

Abstract
Hepatocarcinogenesis is a multistep process involving different genetic alterations that ultimately lead to malignant transformation of the hepatocyte. The liver is one of the main targets for different metastatic foci, but it represents an important and frequent locus of degeneration in the course of chronic disease. In fact, Hepatocellular carcinoma (HCC) represents the outcome of the natural history of chronic liver diseases, from the condition of fibrosis, to cirrhosis and finally to cancer. HCC is the sixth most common cancer in the world, some 630,000 new cases being diagnosed each year. Furthermore, about the 80% of people with HCC, have seen their clinical history developing from fibrosis, to cirrhosis and finally to cancer. The three main causes of HCC development are represented by HBV, HCV infection and alcoholism. Moreover, metabolic disease [starting from Non Alcoholic Fatty Liver Disease (NAFLD), Non Alcoholic Steatohepatitis (NASH)] and, with reduced frequency, some autoimmune disease may lead to HCC development. An additional rare cause of carcinogenetic degeneration of the liver, especially developed in African and Asian Countries, is represented by aflatoxin B1. The mechanisms by which these etiologic factors may induce HCC development involve a wide range of pathway and molecules, currently under investigation. In summary, the hepatocarcionogenesis results from a multifactorial process leading to the common condition of genetic changes in mature hepatocytes mainly characterized by uncontrolled proliferation and cell death.
Advances in understanding the mechanism of action are fundamental for the development of new potential therapies and results primarily from the association of the research activities coming from basic and clinical science.
This review article analyzes the current models used in basic research to investigate HCC activity, and the advances obtained from a basic and clinical point of view.
Keywords: Hepatocellular carcinoma, liver fibrosis, metabolic syndrome, NAFLD, NASH
Mouse models of HCC
In order to deeply investigate the hepatocarcionegenesis and new potential therapies in humans, there is growing interest to recreate experimental models that could be used in basic research, able to resemble the human characteristics of HCC.
In vitro testing of human HCC cell lines is usually an early step in the process of anticancer drug discovery that requires evaluation of viability, cell proliferation, clonogenicity and apoptosis. Several cell lines are currently used in literature: Huh7.5, HepG2, Hep3B and SK-Hep1 (1).
While results obtained using cell cultures provide important information regarding drug efficacy and mechanisms of action, in vitro systems lack the power to recapitulate the complex relationship between the tumor and its microenvironment.
Based on these data, a key role in the study of HCC is played by the in vivo experimental models (2). Concerning experimental models of HCC, genetic models, conditioned knock-out or transgenic models are mainly used to study the involvement of specific protein in the carcinogenetic process (3,4), while chemotoxic agents-induced HCC [such as N-nitrosodiethylamine (DEN) model] may provide a useful technique to study the interactions of different molecules and drugs. However, chemotoxic-induced HCC models do not completely resemble the human disease.
The DEN (N-nitrosodiethylamine) model of HCC is mainly used in basic research and promotes cancer development both in rats and in mice. DEN may be administered at different age of the mouse, however the results may vary in terms of efficacy and efficiency. To reduce the time of HCC development and limit the administration of the carcinogen, several studies adopted the association of promoting-agents to a single dose of DEN administration, the so called “two stage models” of HCC. Among the promoting agents, phenobarbital (PB) needs to be taken in consideration: the effects of PB promotion on DEN-initiated mice also vary considerably depending upon strain, sex and age of the mice (5-7). These models are largely used in literature and represent a good model to define and study the primitive HCC nodule, independently from the condition of cirrhosis.
Other experimental models of HCC involves AFB administration have been used in literature to specifically investigate the mechanisms involved in AFB-induced hepatocarcinogenesis, yet limited to the specific cases in which the AFB mechanisms need to be elucidated (8,9).
An increasing interest has been recently related to the metabolic conditions leading from simple steatosis to HCC in humans. This issue led to the development of additional mouse-model based on the use of diets, such as the choline deficient diet (CDD). In origin, such a diet has been developed to induce steatohepatitis, fibrosis and cirrhosis in mice and rats (10,11). More recently, it has been observed that mice subjected to CDD diet develop HCC formation after 50-52 weeks (10). The effects of CDD have been also evaluated in association with the administration of chemotoxic compounds (12). Ethionine supplementation to CDD diet is able to enhance the oval cells stimulation increasing the carcinogenetic potential (13,14). Similarly, combination of the CDD and DEN results in the earlier induction of HCC (12). A small variation of the current diet is represented by the choline-deficient and iron-supplemented l-amino acid-defined (CDAA) diet that mimics the same effect of the CDD diet in a shorter time frame (11,15).
An additional method used in basic research for the study of cancer is represented by the xenograft models: in xenograft models, the tumors are induced by injecting human cancer cells in immune deficient mice, such as athymic (nude) or severe combined immune deficient (SCID) mice (16). Among the xenograft models, the main ones are (I) the ectopic model, in which human cancer cells are directly injected subcutaneously in the flank of mice, and (II) the orthotopic model, in which tumor cells are injected directly into the mouse liver. These models are largely used in literature for the study of the metastatic spread of the tumor (17).
Finally, a considerable part of the basic research has been conducted in the HCC field by the use of genetically modified models. Genetically modified mouse models (GMM) have the purpose to mimic pathophysiological and molecular features of HCC (18). This approach allows to test the effects of oncogenes in the presence or not of carcinogenic agents. GMMs may be further improved by using cDNA constructs containing a promoter able to target a specific cell type (19). Mice with albumin promoter are often used in this field.
Rather than constitutive tissue-specific deleted expression of genes, an alternative model could be represented by the induction of specific genes, obtained generating transgenic mice. Among them, it is important to consider the transgenic mice models expressing viral genes for hepatitis. Most of the HBV-related transgenic animals express the HBx genes, which are associated with altered hepatocellular functions and HCC development (20). Concerning the HCV infection, transgenic mice expressing core, E1 and E2 structural proteins are mainly used in basic research (21).
