

Abstract
The acute respiratory distress syndrome (ARDS) is a devastating constellation of clinical, radiological and pathological signs characterized by failure of gas exchange and refractory hypoxia. Despite nearly 30 years of research, no specific pharmacological therapy has yet proven to be efficacious in manipulating the pathophysiological processes that underlie this condition. Several in vitro and in vivo animal or human studies suggest a potential role for β2-agonists in the treatment of ARDS. These agents have been shown to reduce pulmonary neutrophil sequestration and activation, accelerate alveolar fluid clearance, enhance surfactant secretion, and modulate the inflammatory and coagulation cascades. They are also used widely in clinical practice and are well tolerated in critically ill patients. The present review examines the evidence supporting a role for β2-agonists as a specific pharmacological intervention in patients with ARDS.
Keywords: acute lung injury, acute respiratory distress syndrome, alveolar epithelium, β2-agonists, pharmacotherapy
Introduction
Acute lung injury (ALI) and its more severe form – the acute respiratory distress syndrome (ARDS) – are common, devastating clinical syndromes of acute respiratory failure that affect all age groups [1]. Recent European [2], American [3] and Australian [4] multicentre studies have estimated the incidence of ALI and ARDS at 34 and 28 cases per 100 000 per year, respectively; otherwise stated, 7.1% of all intensive care admissions are for ALI/ARDS. More than three decades after its first description in 1967 [5], mortality associated with ARDS is still high, with reported rates between 40% and 60% [1]. Morbidity among survivors is also high, with persistent functional limitation 1 year after discharge preventing over half from returning to work [6].
Improvements in general supportive care have contributed toward a trend of decreasing mortality over the past 10 years [7], and recently strategies to reduce the effects of ventilator-associated lung injury have resulted in an important reduction in mortality [8]. However, as yet no specific pharmacological therapies to target the underlying pathological processes have proved efficacious [9]. Recent in vitro and in vivo animal or human studies suggest that β2-agonists – drugs that are well established in the management of patients with chronic bronchitis or asthma – may have an important therapeutic role to play in modulating the initial inflammatory insult and enhancing alveolar fluid clearance in patients with ARDS.
The present review discusses the effects of β2-agonists onneutrophil functions, on inflammatory mediators, and on epithelial and endothelial functions (Fig. 1). It draws on the extensive experimental and clinical literature on the mechanisms of effects of β2-agonists to suggest a potential role for their use as a specific pharmacological intervention in patients with ARDS.
Figure 1.
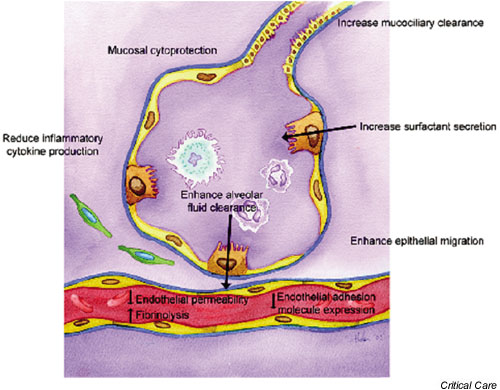
The effects of β-agonists on epithelial and endothelial function.
β-Adrenergic stimulation and neutrophil function
Role of the neutrophil in acute respiratory distress syndrome
Classical descriptions of ARDS, based on lung biopsy and postmortem specimens, have artificially divided the condition into three phases – exudative, proliferative and fibrotic [10] – although in practice these phases often overlap [1]. The early phases are characterized by infiltration with neutrophils, macrophages and inflammatory cytokines, and disruption of the alveolar capillary barrier, leading to an influx of protein-rich oedema fluid into the alveolar spaces [11]. Although controversy still exists regarding the role of polymorphonuclear neutrophils in all causes of ALI [12], it is likely that they play a central role in early stages [13]. Analysis of bronchoalveolar lavage (BAL) fluid from patients with ARDS has revealed increased numbers of activated neutrophils in the early stages of ARDS [13,14]. The number of neutrophils in BAL fluid correlates with the severity of lung injury [15], and persistence of neutrophils in BAL fluid by day 7 is associated with increased mortality [14].
Pulmonary neutrophil sequestration occurs within minutes of exposure to an inflammatory insult [16,17]. The insult causes an increase in neutrophil stiffness and a reduction in deformability [18], leading to sequestration into the pulmonary capillaries followed by emigration into the alveolar space. The process of neutrophil emigration occurs by at least two different pathways. Neutrophil emigration is dependent on CD11/18 adhesion molecule interactions in response to Gram-negative organisms, IL-1α and phorbol 12-myristate 13-acetate. Gram-positive organisms, hyperoxia and the complement anaphylatoxins (C5a) appear to induce neutrophil emigration through a CD11/18 independent pathway [19].
