

Abstract
Background
Sunitinib treatment results in a compensatory increase in plasma VEGF levels. Acute withdrawal of sunitinib results in a proliferative withdrawal flare, primarily due to elevated VEGF levels. Concurrent sunitinib plus bevacizumab is poorly tolerated with high (37 %) incidence of microangiopathic hemolytic anemia (MAHA). We evaluated a sequential design administering bevacizumab during the sunitinib treatment break to suppress the sunitinib withdrawal flare.
Methods
Patients with no prior VEGF treatment were enrolled in this study. All patients had target lesions amenable to serial FLT PET/CT imaging. Sunitinib 37.5 mg was given on days 1–28 every 6 weeks with bevacizumab 5 mg/kg on day 29. If safe and tolerable, sunitinib increased to 50 mg. FLT PET/CT scans would be obtained at baseline (D1), week 4, and week 6 to evaluate pharmacodynamics of the sequential combination. Sunitinib pharmacokinetics and total, free, and bound VEGF levels were obtained on each cycle at D1, pre-bevacizumab (D29), 4 h post-bevacizumab (D29H4), and day 42 (D42).
Results
Six patients enrolled in the safety cohort of sunitinib 37.5 mg plus bevacizumab (see Table). One patient experienced grade 1 MAHA, and after discussion with the Cancer Therapy Evaluation Program (CTEP), the trial was closed to further accrual. No imaging scans were obtained due to early closure.
Total and free VEGF levels during cycle 1
| Cycle 1 | Total VEGF (pg/mL) Mean ± SD |
Free VEGF (pg/mL) Mean ± SD |
|---|---|---|
| D1 | 80 ± 70 | 51 ± 47 |
| D29 | 150 ± 62 | 103 ± 35 |
| D29H4 | 10 ± 12 | 2 ± 5 |
| D42 | 177 ± 34 | 97 ± 18 |
Conclusions
Subclinical MAHA was seen despite using sequential sunitinib with low-dose bevacizumab, and this combination was not feasible for further development. As predicted, VEGF levels increased during sunitinib exposure followed by a rapid decline after bevacizumab. Due to the long half-life of bevacizumab, we expected VEGF ligand suppression through D42, but instead observed a complete rebound in total/free VEGF levels by D42. The increase in VEGF at D42 was unexpected based on sunitinib alone and contrary to the hypothesis that we would block VEGF flare with low-dose bevacizumab. VEGF ligand production may increase as a result of bevacizumab, implying a robust host compensatory mechanism to VEGF signaling pathway inhibition. A greater understanding of the compensatory mechanism would aid future sequencing strategies of new agents.
Keywords: Pharmacodynamic, Phase I clinical trial, Sequential combination, Sunitinib, Bevacizumab
Background and introduction
Renal cell carcinoma affects approximately 64,770 Americans, in which multiple agents have recently been approved for treatment with an improvement in overall survival of 26.4 months [4, 5]. Sunitinib is an oral, multitargeted, small-molecule inhibitor of the receptor tyrosine kinases involved in tumor proliferation and angiogenesis. Sunitinib and other tyrosine kinase inhibitors have been approved for the treatment of advanced metastatic renal cell carcinoma and also other solid malignancies [5–7]. Although sunitinib demonstrated a significant improvement over interferon alfa, the median progression-free survival is still <1 year (11 months) [5]. Despite significant progress in anticancer therapies for both renal cell carcinomas and other solid malignancies, most, if not all, patients eventually develop disease progression and die from their disease. Thus, another strategy is necessary to improve upon the current treatment regimen.
VEGF is one of the most potent and specific proangiogenic factors and has been identified as a crucial regulator of both normal and pathological angiogenesis. In vitro models demonstrate that neutralization of VEGF has inhibited the VEGF-induced proliferation of human endothelial cell and results in decreased microvessel density in vivo [8]. Because sunitinib and bevacizumab inhibit different components of the VEGF signaling pathway, it was believed that the combination of these two agents could potentially provide a more effective inhibition of this pathway and thus improve antitumor activity in a superior fashion.
At our institution, we evaluated the use of FLT PET/CT imaging in patients treated with sunitinib malate, in order to further characterize proliferative changes in tumors during the sunitinib exposure and withdrawal [9]. Sixteen patients were treated in two cohorts: eight patients on a 4/2 schedule (4 weeks on, 2 weeks off) and eight patients on 2/1 schedule who completed all three planned FLT PET/CT scans and were evaluable for pharmacodynamic imaging evaluation. During sunitinib treatment, the plasma VEGF ligand levels increased during sunitinib exposure and returned toward baseline during the treatment withdrawal. During sunitinib withdrawal, the FLT PET standardized uptake value (SUVmean) increased to +15 % in patients on the 4/2 schedule and to +19 % on the 2/1 schedule. In addition, dynamic FLT PET/CT imaging demonstrated similar changes in proliferative rate as the SUV, which implied that FLT PET/CT imaging can be utilized to represent proliferative activity (rather than due to an inflammatory or vascular effect).