In line with the in vitro study identifying the main singalling pathways potentially involved in HCC, several specific transgenic mice have been created and used in literature (22). The Myc transgenic mice are genetically close to human HCC of good prognosis and may be specifically used to study the entire range of pathways involved at this level (23). β-catenin transgenic mice have been used in the study of HCC: β-catenin is involved in the development and regeneration of the liver and β-catenin mutations are an early event in hepatocarcinogenesis (24). Mutations in growth factor genes, such as Tumor Growth Factor α (TGFα), Epidermal growth factor, Fibroblast growth factor 19, Platelet-derived growth factor (PDGF) and TGFβ1, have been recognized as involved in HCC development (25,26). Thus, specific transgenic mice have been created in basic research.
Moreover, transgenic mice expressing a human form of transport-impaired Alpha-1 antitrypsin [transport-impaired Alpha-1 antitrypsin (AAT) (27)] represents a good model for studying the effects of AAT deficiency on the liver. AAT deficiency is an autosomal recessive disorder in which a mutation causes the production of AAT that is unable to be transported (27). This leads to decreased AAT activity in serum and deposition of excessive AAT in the liver. Both heterozygous and homozygous individuals develop cirrhosis and HCC. AAT-deficient mice develop HCC after 52-90 weeks of age (27).
As demonstrated by in vitro studies, PTEN is a tumor suppressor gene that regulates the serine-threonine kinase protein kinase B (PKB⁄akt) pathway. Thus, PTEN knock out mice also develop HCC in vivo, in 66% of male and 30% of female mice by 40-44 weeks of age (28). PTEN deficiency induces cellular hyper-proliferation, anti-apoptosis and oncogenesis (29). Liver-specific PTEN-deficient mice develop hepatic steatosis, inflammation and fibrosis, thus resembling the features of human non-alcoholic steatohepatitis (NASH) (28).
In addition to the pure carcinogenetic mechanism evaluated by the previously described models, recent advances have demonstrated the involvement of inflammation pathways in the process of HCC formation. This issue has been clearly demonstrated by the use of hepatocytes specific NEMO deletion (IKK gamma subunit involved in the regulation of NFκB pathway). Several studies demonstrated that NEMO-mediated NF-kappaB activation in hepatocytes has an essential physiological function to prevent the spontaneous development of steatohepatitis and hepatocellular carcinoma, identifying NEMO as a tumor suppressor in the liver. NEMO specific hepatocytes-deleted mice spontaneously develop tumor after 10 to 12 months (30-32).
Finally, as a proof of the real existance of a link between fibrosis and cancer in the liver, an additional genetically modified mouse model has been used in basic research: as widely demonstrated in literature hepatocyte-specific deletion of TAK1 in mice results in spontaneous hepatocyte death, inflammation, fibrosis, and consequently in the development of HCC with a success rate represented by clear macroscopic nodules of 80% after 9 months (33).
Signalling pathways
In vivo study and the associated in vitro evaluation of specific molecules allow researchers to investigate the main potential mechanisms involved in carcinogenesis, defining several pathways strictly related to the process (Figure 1).
Figure 1.
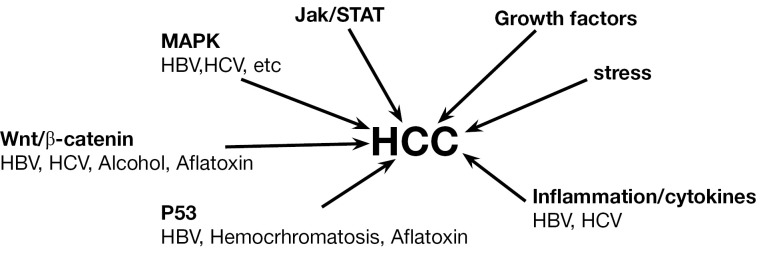
Intracellular pathways involved in the process of hepatocellular carcinoma formation and development.
This approach to basic research provided therefore the tools to discover the involvement of several mechanisms in carcinogenesis: among these, a special mention is related to the Wnt signaling pathway that is significantly deregulated in a number of cancers, including HCC (34). Wnt pathway is involved in HCC arising from HBV/HCV infections and alcoholic liver cirrhosis. Up-regulation of one of its component, frizzled-7, and dephosphorylation of β-catenin is frequently observed in HCC (35,36). The use of transgenic mice for β-catenin allowed to directly demonstrate the molecule involvement in the HCC formation, leading researchers to deeply investigate the mechanism. Based on these models, it has been demonstrated that mutations in β-catenin arise in HCC. This issue has been also demonstrated in patients with increased exposure to HCV infection and aflatoxin (37,38).
In the spectrum of genes related to HCC, a key role is played by p53. Several studies have reported that p53 mutations and inactivation play a critical role in HCC. The studies conducted in vivo on experimental models of HCC have been additionally confirmed in humans. Specifically, mutation of p53 correlates with the HCC developments induced by aflatoxin B1 (AFB1), as demonstrated using mouse models and subsequently confirmed in humans. Thus, detection of mutant p53 in plasma serves as a potential biomarker for AFB1 exposure and presence of HCC.
Human ras proteins H-Ras, N-Ras, K-ras4A, and K-Ras4B are small GTP-binding proteins that function as molecular switches to influence cell growth, differentiation and apoptosis (39). Single point mutations in codon 13 of H-ras, codon 12 of N-ras, and codon 61 of K-ras were originally observed in HCC caused by various chemicals such DEN (40-43). By the use of this model it has been demonstrated that Ras interacts with a downstream serine/threonine kinase Raf-1 leading to its activation and downstream signaling, which includes activation of MAPK kinases MEK1 and MEK2, to regulate proliferation and apoptosis (44). Activation of Ras and expression of Ras pathway proteins such as p21 were also reported in solid tumors as well as in cell lines (45,46). The strategies of inhibiting several kinases and suppressing Ras expression using antisense RNA has been successfully applied in cell line and in animal models (47,48).
