paternal age may suggest a mechanism of protein-driven selfish selection in sperm.
Acknowledgments
We thank Pr Brendan H Lee from the Department of Molecular and Human Genetics, Baylor College of Medicine; Howard Hughes from the Medical Institute, Houston, TX, USA; Dr Lewis B Holmes and Dr Patricia Blakely from the Massachusetts General Hospital; Meaghan Muir from the Brigham and Women's Hospital; and Dr Nicola Brunetti-Pierri from the Telethon Institute of Genetics and Medicine, Naples, Italy. We also would like to thank all the patients and their families.
The authors declare no conflict of interest.
References
- Myhre SA, Ruvalcaba RH, Graham CB. A new growth deficiency syndrome. Clin Genet. 1981;20:1–5. doi: 10.1111/j.1399-0004.1981.tb01798.x. [DOI] [PubMed] [Google Scholar]
- Becerra-Solano LE, Diaz-Rodriguez M, Nastasi-Catanese JA, et al. The fifth female patient with Myhre syndrome: further delineation. Clin Dysmorphol. 2008;17:113–117. doi: 10.1097/MCD.0b013e3282f52828. [DOI] [PubMed] [Google Scholar]
- Burglen L, Heron D, Moerman A, et al. Myhre syndrome: new reports, review, and differential diagnosis. J Med Genet. 2003;40:546–551. doi: 10.1136/jmg.40.7.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos NO, Garcia-Ortiz JE, Garcia-Cruz D, Feria-Velasco A, Sanchez-Corona J. Myhre syndrome: first female case. Clin Dysmorphol. 2003;12:119–121. doi: 10.1097/00019605-200304000-00009. [DOI] [PubMed] [Google Scholar]
- Garcia-Cruz D, Figuera LE, Feria-Velazco A, et al. The Myhre syndrome: report of two cases. Clin Genet. 1993;44:203–207. doi: 10.1111/j.1399-0004.1993.tb03880.x. [DOI] [PubMed] [Google Scholar]
- Lopez-Cardona MG, Garcia-Cruz D, Garcia-Ortiz JE, et al. Second female case of Myhre syndrome. Clin Dysmorphol. 2004;13:91–94. [PubMed] [Google Scholar]
- McGowan R, Gulati R, McHenry P, et al. Clinical features and respiratory complications in Myhre syndrome. Eur J Med Genet. 2011;54:e553–e559. doi: 10.1016/j.ejmg.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Rulli I, Ferrero GB, Belligni E, Delmonaco AG, Defilippi C, Silengo M. Myhre's syndrome in a girl with normal intelligence. Am J Med Genet A. 2005;134A:100–102. doi: 10.1002/ajmg.a.30444. [DOI] [PubMed] [Google Scholar]
- Soljak MA, Aftimos S, Gluckman PD. A new syndrome of short stature, joint limitation and muscle hypertrophy. Clin Genet. 1983;23:441–446. doi: 10.1111/j.1399-0004.1983.tb01979.x. [DOI] [PubMed] [Google Scholar]
- Titomanlio L, Marzano MG, Rossi E, et al. Case of Myhre syndrome with autism and peculiar skin histological findings. Am J Med Genet. 2001;103:163–165. doi: 10.1002/ajmg.1517. [DOI] [PubMed] [Google Scholar]
- van Steensel MA, Vreeburg M, Steijlen PM, de Die-Smulders C. Myhre syndrome in a female with previously undescribed symptoms: further delineation of the phenotype. Am J Med Genet A. 2005;139A:127–130. doi: 10.1002/ajmg.a.30988. [DOI] [PubMed] [Google Scholar]
- Whiteford ML, Doig WB, Raine PA, Hollman AS, Tolmie JL. A new case of Myhre syndrome. Clin Dysmorphol. 2001;10:135–140. doi: 10.1097/00019605-200104000-00011. [DOI] [PubMed] [Google Scholar]
- Lindor NM, Kasperbauer JL, Hoffman AD, Parisi JE, Wang H, Warman M. Confirmation of existence of a new syndrome: LAPS syndrome. Am J Med Genet. 2002;109:93–99. doi: 10.1002/ajmg.10316. [DOI] [PubMed] [Google Scholar]
- Hopkin RJ, Cotton R, Langer LO, Saal HM. Progressive laryngotracheal stenosis with short stature and arthropathy. Am J Med Genet. 1998;80:241–246. doi: 10.1002/(sici)1096-8628(19981116)80:3<241::aid-ajmg12>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Lindor NM. LAPS syndrome and Myhre syndrome: two disorders or one. Am J Med Genet A. 2009;149A:798–799. doi: 10.1002/ajmg.a.32719. [DOI] [PubMed] [Google Scholar]
- Le Goff C, Mahaut C, Abhyankar A, et al. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2012;44:85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- Caputo V, Cianetti L, Niceta M, et al. A restricted spectrum of mutations in the SMAD4 tumor-suppressor gene underlies Myhre syndrome. Am J Hum Genet. 2012;90:161–169. doi: 10.1016/j.ajhg.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindor NM, Gunawardena SR, Thibodeau SN. Mutations of SMAD4 account for both LAPS and Myhre syndromes. Am J Med Genet A. 2012;158A:1520–1521. doi: 10.1002/ajmg.a.35374. [DOI] [PubMed] [Google Scholar]
- Rosser EM, Wilkinson AR, Hurst JA, McGaughran JM, Donnai D. Geleophysic dysplasia: a report of three affected boys – prenatal ultrasound does not detect recurrence. Am J Med Genet. 1995;58:217–221. doi: 10.1002/ajmg.1320580304. [DOI] [PubMed] [Google Scholar]
- Al Ageeli E, Mignot C, Afenjar A, et al. Retinal involvement in two unrelated patients with Myhre syndrome. Eur J Med Genet. 2012;55:541–547. doi: 10.1016/j.ejmg.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Attisano L, Lee-Hoeflich ST.The Smads Genome Biol 20012REVIEWS3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Tan X, Li W, et al. Smad4 is required for the normal organization of the cartilage growth plate. Dev Biol. 2005;284:311–322. doi: 10.1016/j.ydbio.2005.05.036. [DOI] [PubMed] [Google Scholar]
- Yang SM, Hou ZH, Yang G, et al. Chondrocyte-specific Smad4 gene conditional knockout results in hearing loss and inner ear malformation in mice. Dev Dyn. 2009;238:1897–1908. doi: 10.1002/dvdy.22014. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Howe JR, Roth S, Ringold JC, et al. Mutations in the SMAD4/DPC4 gene in juvenile polyposis. Science. 1998;280:1086–1088. doi: 10.1126/science.280.5366.1086. [DOI] [PubMed] [Google Scholar]
- Hahn SA, Schutte M, Hoque AT, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271:350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- Schutte M, Hruban RH, Hedrick L, et al. DPC4 gene in various tumor types. Cancer Res. 1996;56:2527–2530. [PubMed] [Google Scholar]
- Goriely A, Wilkie AO. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am J Hum Genet. 2012;90:175–200. doi: 10.1016/j.ajhg.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]