There was also a trend that with chronic sunitinib exposure, VEGF levels gradually increase. We observed that the tumor flare was due to the higher VEGF ligand levels and also associated with a shorter progression-free survival. We hypothesize that a compensatory increase in VEGF ligand levels may be a key mechanism for antiangiogenic escape and thus an explanation for treatment failure. By combining bevacizumab with sunitinib in a sequential fashion, we wanted to target further suppression of the VEGF ligand with the administration of bevacizumab during the off-treatment period of sunitinib. Thus, we hypothesize that the sequential administration of these two agents would result in continuous VEGF ligand suppression and hopefully would improve progression-free survival.
Sunitinib has been combined with bevacizumab in two phase I clinical trials for either renal cell carcinoma or other advanced solid tumors in order to maximize the inhibition of VEGF and other proangiogenic factors [10]. In the phase I study evaluating patients with metastatic renal cell carcinoma, sunitinib was given on days 1–28 and concurrent bevacizumab at a fixed dose of 10 mg/kg given every 2 weeks, repeated in 6-week cycles. Although the maximally tolerated dose was found to be sunitinib 50 mg plus bevacizumab 10 mg/kg, chronic treatment resulted in significant hypertension, thrombocytopenia, reversible posterior leukoencephalopathy, and microangiopathic hemolytic anemia [10]. The maximum tolerated dose (MTD) was determined to be sunitinib 50 mg and bevacizumab 10 mg/kg, but the dose escalation trial resulted in significant toxicities. Although there was activity with a 4 % complete response and 48 % partial response, the toxicities were significant enough to discontinue further development of the concurrent combination. The use of concurrent treatment with sunitinib and bevacizumab resulted in these adverse events secondary to overlapping toxicities.
The second phase I clinical trial enrolled patients with advanced solid tumors using sunitinib at 25 mg daily for 4 weeks followed by 2 weeks off and bevacizumab given 5 mg/kg on days 1, 15, and 29 of a 42-day cycle [11]. Approximately 18 % of enrolled patients (n = 7 of the 38 patients) had a confirmed partial response with this combination, but 42 % of patients discontinued treatment due to adverse events. Despite a 50 % lower dose of each agent based on the safety signal from the previous clinical trial, there were still notable toxicities of fatigue, thrombocytopenia, and GI symptoms. Fortunately, no thrombotic microangiopathy was noted in this study, but significant toxicities were again noted at the MTD of sunitinib 50 mg in combination with bevacizumab 10 mg/kg.
In order to mitigate these toxicities but still try to capitalize on the anti-VEGF inhibition, we attempted to treat patients with advanced metastatic renal cell carcinoma and other solid malignancies with sunitinib and bevacizumab given in a sequential fashion. Since sunitinib is given on a 4/2 schedule, we attempted to administer bevacizumab on day 29 after completion of the sunitinib therapy. Thus, we hypothesize that the sequential combination of these two agents would increase efficacy while minimizing any overlapping toxicities. We specifically chose a low dose of bevacizumab (5 mg/kg) as it was calculated that this dose should adequately bind free plasma VEGF ligand. We also planned to observe any changes in tumor proliferation and changes in vasculature using novel FLT PET/CT imaging to measure these vascular parameters during active treatment.
In summary, it has been shown that plasma VEGF levels increase with VEGFR TKI therapy and perhaps the upregulation of VEGF eventually results in compensatory sunitinib failure [9, 12]. This may explain why subsequent treatment (or retreatment) with VEGFR TKIs may result in transient clinical benefit [13, 14]. We have shown that the acute VEGFR TKI withdrawal flare during the scheduled treatment break is likely due to upregulated VEGF ligand, which results in a brief period of rapid tumor growth, and that the degree of tumor flare during acute sunitinib withdrawal is inversely related to duration of clinical benefit. Thus, we hypothesize that the degree of the sunitinib withdrawal flare represents the patient’s ability to compensate for sunitinib-induced hypoxia by upregulating proangiogenic factors, in particular VEGF, and that patients with a robust compensatory mechanism will experience early treatment failure. This study was designed to test the hypothesis by combining a VEGF-ligand-binding agent (bevacizumab) with sunitinib utilizing a novel sequential strategy to minimize overlapping toxicity and still achieve flare suppression. With additional blockage of the VEGF flare, we hypothesize that this sequential treatment can improve the progression-free survival.
Materials and methods
Patients
Patients with histologically confirmed renal cell or other solid malignancies were eligible for this clinical trial. For patients with renal cell carcinoma, a component of clear cell histology was required. All patients were required to have measureable disease and also PET-measureable disease. Prior treatment with any other VEGF signaling pathway inhibitors was not allowed. Other eligibility criteria included adequate bone marrow (ANC ≥ 1,500/mcL, hemoglobin ≥ 9 g/dL, platelet count ≥ 100,000/mcL), renal (creatinine ≤ 1.5 × ULN or measured creatinine clearance ≥50 mL/min for patients with creatinine levels ≥1.5 × ULN), and hepatic function (AST/ALT ≤ 2.5 × ULN, unless subjects had liver metastases, in which case ≤5× ULN). Patients who required anticoagulation with therapeutic doses of coumarin-derivative anticoagulants or heparin derivatives were not allowed on the study. Patients were excluded for inadequately controlled blood pressure (≥140/90), significant proteinuria, or any history of brain metastases. Patients with a history of an acute cardiac event or those who underwent intervention for coronary disease or stroke in the prior 12 months were not enrolled.