In vivo mouse models of HCC have been also used to investigate the role of JAK/STAT pathways (49). STAT activation occurs through tyrosine phosphorylation by Janus kinases (JAKs). Activated STATs stimulate the transcription of suppressors of cytokine signaling (SOCS) genes. SOCS proteins, in turn, bind phosphorylated JAKs and their receptors to inhibit this pathway, thereby preventing overactivation of cytokine-stimulated cells (50). Thus, SOCS are part of the negative feedback loop in the JAK/ STAT circuitry. Two other families of STAT inhibitors that are described in the literature include the protein inhibitors of activated STATs and the SH2-containing proteins (51). JAK stimulation of STATs activates cell proliferation, migration, differentiation, and apoptosis, and deregulation of inhibitors leads to human diseases, including cancer (49). Inactivation of SOCS-1 and SSI-1, a JAK-binding protein, in HCC have been reported (49,50) as has the ubiquitous activation of the JAK/STAT pathway (52).
Proteins and cellular factors of other signaling pathways can also influence the molecular dynamics of HCC. For example, vascular endothelial growth factor and fibroblast growth factor play important roles in HCC development (53,54). It was reported recently that inflammation is inherently associated with cancer and a number of cytokines are involved in promoting HCC development and progression, especially during infection with hepatitis viruses (55). In particular, Th2 cytokines are induced and Th1 cytokines decreased in metastases. Therefore, modulating the expression of cytokines and the use of inhibitors of inflammatory cytokines might be critical in alleviating HCC progression. In a recent study, it was shown that the use of inhibitors of epidermal growth factor receptor and transforming growth factor prevented the development of HCC in rat liver, demonstrating the harmful nature of these growth factors if they exist in excessive amounts (56,57).
Clinical relevance and current treatment
Althoguh the enormous amount of data coming from basic research and the interest in developing drugs potentially effective, the clinical and pharmacological treament of HCC is still limited to the advanced stage of the disease.
As well established in the clinical practice, concerning the HCC development and the best treatment, it is mandatory to refer to the Barcelona Clinic Liver Cancer (BCLC) staging system, that represent not only a useful tool for classifying patients according to their prognosis, but also a method for selecting the appropriate treatment (Table 1) (58).
Table 1. Adapted table resuming the BCLC staging system for hepatocellular carcionoma classification and treatment strategy (Bruix, Hepatology 2011).
| Features | Definition | Treatment |
|---|---|---|
| Single HCC nodule <2 cm | Very early stage | Resection, liver transplantation, RadioFrequency |
| Single or 3 nodules <3 cm | Early stage | Liver transplantation, radiofrequency |
| Multinodular | Intermediate | TACE |
| Vascular portal invasion | Advanced stage | Sorafenib |
| Critical conditions | End stage | Symptomatic therapy |
Surgical treatments are the first treatment choice to consider. Resection and Orthotropic liver transplantation (OLT) achieve excellent results in BCLC 0 and A patients. Resection is the treatment of choice in non-cirrhotic patients where major resections are well tolerated. However, liver function impairment limits the feasibility of resection in cirrhotics if aiming at minimal morbidity and mortality. The best results in liver resection are obtained in solitary HCC. Multinodularity is correlated with recurrence and worse patient survival (59-62). Therefore, in multinodular HCC meeting the Milan criteria, OLT is a preferable option. In fact, the best results in liver transplantation are obtained applying the so-called Milan criteria (solitary ≤5 cm or if multiple, a maximum of 3 nodules ≤3 cm, without vascular invasion or extrahepatic spread). Meeting these criteria, the 5-year survival exceeds 70%, with recurrence ranging from 5% to 15% (63,64).
If OLT is not available, resection can still be considered in selected cases and optimally within prospective cohort investigations. However, since there are a growing number of publications reporting excellent results for early tumors treated with percutaneous ablation (65,66) or Transarterial chemoembolization (TACE) (65), with a lower rate of complications than with surgical resection, patients with multinodular HCC not suitable for OLT may be equally well served by percutaneous ablation or chemoembolization. To date, the optimal candidates for TACE are patients with preserved liver function (Child-Pugh A), without extrahepatic spread or vascular invasion (BCLC B). These patients have an estimated median survival of 16 months without treatment, while TACE expands this to >24 months (67,68). In contrast, performing TACE in patients with deteriorated liver function may lead to severe complications and death due to liver failure (69).
In very early tumors (≤2 cm), whose probability of dissemination is very low, and in which the probability of complete response with a safe margin with radiofrequency ablation (RFA) is high (90-100%), it is likely that resection and RFA are similar in terms of outcome. Thus, as stated recently, resection will not offer better survival than ablation in BCLC 0 patients and RFA would become the first-line option, leaving surgery for those patients with treatment failure.
Differently from the others, patients with advanced HCC fitting into BCLC C (extrahepatic dissemination or vascular invasion, or mild tumor-related symptoms, preserved liver function) have a median survival of about 6-8 months. Until recently there was no effective treatment for these patients. Neither chemotherapy, nor agents such as antiandrogens, antiestrogens or interferon induced any survival benefit (70). The growing knowledge in the field of molecular pathways involved in hepatocarcinogenesis led to the development of multiple molecules targeted to block those pathways (71). Currently, the multikinase inhibitor Sorafenib represents the drug that is recognized effective for the treatment of advanced HCC in human. Sorafenib has antiangiogenic and antiproliferative effects and has been shown to improve survival patients with advanced HCC compared with placebo (72). As observed in the SHARP trial, median survival for the placebo arm was 7.9 months, whereas it was 10.7 months for the group of patients treated with sorafenib [HR (sorafenib/placebo): 0.69 (95% CI: 0.55-0.88)]. This increase in survival was obtained without a significant radiological response, but with a significant difference in time to progression between the placebo and sorafenib groups that was 2.8 and 5.5 months respectively with a HR (sorafenib/placebo) of 0.58 (95% CI: 0.45-0.74). In the trials where the evidence was provided, treatment was maintained until symptomatic progression and not just until tumor progression as per radiology. Hence, in clinical practice, treatment might be maintained until symptomatic progression unless there are second-line options to be offered.