Neutrophils are a potent source of reactive oxygen and nitrogen species, inflammatory cytokines, proteolytic enzymes and lipid mediators. A recent study examining ARDS BAL fluid [20] demonstrated a positive correlation between neutrophil myeloperoxidase and oxidatively modified amino acids, suggesting an association between pulmonary neutrophil activation and oxidative protein damage. Carden and coworkers [20] reported that damage to human surfactant protein A in BAL fluid from patients with ARDS resembled the damage caused when it is cleaved by neutrophil elastase in patients with ARDS. Therapeutic interventions with neutrophil elastase inhibitors in animal models of ARDS have shown that inhibition of neutrophil function can limit the degree of lung injury caused by ischaemia–reperfusion [21] and lipopolysaccharide (LPS) [22].
The importance of regulation of neutrophil apoptosis in ARDS was recently reviewed in detail [23]. It is known that ARDS BAL fluid delays neutrophil apoptosis in vitro [24]. At present the relationship between neutrophil apoptosis and survival from ARDS has not been clearly defined, although it has been suggested that increasing neutrophil apoptosis could be beneficial in aiding resolution of ARDS [23]. Apoptotic neutrophils are cleared from the alveolar space by alveolar macrophages. Interestingly, this process changes the inflammatory cytokine profile produced by the macrophage from an inflammatory to anti-inflammatory phenotype [25]. Furthermore, in a recent study conducted in mice [26], stimulating neutrophil apoptosis led to reduced lung injury and improved survival. This suggests that acceleration of neutrophil apoptosis could be beneficial in the treatment of ARDS. Modulation of neutrophil recruitment, activation and apoptosis are thus potential therapeutic targets for the treatment of patients with ARDS.
Effects of β-adrenergic stimulation on neutrophil sequestration
β-Adrenergic stimulation has been shown to reduce pulmonary neutrophil sequestration in several different models of lung injury. Using a murine model of direct lung injury (endotoxin inhalation), Dhingra and coworkers [27] showed that pretreatment with intravenous dobutamine reduced BAL fluid neutrophilia by 30% in parallel with reduced pulmonary IL-6, IL-10 and macrophage inflammatory protein-2 productions. Similarly, in a rodent model of indirect lung injury following endotoxic shock, pretreatment with intravenous terbutaline before exposure to endotoxin blocked pulmonary neutrophil accumulation, prevented circulatory failure and reduced mortality [28]. In normal human volunteers, in a placebo-controlled trial, treatment with 300 μg inhaled salbutamol was able to prevent platelet-activating factor induced pulmonary sequestration of radio-labelled neutrophils [29].
The precise mechanisms of reduced pulmonary neutrophil sequestration have not fully been elucidated, although they may involve modulation of adhesion and emigration, accelerated apoptosis and reduced generation of inflammatory mediators.
Adhesion and migration
β2-Agonists reduce in vitro neutrophil adhesion to human bronchial epithelial cells [30] and endothelial cells [31,32]. This occurred through elevation in intracellular cAMP and reduction in CD11b/18 adhesion molecule expression [30,32]. Whether this was due a direct effect on CD11b/18 synthesis and release, or indirectly through reducing tumour necrosis factor (TNF)-α expression (which causes CD11b/18 upregulation) remains to be determined [33].
Chemotaxis is the phenomenon of cell migration toward a chemoattractant stimulus such as bacterial peptides (formyl-methionyl-leucyl-phenylalanine [fMLP]) and complement (C5a), and it is an important step in the migration of neutrophils toward sites of inflamed or damaged tissues. Most studies investigating the effects of β2-agonists on neutrophil chemotaxis have shown a reduction in neutrophil chemotaxis [34-37]] at doses equivalent to levels reported in oedema fluid following nebulized salbutamol administration (10-6 mol/l) [38]. However Llewellyn-Jones and coworkers [39] reported a biphasic response with increased neutrophil chemotaxis toward fMLP after incubation with 10-5 mol/l terbutaline, and a reduction in chemotaxis when supraphysiological concentrations (10-3 mol/l) were used. At higher doses of β2-agonists, stimulation of β1- and β2-adrenergic receptors occurs and it is possible that this might contribute to the biphasic effect.