Study design
This was a multicenter, investigator-initiated, phase I clinical trial that implemented a dose escalation portion of increasing sunitinib doses with two cohorts (sunitinib 37.5 mg and 50 mg) in combination with bevacizumab 5 mg/kg. Each cohort consisted of up to 12 patients each. Within each cohort, six patients with renal cell cancer were enrolled in the safety cohort and six additional patients with renal or other solid malignancies were enrolled in the imaging cohort. Once the recommended phase II dose was determined and identified, an additional 15 evaluable patients with renal cell cancer were to be enrolled for further safety evaluation (Fig. 1). We specifically targeted the renal cell cancer patient population for the recommended phase II dose cohort in anticipation of obtaining additional safety data for the use of this novel sequential strategy in the treatment for kidney cancer.
Fig. 1.
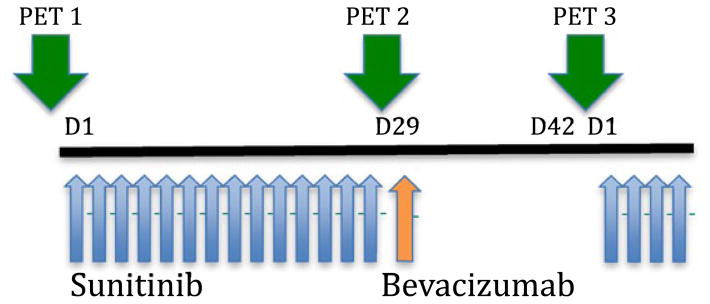
Schema for dosing and imaging. Each cycle consisted of sunitinib given on days 1–28, and bevacizumab given on day 29. PET scans were performed only during cycle 1 at baseline, prior to the bevacizumab dose, and on day 42 (prior to sunitinib dose)
Based on the previous clinical trials evaluating sunitinib plus bevacizumab in both renal cancers and advanced solid malignancies, there were significant toxicities when these two agents were given concurrently, and thus, the combination was not tolerable [1, 2, 10]. This clinical study was designed with a novel sequential strategy in order to reduce toxicities from overlapping side effects of each individual agent.
Based on a previously conducted trial evaluating FLT PET/CT imaging in patients with renal cell and other solid malignancies treated with sunitinib alone, we demonstrated that the temporary discontinuation of sunitinib did result in a proliferative flare ranging from +40 to 180 % (mean 85 %) [3]. In this prior study evaluating FLT PET/CT imaging in patients with solid malignancies, the tumor flare phenomenon was seen and demonstrated across multiple various tumor types. The degree of flare appeared to correlate with early treatment failure (e.g., higher flare resulted in shorter progression-free survival). Thus, we designed this clinical trial using similar patient populations to evaluate the changes in the flare with the addition of a VEGF-ligand-binding agent such as bevacizumab.
Treatment and dose escalation plan
Treatment was administered in 42-day cycles, during which patients were given sunitinib daily for 4 consecutive weeks (days 1–28) followed by 2 weeks of rest (drug holiday) with no sunitinib. Bevacizumab was given by intravenous infusion on day 29. Each cycle was defined as a six-week period. Two cohorts were planned during this clinical trial. Patients in the first cohort were given sunitinib 37.5 mg and bevacizumab 5 mg/kg. At least six patients with renal cell cancer were enrolled in cohort 1 for safety and toxicity assessment. If no significant toxicity was observed after three cycles of observation, then cohort 2 would accrue patients. Patients in the safety cohort with tumor-targeted lesions amenable to FLT PET/CT imaging were to be concurrently enrolled onto the imaging cohort for PET/CT analysis. If the dose was deemed to be safe and tolerable, then patients would be enrolled in cohort 2 of sunitinib 50 mg and bevacizumab 5 mg/kg.
Patients were evaluated for adverse events on the basis of National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0. Escalation to cohort 2 was based on safety evaluation of cohort 1 after three cycles of observation. In contrast to the other combination trials evaluating sunitinib and bevacizumab, we chose to observe patients for a longer period of time in order to assess for dose-limiting toxicities (DLTs). Due to the observation of microangiopathic hemolytic anemia (MAHA) occurring in a previous phase I clinical study evaluating the combination in advanced renal cell carcinoma, there was concern about duplicating similar toxicities [10]. Because MAHA was noted to occur during cycle 3, we proposed to extend the observation period for safety and toxicity for three cycles.
Dose-limiting toxicity (DLT) is defined as probably or definitely related to sunitinib or bevacizumab. Hematologic DLTs included grade ≥3 neutropenia for ≥7 days or neutropenic fever, and grade ≥3 thrombocytopenia. Non-hematologic DLTs included symptomatic hypertension with BP > 150/100, grade 4 hypertension, or uncontrolled hypertension (SBP > 160/100) despite maximal antihypertensive medications, or any grade thrombotic microangiopathy. During cycle 1 of therapy, grade 2 subjective intolerable or grade 3 gastrointestinal toxicity despite maximal use of supportive measures or any other grade ≥3 toxicities (except alopecia) related to treatment with sunitinib or bevacizumab was also considered DLT.
Antitumor activity assessment
Response was assessed by Response Evaluation Criteria in Solid Tumors version 1.1 every even cycle of therapy [15]. In addition to a baseline scan, confirmatory scans were also obtained 4–12 weeks following initial documentation of objective response.