Future perspectives
Role of stem/progenitor cells in HCC
Over the years, it has been well established that both hepatocytes and cholangiocytes are capable of repopulating liver tissue following injury (73). Therefore, the concept of stem/progenitor cell existence in the liver did not gain much recognition until the past decade. Furthermore, growing evidence also demonstrated that the capacity to sustain tumor formation and growth resides in a small proportion of cancer stem cells (CSCs) (74,75). Subsequent identification of CSCs in a number of tissues including brain (76-78), prostate (79), breast (80), myeloid (81), gastric (82), colon (83,84), and lung (85), has reinforced the notion that stem cells might also exist in the liver. In the early studies, embryonic stem cells from murine embryos were shown to differentiate into functional hepatocytes in vitro (86,87). It was later shown that murine as well as human bone marrow-derived mesenchymal stem cells could differentiate into hepatocytes both in vitro and in vivo (88,89). Studies of bone marrow transplant recipients have shown that these cells could home to liver and differentiate into normal hepatocytes (90). One of the most common liver stem cells is the oval cell (91). Oval cells express markers common to hepatocytes and cholangiocytes, suggesting that they are bipotential. In fact, they differentiate into hepatocytes and cholangiocytes in vitro under the appropriate culture conditions (92). In diseases such as alcoholic liver disease and HCV infection, oval cell numbers increase and correlate with the severity of the disease (93). Several groups have isolated liver progenitor cell lines using oval cells from choline-deficient diet-fed rats (92), c-met transgenic mice (93), p53 null mice (94), and murine embryonic liver cells (95). Successful isolation of oval cells and establishment of liver progenitor cell lines from human liver tumors (96) and isolation of CSCs from human cell lines have been reported (97). The presence of CSCs and successful isolation of oval cells from cancerous tissue suggests that stem/progenitor cells play a key role in tumor formation. Recently, a novel cell type, the liver-derived progenitor cell, was also discovered and was isolated from healthy, uninjured rat livers (98). Further studies with these progenitor cells may provide insight to understand the molecular events that regulate cellular differentiation of the liver and those that lead to tumor progression.
Role of MicroRNAs in HCC
Identification of small, noncoding RNAs in the early 1990s has led to the development of a new research area (99).Several different classes of noncoding RNAs have been discovered in mammalian cells. These include small interfering RNAs (100), small nucleolar RNAs (101), and microRNAs (miRNAs) (102). miRNA complexes bind to imperfect complementary sequences in the 3'untranslated region of target mRNAs and negatively regulate gene expression either through mRNA degradation or translational inhibition (102,103). Recent studies have demonstrated that alterations in miRNA genes lead to tumor formation, and several miRNAs that regulate either the tumor suppression or promote tumor formation have been identified (104). For example, down-regulation of miR-15 and miR-16 results in overexpression of bcl2, cdk6, and cdc27, whereas overexpression of miR-21 causes suppression of PTEN and TPN1 (105). Several miRNAs that regulate the tumor suppressor p53 and p53-responsive genes have also been identified. Among these, miR-34 regulates p53 function in cell cycle arrest, cellular senescence, and apoptosis (106).
Thus miRNA expression profiles serve as signatures to determine not only the stages of a cancer but also a potential therapeutic strategy (107). The most abundant miRNA currently known in the liver, miR-122, is involved in cellular stress response, hepatocarcinogenesis, and inhibition of HCV replication [reviewed by Girard et al. (108)]. Therefore it has been suggested that downregulation of miR-122 could be a potential biomarker for liver cancers (109).
Other studies in literature, by examining microarray profile, found that miR-21 is highly overexpressed in HCC and cell lines. Inhibition of miR-21 in cultured HCC cells is able to increase the expression of the PTEN tumor suppressor and to decrease tumor cell proliferation, migration, and invasion; in contrast, enhanced miR-21 expression shows the opposite effect. These data reveal a correlation among miR-21 and PTEN, suggesting a direct involvement of miR-21 in carcionegensis (110). Further comparison of miRNA expression profile in the HCC tumors with patient’s survival time showed that a set of 19 miRNAs, involved in biological processes such as cell division, mitosis, and G1-S transition, significantly correlated with disease outcome (111). Based on these data, it could be easily stated that miRNAs may be useful to screen patients with cancer and identify those with a high likelihood of developing metastases/reoccurrence.
Conclusions
Animal models represent essential tools in cancer research, since they allow scientists to reproduce genetic, pathological or environmental abnormalities thought to be important for cancer development. Over the last few years, a number of rodent models have been developed allowing to study the different aspects of liver cancer. The cooperation of basic and clinical research has been able to promote an important development in the field of liver cancer, leading to the definition of the best diagnostic and therapeutic approach, as provided and elegantly resumed in the BCLC staging system. In many cases it is difficult to determine to what extent mouse models reproduce features observed in corresponding human conditions, but they could certainly provide a useful and unique approach in understanding novel pathways, unknown mechanisms and potential effective therapies for clinical use. Thus, future research and the use of novel tools and pathways may lead to the development of new drugs able to better interfere with the process of HCC development.
Acknowledgements
Financial support: this work was supported by a MIUR grant PRIN 2009 - prot. 2009X84L84_003 and Ministero della Salute grant GR-2010-2306996 to Dr. Marzioni; by MIUR grant PRIN 2009 - prot. 2009YNERCE_002, FIRB 2010 - prot RBAP10MY35_001 to Dr. Svegliati Baroni; by grants P 19118 -B05, F3008-B05 and F3517 from the Austrian Science Foundation and a GEN-AU project grant from the Austrian Ministry for Science to Dr. Trauner.