Apoptosis
β2-Agonists induce apoptosis in several different cell typesincluding the human neutrophil [40]. Although this may have potentially beneficial effects by promoting neutrophil apoptosis, this needs to be balanced against the potentially deleterious effects of β2-agonist enhanced alveolar cell apoptosis leading to a worsening of lung injury [41].
Neutrophil mediator release
β2-Agonists reduce oxygen free radical production from neutrophils and other inflammatory cells [42,43]. This effect appears to occur because of both β-receptor dependent and independent mechanisms [44]. Although β-receptor independent mechanisms may occur because of a direct effect on cellular metabolism, Gillissen and coworkers [45] recently showed that it may in part be due to an intrinsic scavenger function of β2-agonists for reactive oxygen species. In contrast, these agents have little effect on neutrophil degranulation [39], phagocytosis, or bacterial killing [36].
β-Adrenergic stimulation and inflammatory mediators
Inflammatory cascade
A complex network of cytokines, proinflammatory and anti-inflammatory substances are involved in the inflammatory response in ARDS. Inflammatory cytokines such as IL-8, TNF-α and IL-1β have been found in high concentrations in the early phase of ARDS [46,47]. The balance between proinflammatory and anti-inflammatory cytokines is likely to be critical in the development and persistence ARDS [48]. High initial titres and persistence of inflammatory cytokines have been shown to be predictors of poor outcome [49]. IL-8, a cytokine that is seen early in the inflammatory response, is important in pulmonary neutrophil recruitment and activation [50]. Treatment with anti-IL-8 monoclonal antibody in experimental animal models of ARDS has been shown to decrease the magnitude of ALI [50-52]], suggesting that modulation of cytokine production may have a role to play in ameliorating lung injury.
Effects of β-adrenergic stimulation on inflammatory mediators
β-Adrenergic stimulation in vitro reduces inflammatory cytokine production (IL-1β [53], TNF-α [54-57]], IL-6 [58] and IL-8 [59,60]) and enhances release of the anti-inflammatory cytokine IL-10 [61] from whole blood, monocytes and macrophages. In an in vivo mouse model of LPS-induced septic shock, Wu and coworkers [28] demonstrated that treatment with terbutaline was able to reduce TNF-α production, enhance IL-10 production and improve survival. In an ex vivo model using human lung explants in culture, treatment with 1 ng/ml isoproterenol attenuated LPS-induced release of TNF-α and reduced lipid peroxidation, which was associated with an increase in intracellular cAMP levels [62]. Van der Poll and coworkers [63] extended these findings in vivo in human volunteers using adrenaline before LPS exposure. That study confirmed that adrenaline reduced LPS-induced TNF-α release in vivo and in whole blood ex vivo. This occurred in parallel with an increase in the release of the anti-inflammatory cytokine IL-10. In addition β-adrenergic stimulation, in contrast to α-receptor stimulation, caused an increase in IL-10 similar to that with adrenaline. These data suggest that treatment with β2-agonists may have a role to play in reducing the excessive proinflammatory effects of the cytokine network during the early phases of ARDS.
β-Adrenergic stimulation and endothelial and epithelial function
Effects of β-adrenergic stimulation on endothelial permeability
Extensive damage to the alveolar–capillary barrier and microvascular thrombosis are prominent features in the early stages of ARDS [64]. This leads to alveolar flooding and the development of noncardiogenic pulmonary oedema, which impairs gas exchange and contributes to the refractory hypoxia that characterizes ARDS.
In vitro studies using pulmonary artery endothelial cells have shown that incubation with isoprotenerol reduces baseline monolayer permeability to albumin and can block the effects of thrombin-induced increase in permeability [65,66]. These findings have been confirmed in vivo in a sheep ARDS model using terbutaline [67] and a rat ARDS model using isoproten-erol [68]. In a small nonrandomized study conducted in humans, administration of intravenous terbutaline to 10 patients with ARDS was associated with a significant reduction in lung vascular permeability (measured by radio-labelled transferrin) in six patients, which was associated with an increased probability of survival [69]. The mechanism appears to be related to inhibition of endothelial cell contraction and increased force between endothelial cell tight junctions.
Alterations to the coagulation/fibrinolysis pathways may be important in the pathogenesis of ARDS [70]. Two recent studies from Matthay and coworkers [71,72] showed that plasma and oedema fluid levels of protein C and oedema fluid levels of thrombomodulin and plasminogen activator inhibitor-1 are associated with increased mortality in patients with ARDS. There is some preliminary evidence from studies in healthy volunteers that the intravenous administration of isoproterenol increases the release of tissue plasminogen activator and urokinase plasminogen activator, which may enhance fibrinolysis and vessel patency [73,74]. The effects of β-adrenergic stimulation on the coagulation–fibrinolysis cascade in ARDS, however, remains to be determined.