Sample collection
Plasma for VEGF and bevacizumab PK was obtained each cycle on day 1 prior to dosing and on day 29 prior to bevacizumab administration and 4 h after the dose. At each time point specified, two 4-mL purple top (EDTA) vacutainers were collected for VEGF analysis and two 4-mL purple top (EDTA) vacutainers were collected for bevacizumab analysis. Plasma was separated by centrifugation at approximately 1200 g × 15 min, aliquoted into two cryovials, and stored at −70 °C until analysis.
Detection of free and bound VEGF
Plasma was immunodepleted using Protein G-Sepharose 4 Fast Flow beads (GE Healthcare, Piscataway, NJ) as has been described previously [16]. The beads were prepared by washing twice with phosphate-buffered saline (PBS), and a slurry (50 % v/v beads in PBS) was made. Two hundred microliters of plasma was incubated with 100 μL of the Protein G-Sepharose bead slurry for 4 h at 4 °C. After centrifugation to pellet the beads, 200 μL of the plasma supernatant was removed and another 100 μL of the Protein-G Sepharose slurry was added. Samples were incubated overnight at 4 °C. Two hundred microliters of plasma supernatant was removed from each tube after centrifugation and analyzed for VEGF using the human VEGF Quantikine ELISA kit (R&D Systems, Minneapolis MN), following the instructions of the manufacturer. In parallel, plasma was subjected to the same treatment, substituting 50 μL of PBS for the Protein-G Sepharose slurry, for analysis of VEGF levels without immunodepletion. Each sample was analyzed in duplicate using the VEGF ELISA described above.
Detection of free bevacizumab
An ELISA was developed to detect free bevacizumab (bound to zero or one molecule of VEGF), using the protocol described by Ternant et al. [17] with some modifications. High-binding 96-well ELISA plates (Grainger Bio-One, through VWR, Radnor, PA) were coated with 100 μL of 0.15 mg/ml VEGF165 peptide (R&D Systems, Minneapolis MN) in a 50 mM carbonate/bicarbonate buffer, pH 9.6, overnight at 4 °C. Plates were washed four times with 200 μL PBS with 0.05 % Tween 20. Blocking was performed for 2 h at room temperature with 200 μL PBS with 1 % BSA, after which the plates were washed as described. One hundred microliters of PBS with 1 % BSA was added to each well, followed by addition of 100 μL of samples, standards, and QC samples, all of which were first diluted 1:500 in PBS with 1 % BSA. Plates were incubated for 1 h at 37 °C and then washed as described. One hundred microliters of a peroxidase-conjugated rabbit anti-human IgG Fc fragment-specific secondary (Jackson ImmunoResearch, West Grove, PA) diluted 1:100,000 in PBS with 1 % BSA was then added, and plates were incubated for 1 h at room temperature. After washing, the HRP was developed by adding 100 μL TMB (Sigma-Aldrich, St. Louis, MO) to each well and incubating in the dark for 8 min. One hundred microliters of 1N sulfuric acid was added to stop the reaction, and plates were read at 450, with wavelength correction at 540. A blank reading was also subtracted. All samples, standards, and QCs were assayed in duplicate, and results were rejected if CV values were greater than 20 %.
Assay validation
For the bevacizumab assay, intraday and interday variability testing was performed by running the standard curve and three QC samples (high, medium, and low concentrations) at least three times on the same and different days. The standard curve was created using concentrations of 250, 100, 50, 20, 10, 3, 1, and 0 mg/L, and QC samples used were as follows: high, 75 mg/L; medium, 30 mg/L; and low, 5 mg/L. Accuracy for QC samples ranged from 94 to 122 % for the low sample (average 114 %), 90–105 % for the medium sample (average 94 %), and 83–99 % for the high sample (average 91 %), with average CVs under 10 %. For the commercial VEGF assay, interday variability testing was performed by running the standard curve at least three times on different days. The standard curve was generated following the instructions of the manufacturer. The average percentage recovery for all points on the standard curve was 102 %, with the CV averaging less than 10 %.
Statistical analysis
The primary objectives were to assess both the safety and objective response rate of sequential sunitinib with bevacizumab or sunitinib malate alone at the pharmacodynamically active dose suggested by the phase I study in the clear cell kidney cancer patient group. The secondary objectives were to assess the pharmacodynamic change in SUVpeak and tumor perfusion using FLT PET/CT scans at baseline, during sunitinib exposure, and during bevacizumab exposure (during sunitinib withdrawal period), and to characterize changes in the total and free VEGF levels in patients treated with sequential sunitinib with bevacizumab or sunitinib malate alone. Due to early termination of the trial, only six patients were enrolled on the trial and no FLT PET/CT imaging results were obtained. The plasma VEGF levels were summarized using descriptive statistics such as the mean and standard deviation (SD) for each assessment time point.
Results
This study enrolled six patients with advanced renal cell carcinoma in the first safety cohort of sunitinib 37.5 mg with bevacizumab 5 mg/kg from November 1, 2010, until November 29, 2011 (see Table 1). A total of 20 cycles (median 2 cycles per patient) were administered to all six patients who were evaluable for toxicity and response. At best response, two patients had a partial response, three patients exhibited stable disease, and one patient had disease progression with sequential treatment of sunitinib plus bevacizumab.