The research leading to these results has received funding from the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement n° HEALTH-F2-2009-241762 for the project FLIP.
Disclosure: The authors declare no conflict of interest.
References
- 1.Pang RW, Poon RT. From molecular biology to targeted therapies for hepatocellular carcinoma: the future is now. Oncology 2007;72:30-44. [DOI] [PubMed] [Google Scholar]
- 2.De Minicis S, Kisseleva T, Francis H, et al. Liver carcinogenesis: Rodent models of hepatocarcinoma and cholangiocarcinoma. Dig Liver Dis 2013;45:450-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feng H, Cheng AS, Tsang DP, et al. Cell cycle-related kinase is a direct androgen receptor-regulated gene that drives β-catenin/T cell factor-dependent hepatocarcinogenesis. J Clin Invest 2011;121:3159-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koch KS, Maeda S, He G, et al. Targeted deletion of hepatocyte Ikkbeta confers growth advantages. Biochem Biophys Res Commun 2009;380:349-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Awuah PK, Rhieu BH, Singh S, et al. β-Catenin loss in hepatocytes promotes hepatocellular cancer after diethylnitrosamine and phenobarbital administration to mice. PLoS One 2012;7:e39771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kamino H, Yamazaki Y, Saito K, et al. Nuclear receptor CAR-regulated expression of the FAM84A gene during the development of mouse liver tumors. Int J Oncol 2011;38:1511-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang M, Halasi M, Kabirov K, et al. Combination treatment with bortezomib and thiostrepton is effective against tumor formation in mouse models of DEN/PB-induced liver carcinogenesis. Cell Cycle 2012;11:3370-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hulla JE, Chen ZY, Eaton DL. Aflatoxin B1-induced rat hepatic hyperplastic nodules do not exhibit a site-specific mutation within the p53 gene. Cancer Res 1993;53:9-11. [PubMed] [Google Scholar]
- 9.McGlynn KA, Hunter K, LeVoyer T, et al. Susceptibility to aflatoxin B1-related primary hepatocellular carcinoma in mice and humans. Cancer Res 2003;63:4594-601. [PubMed] [Google Scholar]
- 10.Knight B, Yeoh GC, Husk KL, et al. Impaired preneoplastic changes and liver tumor formation in tumor necrosis factor receptor type 1 knockout mice. J Exp Med 2000;192:1809-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakano T, Cheng YF, Lai CY, et al. Impact of artificial sunlight therapy on the progress of non-alcoholic fatty liver disease in rats. J Hepatol 2011;55:415-25. [DOI] [PubMed] [Google Scholar]
- 12.de Lima VM, Oliveira CP, Alves VA, et al. A rodent model of NASH with cirrhosis, oval cell proliferation and hepatocellular carcinoma. J Hepatol 2008;49:1055-61. [DOI] [PubMed] [Google Scholar]
- 13.Guest I, Ilic Z, Sell S.Age dependence of oval cell responses and bile duct carcinomas in male fischer 344 rats fed a cyclic choline-deficient, ethionine-supplemented diet. Hepatology 2010;52:1750-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhong B, Zhou Q, Toivola DM, et al. Organ-specific stress induces mouse pancreatic keratin overexpression in association with NF-kappaB activation. J Cell Sci 2004;117:1709-19. [DOI] [PubMed] [Google Scholar]
- 15.Kodama Y, Kisseleva T, Iwaisako K, et al. c-Jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology 2009;137:1467-1477.e5. [DOI] [PMC free article] [PubMed]
- 16.Newell P, Villanueva A, Friedman SL, et al. Experimental models of hepatocellular carcinoma. J Hepatol 2008;48:858-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun FX, Tang ZY, Lui KD, et al. Establishment of a metastatic model of human hepatocellular carcinoma in nude mice via orthotopic implantation of histologically intact tissues. Int J Cancer 1996;66:239-43. [DOI] [PubMed] [Google Scholar]
- 18.Frese KK, Tuveson DA. Maximizing mouse cancer models. Nat Rev Cancer 2007;7:645-58. [DOI] [PubMed] [Google Scholar]
- 19.Tuveson DA, Jacks T. Technologically advanced cancer modeling in mice. Curr Opin Genet Dev 2002;12:105-10. [DOI] [PubMed] [Google Scholar]
- 20.Xiong J, Yao YC, Zi XY, et al. Expression of hepatitis B virus X protein in transgenic mice. World J Gastroenterol 2003;9:112-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naas T, Ghorbani M, Alvarez-Maya I, et al. Characterization of liver histopathology in a transgenic mouse model expressing genotype 1a hepatitis C virus core and envelope proteins 1 and 2. J Gen Virol 2005;86:2185-96. [DOI] [PubMed] [Google Scholar]
- 22.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene 2003;22:9007-21. [DOI] [PubMed] [Google Scholar]
- 23.Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet 2004;36:1306-11. [DOI] [PubMed] [Google Scholar]
- 24.Laurent-Puig P, Zucman-Rossi J.Genetics of hepatocellular tumors. Oncogene 2006;25:3778-86. [DOI] [PubMed] [Google Scholar]
- 25.Baek HJ, Lim SC, Kitisin K, et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology 2008;48:1128-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholes K, Guillet S, Tomlinson E, et al. A mouse model of hepatocellular carcinoma: ectopic expression of fibroblast growth factor 19 in skeletal muscle of transgenic mice. Am J Pathol 2002;160:2295-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geller SA, Nichols WS, Kim S, et al. Hepatocarcinogenesis is the sequel to hepatitis in Z#2 alpha 1-antitrypsin transgenic mice: histopathological and DNA ploidy studies. Hepatology 1994;19:389-97. [PubMed] [Google Scholar]
- 28.Watanabe S, Horie Y, Kataoka E, et al. Non-alcoholic steatohepatitis and hepatocellular carcinoma: lessons from hepatocyte-specific phosphatase and tensin homolog (PTEN)-deficient mice. J Gastroenterol Hepatol 2007;22:S96-S100. [DOI] [PubMed] [Google Scholar]
- 29.Horie Y, Suzuki A, Kataoka E, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest 2004;113:1774-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beraza N, Malato Y, Sander LE, et al. Hepatocyte-specific NEMO deletion promotes NK/NKT cell- and TRAIL-dependent liver damage. J Exp Med 2009;206:1727-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luedde T, Beraza N, Kotsikoris V, et al. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007;11:119-32. [DOI] [PubMed] [Google Scholar]
- 32.Seki E, Brenner DA. The role of NF-kappaB in hepatocarcinogenesis: promoter or suppressor? J Hepatol 2007;47:307-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inokuchi S, Aoyama T, Miura K, et al. Disruption of TAK1 in hepatocytes causes hepatic injury, inflammation, fibrosis, and carcinogenesis. Proc Natl Acad Sci U S A 2010;107:844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herbst A, Kolligs FT. Wnt signaling as a therapeutic target for cancer. Methods Mol Biol 2007;361:63-91. [DOI] [PubMed] [Google Scholar]
- 35.Merle P, Kim M, Herrmann M, et al. Oncogenic role of the frizzled-7/beta-catenin pathway in hepatocellular carcinoma. J Hepatol 2005;43:854-62. [DOI] [PubMed] [Google Scholar]
- 36.Terris B, Pineau P, Bregeaud L, et al. Close correlation between beta-catenin gene alterations and nuclear accumulation of the protein in human hepatocellular carcinomas. Oncogene 1999;18:6583-8. [DOI] [PubMed] [Google Scholar]
- 37.Devereux TR, Stern MC, Flake GP, et al. CTNNB1 mutations and beta-catenin protein accumulation in human hepatocellular carcinomas associated with high exposure to aflatoxin B1. Mol Carcinog 2001;31:68-73. [DOI] [PubMed] [Google Scholar]
- 38.Satoh S, Daigo Y, Furukawa Y, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet 2000;24:245-50. [DOI] [PubMed] [Google Scholar]
- 39.Clark GJ, Quilliam LA, Hisaka MM, et al. Differential antagonism of Ras biological activity by catalytic and Src homology domains of Ras GTPase activation protein. Proc Natl Acad Sci U S A 1993;90:4887-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baba M, Yamamoto R, Iishi H, et al. Ha-ras mutations in N-nitrosomorpholine-induced lesions and inhibition of hepatocarcinogenesis by antisense sequences in rat liver. Int J Cancer 1997;72:815-20. [DOI] [PubMed] [Google Scholar]
- 41.Bai F, Nakanishi Y, Takayama K, et al. Codon 64 of K-ras gene mutation pattern in hepatocellular carcinomas induced by bleomycin and 1-nitropyrene in A/J mice. Teratog Carcinog Mutagen 2003;Suppl 1:161-70. [DOI] [PubMed] [Google Scholar]
- 42.Challen C, Guo K, Collier JD, et al. Infrequent point mutations in codons 12 and 61 of ras oncogenes in human hepatocellular carcinomas. J Hepatol 1992;14:342-6. [DOI] [PubMed] [Google Scholar]
- 43.Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol 1997;9:174-9. [DOI] [PubMed] [Google Scholar]
- 44.Jagirdar J, Nonomura A, Patil J, et al. ras oncogene p21 expression in hepatocellular carcinoma. J Exp Pathol 1989;4:37-46. [PubMed] [Google Scholar]
- 45.Calvisi DF, Ladu S, Gorden A, et al. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006;130:1117-28. [DOI] [PubMed] [Google Scholar]
- 46.Liao Y, Tang ZY, Liu KD, et al. Apoptosis of human BEL-7402 hepatocellular carcinoma cells released by antisense H-ras DNA--in vitro and in vivo studies. J Cancer Res Clin Oncol 1997;123:25-33. [DOI] [PubMed] [Google Scholar]
- 47.Grisham JW. Interspecies comparison of liver carcinogenesis: implications for cancer risk assessment. Carcinogenesis 1997;18:59-81. [DOI] [PubMed] [Google Scholar]
- 48.Liao Y, Tang ZY, Ye SL, et al. Modulation of apoptosis, tumorigenesity and metastatic potential with antisense H-ras oligodeoxynucleotides in a high metastatic tumor model of hepatoma: LCI-D20. Hepatogastroenterology 2000;47:365-70. [PubMed] [Google Scholar]
- 49.Yoshikawa H, Matsubara K, Qian GS, et al. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat Genet 2001;28:29-35. [DOI] [PubMed] [Google Scholar]
- 50.Wormald S, Hilton DJ. Inhibitors of cytokine signal transduction. J Biol Chem 2004;279:821-4. [DOI] [PubMed] [Google Scholar]
- 51.Nagai H, Kim YS, Konishi N, et al. Combined hypermethylation and chromosome loss associated with inactivation of SSI-1/SOCS-1/JAB gene in human hepatocellular carcinomas. Cancer Lett 2002;186:59-65. [DOI] [PubMed] [Google Scholar]
- 52.Yasuda E, Kumada T, Takai S, et al. Attenuated phosphorylation of heat shock protein 27 correlates with tumor progression in patients with hepatocellular carcinoma. Biochem Biophys Res Commun 2005;337:337-42. [DOI] [PubMed] [Google Scholar]
- 53.Budhu A, Forgues M, Ye QH, et al. Prediction of venous metastases, recurrence, and prognosis in hepatocellular carcinoma based on a unique immune response signature of the liver microenvironment. Cancer Cell 2006;10:99-111. [DOI] [PubMed] [Google Scholar]