Table 1.
Patient demographics and clinical characteristics
| Characteristic | No. | % |
|---|---|---|
| Age (years) | ||
| Median | 65 | |
| Range | 62–67 | |
| Sex | ||
| Male | 4 | 67 |
| Female | 2 | 33 |
| Histology | ||
| Clear cell | 6 | 100 |
| Prior therapy | ||
| Nephrectomy | 5 | 83 |
| Radiation | 0 | 0 |
| Cytokine treatment | 1 | 17 |
| No. of metastatic sites | ||
| 1 | 0 | 0 |
| 2 | 3 | 50 |
| ≥3 | 3 | 50 |
| Select sites of metastatic sites | ||
| Lungs | 2 | |
| Pancreas | 2 | |
| Lymph node | 3 | |
| Bone | 2 | |
| Adrenal gland | 2 | |
| Liver | 1 | |
| Best response | ||
| Partial response | 2 | 33 |
| Stable disease | 3 | 50 |
| Disease progression | 1 | 17 |
Safety and tolerability
The most frequently reported adverse events for all patients included fatigue, mucositis, diarrhea, hypertension, hand–foot syndrome, diarrhea, constipation, and nausea/vomiting. All adverse events were grade ≤3 (see Table 2).
Table 2.
Adverse events
| Toxicity | Grade 1 | Grade 2 | Grade 3 |
|---|---|---|---|
| Hypertension | 2 | 1 | |
| Leukopenia | 2 | 1 | |
| Diarrhea | 3 | 1 | 1 |
| Anemia | 1 | 1 | 1 |
| Fatigue | 2 | 3 | |
| Anorexia | 2 | ||
| Mucositis | 3 | 1 | |
| Hand Foot Syndrome | 3 | 1 | |
| Proteinuria | 1 | 1 | |
| Dry mouth | 1 | 1 | |
| Skin changes | 4 | ||
| Pain | 4 | ||
| Nausea/Vomiting | 3 | ||
| Dysgeusia | 3 | ||
| Epistaxis | 3 | ||
| Constipation | 2 | ||
| MAHA | 1 | ||
| Myalgia | 1 | ||
| Edema | 1 |
One patient experienced asymptomatic grade 1 MAHA, which occurred during cycle 2 of treatment with sunitinib 37.5 mg in combination with bevacizumab 5 mg/kg. She was noted to have thrombocytopenia with evidence of anisocytosis, poikilocytosis, and schistocytes. Her haptoglobin was noted to be 15 mg/dL (decreased from 179 mg/dL). The thrombocytopenia recovered after 8 days with the resolution of haptoglobin, schistocytes, hemoglobin, and thrombocytopenia back to normal limits.
No deaths occurred during treatment on this clinical trial. Based on prior clinical studies of sunitinib plus bevacizumab that demonstrated MAHA, the design of this clinical trial utilized conservative monitoring of any hematologic toxicities. The development of MAHA on this trial warranted a discussion with the Cancer Therapy Evaluation Program (CTEP). Based on the high percentage of MAHA (25 %) in other studies using these agents, and the development of MAHA on our study, the consensus opinion was that the sequential strategy of using sunitinib plus bevacizumab was not feasible or tolerable [10]. Therefore, the trial was closed to further accrual. No imaging scans were obtained due to early closure.
Response
Of the six patients who were treated on this clinical study, two patients had a partial response, and three patients had stable disease at best response. The most common reason for discontinuation of treatment was disease progression (n = 3), MD discretion (n = 1), patient wishes (n = 1), and toxicity (n = 1). One patient is still being treated on this clinical study with sunitinib alone, as the protocol was amended to allow for standard of treatment with sunitinib.
VEGF
Our previous work demonstrated that the tumor flare seen with sunitinib is mediated by increased plasma VEGF levels and that patients with a larger flare were less likely to benefit from sunitinib. This study sought to ameliorate the tumor flare by administering low-dose bevacizumab after sunitinib. We evaluated the levels of free and bound VEGF at baseline, on day 29 after 28 days of sunitinib administration and prior to bevacizumab, and on day 29 4 h after bevacizumab administration to evaluate whether low-dose bevacizumab would be adequate to block the VEGF ligand.
As expected, there were significant increases in free VEGF levels with subsequent cycles of therapy, suggesting that sunitinib administration increases both free and total VEGF (see Table 3). We also noted a significant decline in VEGF levels 4 h after the dosing of bevacizumab. In cycle 1, bevacizumab administration was able to decrease both free and total VEGF from 103 and 150 pg/mL on day 29 prior to bevacizumab administration to 3 and 10 pg/mL 4 h after administration, respectively. We also observed that the initial decline in free and bound VEGF 4 h after bevacizumab administration was greatest during the first cycle, with significant increases in post-bevacizumab free and bound VEGF in cycles 2 and 3 compared with cycle 1. Additionally, by day 42, both free VEGF and total VEGF were significantly higher than at baseline and similar to pre-bevacizumab levels on day 29.
Table 3.