- 54.Huang X, Yu C, Jin C, et al. Ectopic activity of fibroblast growth factor receptor 1 in hepatocytes accelerates hepatocarcinogenesis by driving proliferation and vascular endothelial growth factor-induced angiogenesis. Cancer Res 2006;66:1481-90. [DOI] [PubMed] [Google Scholar]
- 55.Schiffer E, Housset C, Cacheux W, et al. Gefitinib, an EGFR inhibitor, prevents hepatocellular carcinoma development in the rat liver with cirrhosis. Hepatology 2005;41:307-14. [DOI] [PubMed] [Google Scholar]
- 56.Breitkopf K, Haas S, Wiercinska E, et al. Anti-TGF-beta strategies for the treatment of chronic liver disease. Alcohol Clin Exp Res 2005;29:121S-131S. [DOI] [PubMed] [Google Scholar]
- 57.Schulze-Bergkamen H, Fleischer B, Schuchmann M, et al. Suppression of Mcl-1 via RNA interference sensitizes human hepatocellular carcinoma cells towards apoptosis induction. BMC Cancer 2006;6:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Lope CR, Tremosini S, Forner A, et al. Management of HCC. J Hepatol 2012;56:S75-87. [DOI] [PubMed] [Google Scholar]
- 59.Arii S, Tanaka S, Mitsunori Y, et al. Surgical strategies for hepatocellular carcinoma with special reference to anatomical hepatic resection and intraoperative contrast-enhanced ultrasonography. Oncology 2010;78:125-30. [DOI] [PubMed] [Google Scholar]
- 60.Imamura H, Matsuyama Y, Tanaka E, et al. Risk factors contributing to early and late phase intrahepatic recurrence of hepatocellular carcinoma after hepatectomy. J Hepatol 2003;38:200-7. [DOI] [PubMed] [Google Scholar]
- 61.Ishizawa T, Hasegawa K, Aoki T, et al. Neither multiple tumors nor portal hypertension are surgical contraindications for hepatocellular carcinoma. Gastroenterology 2008;134:1908-16. [DOI] [PubMed] [Google Scholar]
- 62.Huang J, Yan L, Cheng Z, et al. A randomized trial comparing radiofrequency ablation and surgical resection for HCC conforming to the Milan criteria. Ann Surg 2010;252:903-12. [DOI] [PubMed] [Google Scholar]
- 63.Cillo U, Vitale A, Bassanello M, et al. Liver transplantation for the treatment of moderately or well-differentiated hepatocellular carcinoma. Ann Surg 2004;239:150-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mazzaferro V, Llovet JM, Miceli R, et al. Predicting survival after liver transplantation in patients with hepatocellular carcinoma beyond the Milan criteria: a retrospective, exploratory analysis. Lancet Oncol 2009;10:35-43. [DOI] [PubMed] [Google Scholar]
- 65.Bargellini I, Sacco R, Bozzi E, et al. Transarterial chemoembolization in very early and early-stage hepatocellular carcinoma patients excluded from curative treatment: a prospective cohort study. Eur J Radiol 2012;81:1173-8. [DOI] [PubMed] [Google Scholar]
- 66.Livraghi T, Meloni F, Di Stasi M, et al. Sustained complete response and complications rates after radiofrequency ablation of very early hepatocellular carcinoma in cirrhosis: Is resection still the treatment of choice? Hepatology 2008;47:82-9. [DOI] [PubMed] [Google Scholar]
- 67.Llovet JM, Real MI, Montaña X, et al. Arterial embolisation or chemoembolisation versus symptomatic treatment in patients with unresectable hepatocellular carcinoma: a randomised controlled trial. Lancet 2002;359:1734-9. [DOI] [PubMed] [Google Scholar]
- 68.Malagari K, Alexopoulou E, Chatzimichail K, et al. Transcatheter chemoembolization in the treatment of HCC in patients not eligible for curative treatments: midterm results of doxorubicin-loaded DC bead. Abdom Imaging 2008;33:512-9. [DOI] [PubMed] [Google Scholar]
- 69.Paleri V, Carding P, Chatterjee S, et al. Voice outcomes after concurrent chemoradiotherapy for advanced nonlaryngeal head and neck cancer: a prospective study. Head Neck 2012;34:1747-52. [DOI] [PubMed] [Google Scholar]
- 70.Bruix J, Sherman M, American Association for the Study of Liver Diseases . Management of hepatocellular carcinoma: an update. Hepatology 2011;53:1020-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646-74. [DOI] [PubMed] [Google Scholar]
- 72.Cheng AL, Kang YK, Chen Z, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol 2009;10:25-34. [DOI] [PubMed] [Google Scholar]
- 73.Michalopoulos GK, DeFrances MC. Liver regeneration. Science 1997;276:60-6. [DOI] [PubMed] [Google Scholar]
- 74.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer 2003;3:895-902. [DOI] [PubMed] [Google Scholar]
- 75.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature 2001;414:105-11. [DOI] [PubMed] [Google Scholar]
- 76.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A 2003;100:15178-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Singh SK, Clarke ID, Terasaki M, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821-8. [PubMed] [Google Scholar]
- 78.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396-401. [DOI] [PubMed] [Google Scholar]
- 79.Collins AT, Berry PA, Hyde C, et al. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005;65:10946-51. [DOI] [PubMed] [Google Scholar]
- 80.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A 2003;100:3983-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645-8. [DOI] [PubMed] [Google Scholar]
- 82.Houghton J, Stoicov C, Nomura S, et al. Gastric cancer originating from bone marrow-derived cells. Science 2004;306:1568-71. [DOI] [PubMed] [Google Scholar]