VEGF levels during multiple treatment cycles
| Baseline | D29 pre-Bev | D29 4 h post-Bev | Day 42 | |
|---|---|---|---|---|
| Free VEGF levels (pg/mL) | ||||
| Cycle 1 | 51.2 ± 46.6 | 103.1 ± 35.0 | 3.0 ± 5.2 | 97.6 ± 18.0 |
| Cycle 2 | 79.0 ± 27.0 | 93.9 ± 67.5 | 39.4 ± 16.5 | |
| Cycle 3 | 103.6 ± 26.6 | 172.5 ± 38.5 | 64.4 ± 0.0 | |
| Total VEGF levels (pg/mL) | ||||
| Cycle 1 | 80.3 ± 70.0 | 150.0 ± 62.2 | 10.0 ± 12.1 | 177.5 ± 33.5 |
| Cycle 2 | 145.4 ± 11.1 | 221.8 ± 141.4 | 68.2 ± 36.7 | |
| Cycle 3 | 195.0 ± 61.5 | 386.7 ± 80.2 | 137.3 ± 0.0 | |
Discussion
The use of antiangiogenic agents has been an area of rapid development, with new agents being developed and approved by the FDA for the treatment of multiple solid malignancies. Although each subsequent generation of VEGFR TKI is more potent and deemed to be more efficacious than the previous generation, the unfortunate reality is that patients still develop disease progression on these agents and ultimately die of their disease.
Combination trials with VEGFR TKIs and bevacizumab have been performed with the hope of increasing the efficacy and enhancing antitumor activities. However, two phase I trials using sunitinib plus bevacizumab in concurrent combination demonstrated significant cardiovascular and hematologic toxicities. Although some patients experienced partial responses and even one complete response, the concurrent combination of sunitinib plus bevacizumab was deemed to be too toxic for further development [10, 11].
Based on our experience with sunitinib, we hypothesized that sunitinib treatment failure was due to a compensatory increase in VEGF ligand, thus driving the tumor proliferative flare during sunitinib withdrawal. By altering the dosing strategy to sequential, we hoped to suppress the VEGF ligand levels by a sequential addition of bevacizumab during the sunitinib withdrawal period. Thus, with continued suppression of VEGF levels, we hypothesized that the proliferative flare seen in the sunitinib withdrawal period would be muted, resulting in an improved clinical benefit and efficacy such as a prolonged progression-free survival. Unexpectedly, this clinical trial definitively disproved our hypothesis that continued VEGF suppression would occur with the sequential combination of these two agents, as VEGF levels continued to rise with subsequent and continued treatment with sunitinib and bevacizumab.
In this clinical trial, we observed three specific features of the sequential treatment using sunitinib plus bevacizumab: potential efficacy of this sequential combination, safety of this treatment design, and further establishment of the pharmacokinetics of sunitinib and VEGF levels. Although only six patients were enrolled in this clinical study, we found several interesting features that potentially can shape future design of clinical trials and other studies involving VEGFR TKIs. Although this is an extremely small and limited sample size, we believe that our findings are hypothesis generating and can spark further discussion and exploration for the drug development of future tyrosine kinase inhibitors.
In terms of clinical outcomes, four patients on the study had a partial response with two patients having stable disease (67 % overall response rate), which is similar and consistent with the known objective response rate of sunitinib at 31 % as previously published [5]. However, we acknowledge that due to the low sample size, we are unable to document any statistical significance of the efficacy.
In the sequential use of sunitinib with bevacizumab, we noted toxicities that were similar to those seen in treatment with each individual agent such as hypertension, proteinuria, palmar plantar erythrodysesthesia, and diarrhea. All of the adverse events were ≤ grade 3, and thus, the sequential use of sunitinib plus bevacizumab appeared to be tolerated well by the six patients on study.
Subclinical MAHA was seen in one patient despite using the sequential strategy of sunitinib with low-dose bevacizumab. Based on the clinical protocol criteria, the patient did not meet any specific criteria for discontinuation from the study, especially in light of the fact that she remained asymptomatic. Although this more rational approach toward combining these two agents sought to reduce the risk of developing MAHA, and the single patient was completely asymptomatic during treatment with both agents, this combination was deemed not to be feasible for further development. In one phase I clinical trial with concurrent sunitinib plus bevacizumab, five out of 26 patients (19 %) developed MAHA with schistocytes on peripheral smear, and two patients on the study were symptomatic with neurologic symptoms and symptoms consistent with microangiopathic hemolytic anemia [10]. On the other hand, another phase I clinical trial with sunitinib plus bevacizumab did not note any evidence of MAHA [11]. It was hypothesized that in the clinical trial that treated only patients with renal cell carcinoma (rather than all solid malignancies), there may be some components associated with metastatic renal cell carcinoma that may predispose to the development of MAHA. Our clinical trial may give support to this hypothesis, but further evaluation would be required to make this conclusion. Given the history of MAHA in patients treated with the sunitinib and bevacizumab in other dosing strategies, we elected to discontinue this patient from further treatment and halted further accrual to this study. Thus, this combination is not feasible for further development due to safety concerns.
In our patient with MAHA, there was evidence of schistocytes on the peripheral blood smear, a significant decline in the haptoglobin level, as well as anemia. However, the patient was completely asymptomatic during the course of therapy. Other patients enrolled on the study were closely monitored for signs and symptoms of MAHA, and no other patients developed hemolytic anemia. No additional peripheral smears were obtained on other enrolled patients as they did not exhibit signs and symptoms of MAHA.
Lastly, we also evaluated the changes in VEGF levels as a result of treatment with sunitinib and bevacizumab. Because of the long half-life of bevacizumab of 20 days, we expected and hypothesized that VEGF ligand suppression would be long-lasting and thus should persist beyond each 42-day cycle. We hypothesized that with continued suppression of VEGF, the VEGF levels should continue to decline over a prolonged period of therapy using the sequential strategy. As predicted, the VEGF levels declined dramatically immediately after treatment with bevacizumab during each cycle of therapy. Measuring the VEGF levels at 4 h after the bevacizumab infusion demonstrated the significant decline. However, we found a complete rebound in total and free VEGF levels by day 42 of the first cycle. In fact, the VEGF levels returned at a higher level than at the baseline. Another intriguing finding is that the robust rebound of VEGF levels not only was seen during the first cycle of therapy, but it appeared to be persistent and increasing with each subsequent cycle. The increase in VEGF levels appeared to occur in a stepwise fashion, with each cycle having a higher VEGF level at the start of treatment (day 1 of each cycle) when compared with day 1 of the previous cycle.
It is unclear what the precise mechanism is by which VEGF ligand production may increase as a result of treatment with bevacizumab. We believe that there is a robust host compensatory mechanism to VEGF signaling pathway inhibition, but this is a field that warrants further discussion and evaluation. One explanation for the increasing VEGF levels is that this may be due to an increase in VEGF synthesis in these patients. In the original phase I trial of bevacizumab, increases in serum total VEGF concentrations were also seen, and it was attributed to an increase in VEGF synthesis and distribution [18].
Another potential hypothesis is that the host compensatory mechanism could also affect other proangiogenic factors in the host that we have not evaluated in this particular clinical trial. In a mouse model, Ebos et al. [19] demonstrated that there were also other plasma proteins with elevated levels during treatment with sunitinib, suggesting systemic collateral or indirect drug effects. In addition, these other plasma proteins include cytokines or chemokines that have been implicated as having proangiogenic activities such as granulocyte colony-stimulating factor (G-CSF), stromal-derived factor 1α (SDF-1α), and osteopontin. Thus, measuring other angiogenic factors during therapy would be worthwhile.
Both the free and total VEGF levels were measured during the course of therapy for all enrolled patients, and we found that the free and total VEGF levels mirrored each other in terms of the rise during each time point of treatment. We believe that these levels do not need to be measured separately, but that measuring the total VEGF level is sufficient in elucidating the changes in pharmacokinetics of VEGF and changes during therapy.
A limitation of the study is the small number of patients that were enrolled prior to study closure, which prevents any definitive conclusions to be made. Due to the small number of patients, we were unable to correlate changes in the VEGF levels with tumor response. However, we believe that VEGF levels could potentially be used as a biomarker and warrant further study in future trials. We also hoped to correlate changes in the VEGF levels with FLT PET/CT imaging for further elucidation of this phenomenon. Investigators of a sunitinib trial used FLT PET/CT imaging to demonstrate that nearly all patients had some initial reduction in tumor proliferation by FLT PET/CT after 4 weeks of sunitinib treatment [9]. During the treatment break, patients experienced a relative increase in FLT uptake consistent with an increase in tumor proliferation during the treatment withdrawal period. Although we had initially planned to perform FLT PET/CT imaging on patients using this sequential combination, this study was closed to accrual prior further evaluation on the pharmacodynamics aspect of this trial. However, correlation of VEGF levels with PET/CT imaging could potentially provide additional information to further elucidate this phenomenon.
Acknowledgments
The authors would like to thank the University of Wisconsin Carbone Cancer Center (UWCCC) for use of its Shared Services to complete this research. This work was supported in part by NIH/NCI P30 CA014520—UW Comprehensive Cancer Center Support, NIH/NCI U01 CA062491, Early Clinical Trials of New Anti-Cancer Agents with Phase I Emphasis [PI: Glenn Liu], and the Pfizer Investigator-Initiated Research Award GA4061YX. The UWCCC is a center of research and hope inspired by our patients and their families whose time and effort made this research possible. The authors would also like to thank the nurses and research specialists of the UWCCC Cancer Therapy Discovery and Development program for their efforts in conducting and managing this trial.
Contributor Information
Justine Yang Bruce, Email: jybruce@medicine.wisc.edu, Section of Hematology/Oncology, University of Wisconsin Carbone Cancer Center, 1111 Highland Avenue, Rm 7105, Madison, WI 53705, USA.
Jill M. Kolesar, School of Pharmacy, University of Wisconsin, K4/554 Clinical Science Center, 600 Highland Avenue, Madison, WI 53792, USA
Hans Hammers, Johns Hopkins Sidney Kimmel Comprehensive Cancer Center, 1650 Orleans St, CRB I, 1M46, Baltimore, MD 21231, USA.
Mark N. Stein, Section of Hematology/Oncology, Cancer Institute of New Jersey, 195 Little Albany St, New Brunswick, NJ 08903, USA
Lakeesha Carmichael, Department of Biostatistics and Medical Informatics, University of Wisconsin, Box 4675, Clinical Science Center K6/422, 600 Highland Avenue, Madison, WI 53792, USA.