- 83.O’Brien CA, Pollett A, Gallinger S, et al. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007;445:106-10. [DOI] [PubMed] [Google Scholar]
- 84.Ricci-Vitiani L, Lombardi DG, Pilozzi E, et al. Identification and expansion of human colon-cancer-initiating cells. Nature 2007;445:111-5. [DOI] [PubMed] [Google Scholar]
- 85.Kim CF, Jackson EL, Woolfenden AE, et al. Identification of bronchioalveolar stem cells in normal lung and lung cancer. Cell 2005;121:823-35. [DOI] [PubMed] [Google Scholar]
- 86.Chinzei R, Tanaka Y, Shimizu-Saito K, et al. Embryoid-body cells derived from a mouse embryonic stem cell line show differentiation into functional hepatocytes. Hepatology 2002;36:22-9. [DOI] [PubMed] [Google Scholar]
- 87.Hamazaki T, Iiboshi Y, Oka M, et al. Hepatic maturation in differentiating embryonic stem cells in vitro. FEBS Lett 2001;497:15-9. [DOI] [PubMed] [Google Scholar]
- 88.Anjos-Afonso F, Siapati EK, Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J Cell Sci 2004;117:5655-64. [DOI] [PubMed] [Google Scholar]
- 89.Lee KD, Kuo TK, Whang-Peng J, et al. In vitro hepatic differentiation of human mesenchymal stem cells. Hepatology 2004;40:1275-84. [DOI] [PubMed] [Google Scholar]
- 90.Alison MR, Poulsom R, Jeffery R, et al. Hepatocytes from non-hepatic adult stem cells. Nature 2000;406:257. [DOI] [PubMed] [Google Scholar]
- 91.Menthena A, Deb N, Oertel M, et al. Bone marrow progenitors are not the source of expanding oval cells in injured liver. Stem Cells 2004;22:1049-61. [DOI] [PubMed] [Google Scholar]
- 92.Lázaro CA, Rhim JA, Yamada Y, et al. Generation of hepatocytes from oval cell precursors in culture. Cancer Res 1998;58:5514-22. [PubMed] [Google Scholar]
- 93.Lowes KN, Brennan BA, Yeoh GC, et al. Oval cell numbers in human chronic liver diseases are directly related to disease severity. Am J Pathol 1999;154:537-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spagnoli FM, Amicone L, Tripodi M, et al. Identification of a bipotential precursor cell in hepatic cell lines derived from transgenic mice expressing cyto-Met in the liver. J Cell Biol 1998;143:1101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dumble ML, Croager EJ, Yeoh GC, et al. Generation and characterization of p53 null transformed hepatic progenitor cells: oval cells give rise to hepatocellular carcinoma. Carcinogenesis 2002;23:435-45. [DOI] [PubMed] [Google Scholar]
- 96.Strick-Marchand H, Morosan S, Charneau P, et al. Bipotential mouse embryonic liver stem cell lines contribute to liver regeneration and differentiate as bile ducts and hepatocytes. Proc Natl Acad Sci U S A 2004;101:8360-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Parent R, Marion MJ, Furio L, et al. Origin and characterization of a human bipotent liver progenitor cell line. Gastroenterology 2004;126:1147-56. [DOI] [PubMed] [Google Scholar]
- 98.Sahin MB, Schwartz RE, Buckley SM, et al. Isolation and characterization of a novel population of progenitor cells from unmanipulated rat liver. Liver Transpl 2008;14:333-45. [DOI] [PubMed] [Google Scholar]
- 99.Hüttenhofer A, Kiefmann M, Meier-Ewert S, et al. RNomics: an experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. EMBO J 2001;20:2943-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lagos-Quintana M, Rauhut R, Lendeckel W, et al. Identification of novel genes coding for small expressed RNAs. Science 2001;294:853-8. [DOI] [PubMed] [Google Scholar]
- 101.Kiss T.Small nucleolar RNAs: an abundant group of noncoding RNAs with diverse cellular functions. Cell 2002;109:145-8. [DOI] [PubMed] [Google Scholar]
- 102.Kato M, Slack FJ. microRNAs: small molecules with big roles - C. elegans to human cancer. Biol Cell 2008;100:71-81. [DOI] [PubMed] [Google Scholar]
- 103.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281-97. [DOI] [PubMed] [Google Scholar]
- 104.Kent OA, Mendell JT. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene 2006;25:6188-96. [DOI] [PubMed] [Google Scholar]
- 105.Calin GA, Croce CM. Chromosomal rearrangements and microRNAs: a new cancer link with clinical implications. J Clin Invest 2007;117:2059-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.He X, He L, Hannon GJ. The guardian’s little helper: microRNAs in the p53 tumor suppressor network. Cancer Res 2007;67:11099-101. [DOI] [PubMed] [Google Scholar]
- 107.Iorio MV, Visone R, Di Leva G, et al. MicroRNA signatures in human ovarian cancer. Cancer Res 2007;67:8699-707. [DOI] [PubMed] [Google Scholar]
- 108.Girard M, Jacquemin E, Munnich A, et al. miR-122, a paradigm for the role of microRNAs in the liver. J Hepatol 2008;48:648-56. [DOI] [PubMed] [Google Scholar]
- 109.Gramantieri L, Ferracin M, Fornari F, et al. Cyclin G1 is a target of miR-122a, a microRNA frequently down-regulated in human hepatocellular carcinoma. Cancer Res 2007;67:6092-9. [DOI] [PubMed] [Google Scholar]
- 110.Meng F, Henson R, Wehbe-Janek H, et al. MicroRNA-21 regulates expression of the PTEN tumor suppressor gene in human hepatocellular cancer. Gastroenterology 2007;133:647-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang J, Gusev Y, Aderca I, et al. Association of MicroRNA expression in hepatocellular carcinomas with hepatitis infection, cirrhosis, and patient survival. Clin Cancer Res 2008;14:419-27. [DOI] [PMC free article] [PubMed] [Google Scholar]