Jens Eickhoff, Department of Biostatistics and Medical Informatics, University of Wisconsin, Box 4675, Clinical Science Center K6/434, 600 Highland Avenue, Madison, WI 53592, USA.
Susan A. Johnston, Pharmaceutical Research Center, University of Wisconsin, 600 Highland Avenue, Madison, WI 53792, USA
Kimberly A. Binger, University of Wisconsin Carbone Cancer Center, Box 5666, Clinical Science Center, 600 Highland Avenue, Madison, WI 53792, USA
Jennifer L. Heideman, University of Wisconsin Carbone Cancer Center, Box 5666, Clinical Science Center, 600 Highland Avenue, Madison, WI 53792, USA
Scott B. Perlman, Department of Radiology, University of Wisconsin, Box 3252, Clinical Science Center, 600 Highland Avenue, Madison, WI 53792, USA
Robert Jeraj, Department of Medical Physics, Wisconsin Institutes for Medical Research, University of Wisconsin, Box 1005, 1111 Highland Avenue, Madison, WI 53705, USA.
Glenn Liu, Section of Hematology/Oncology, University of Wisconsin Carbone Cancer Center, 1111 Highland Avenue, Rm 7105, Madison, WI 53705, USA.
References
- 1.Rini BI, Garcia JA, Cooney MM, et al. Toxicity of sunitinib plus bevacizumab in renal cell carcinoma. J Clin Oncol. 2010;28:e284–e285. doi: 10.1200/JCO.2009.27.1759. author reply e6–e7. [DOI] [PubMed] [Google Scholar]
- 2.Thompson Coon JS, Liu Z, Hoyle M, et al. Sunitinib and bevacizumab for first-line treatment of metastatic renal cell carcinoma: a systematic review and indirect comparison of clinical effectiveness. Br J Cancer. 2009;101:238–243. doi: 10.1038/sj.bjc.6605167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G, Jeraj R, Perlman S, et al. Pharmacodynamic study of FLT-PET imaging in patients treated with sunitinib. J Clin Oncol. 2008;26(Suppl):abstr 3515. [Google Scholar]
- 4.American Cancer Society. Cancer facts and figures 2012. American Cancer Society; Atlanta: 2012. p. 2012. [Google Scholar]
- 5.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 6.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 7.Rini BI, Escudier B, Tomczak P, et al. Axitinib versus sorafenib as second-line therapy for metastatic renal cell carcinoma (mRCC): results of phase III AXIS trial. J Clin Oncol. 2011;29(Suppl):Abstr 4503. [Google Scholar]
- 8.Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–147. doi: 10.1038/nm988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu G, Jeraj R, Vanderhoek M, et al. Pharmacodynamic study using FLT PET/CT in patients with renal cell cancer and other solid malignancies treated with sunitinib malate. Clin Cancer Res. 2011;17:7634–7644. doi: 10.1158/1078-0432.CCR-11-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feldman DR, Baum MS, Ginsberg MS, et al. Phase I trial of bevacizumab plus escalated doses of sunitinib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:1432–1439. doi: 10.1200/JCO.2008.19.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rini BI, Garcia JA, Cooney MM, et al. A phase I study of sunitinib plus bevacizumab in advanced solid tumors. Clin Cancer Res. 2009;15:6277–6283. doi: 10.1158/1078-0432.CCR-09-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deprimo SE, Bello CL, Smeraglia J, et al. Circulating protein biomarkers of pharmacodynamic activity of sunitinib in patients with metastatic renal cell carcinoma: modulation of VEGF and VEGF-related proteins. J Transl Med. 2007;5:32. doi: 10.1186/1479-5876-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Motzer RJ, Escudier B, Tomczak P, et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013;14:552–562. doi: 10.1016/S1470-2045(13)70093-7. [DOI] [PubMed] [Google Scholar]
- 14.Rini BI, Escudier B, Tomczak P, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011;378:1931–1939. doi: 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 15.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 16.Loupakis F, Falcone A, Masi G, et al. Vascular endothelial growth factor levels in immunodepleted plasma of cancer patients as a possible pharmacodynamic marker for bevacizumab activity. J Clin Oncol. 2007;25:1816–1818. doi: 10.1200/JCO.2006.10.3051. [DOI] [PubMed] [Google Scholar]
- 17.Ternant D, Ceze N, Lecomte T, et al. An enzyme-linked immunosorbent assay to study bevacizumab pharmacokinetics. Ther Drug Monit. 2010;32:647–652. doi: 10.1097/FTD.0b013e3181ef582a. [DOI] [PubMed] [Google Scholar]
- 18.Gordon MS, Margolin K, Talpaz M, et al. Phase I safety and pharmacokinetic study of recombinant human anti-vascular endothelial growth factor in patients with advanced cancer. J Clin Oncol. 2001;19:843–850. doi: 10.1200/JCO.2001.19.3.843. [DOI] [PubMed] [Google Scholar]
- 19.Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc Natl Acad Sci USA. 2007;104:17069–17074. doi: 10.1073/pnas.0708148104. [DOI] [PMC free article] [PubMed] [Google Scholar]