Effects of β-adrenergic stimulation on alveolar fluid clearance
Clearance of fluid from the alveolar space is dependent on active sodium and chloride transport. The alveolar type II cell appears to be responsible for the majority of ion transport via the apical sodium and chloride conductive pathways and the basolateral Na/K-ATPase, although the alveolar type I cell and distal airway epithelium may also contribute [75]. Experimental studies in animals, as well as in the ex vivo human lung, have demonstrated that β-adrenergic agonists accelerate the rate of alveolar fluid clearance [76,77]. The mechanism underlying increased alveolar fluid clearance is proposed to be due to an increase in intracellular cAMP, resulting in increased sodium transport across alveolar type II cells by upregulation of the apical sodium and chloride pathways and Na/K-ATPase and probably cystic fibrosis transmembrane conductance regulator [75]. β2-Adrenergic stimulation is more important than β1-adrenergic stimulation in mediating alveolar epithelialsodium and fluid transport. Dopamine, at doses associated with only a β1 effect, whether by intra-alveolar or intravenous route of administration, had no effect on alveolar fluid clearance in vivo in rats. Moreover, the increase in alveolar fluid clearance caused by dobutamine is blocked by selective β2-adrenergic antagonists [78]. Finally, β1-adrenergic stimulation by high-dose terbutaline has been found to downregulate alveolar fluid clearance in the ex vivo rat lung [79].
Impaired ability of the alveolar epithelium to remove alveolar oedema fluid is associated with increased mortality in ARDS [80,81]. This has important implications for the potential use of β2-agonists in the treatment of ALI/ARDS. If the alveolar epithelium is extensively injured, then pharmacological intervention aimed at improving epithelial function may be difficult because of the extent of injury. Alveolar epithelial fluid clearance mechanisms are intact after mild to moderate lung injury and can be upregulated by β-adrenergic agonists [82,83]. However, in some experimental models neutrophil-dependent oxidant injury to the alveolar epithelium is more resistant to β-adrenergic upregulation of alveolar fluid clearance [84-86]]. β-Agonists have also been shown to upregulate fluid transport in hydrostatic oedema [87-89]], hyperoxic lung injury [83,90,91] and ventilator-associated lung injury [92]. In addition, β2-agonists can overcome the depressant effects of hypoxia on alveolar fluid clearance [93,94]. In a randomized, placebo-controlled clinical trial [95], inhaled salmeterol (a long-acting β2-agonist) reduced the incidence of high-altitude pulmonary oedema in volunteers who were known to be at risk for this condition. The authors postulated that this may be due to an increase in alveolar fluid clearance, although beneficial effects of salmeterol on minute ventilation and pulmonary artery pressures could not be excluded. On the basis of these experimental data augmentation of alveolar epithelial fluid clearance with β2-adrenergic agonists may accelerate resolution of pulmonary oedema and improve outcome in ALI/ARDS.
Effects of β2-agonists on surfactant
Surfactant, a mixture of dipalmitoyl-phosphatidylcholine and other lipids and proteins, is produced by type II alveolar epithelial cells. Surfactant is a lipid surface-tension-lowering agent and it helps to prevent pulmonary oedema. Surfactant plays an increasingly recognized role in immune defence. Surfactant protein (SP)-A is known to promote phagocytosis of bacteria by alveolar macrophages, and SP-D also has antimicrobial properties [96,97]. Deficiency in these specific proteins may well contribute to the increase risk for infection in ARDS patients.
Short-acting and long-acting β2-agonists augment total surfactant secretion from alveolar type II cells through activation of β-adrenergic receptors and a cAMP-dependent protein kinase. Several β2-agonists stimulate secretion of phophatidylcholine, the principal lipid component of surfactant [98,99]. In particular, terbutaline is a potent secretagogue [100]. β2-Agonists also stimulate secretion of SP-Band SP-C, the two hydrophobic proteins that are involved in the main biophysical functions of surfactant [101]. Fenoterol has been shown to restore lung phospholipid metabolism, which was altered by sepsis, toward normal [99]. These studies suggest a potential role for β2-agonists as a treatment for surfactant abnormalities in ARDS.
Effects of β2-agonists on epithelial resistance to infection
Nosocomial pneumonia contributes to morbidity and mortality on the intensive care unit [102]. Central to the development of these infections is colonization followed by invasion of the epithelial cell layer. Several studies have investigated the effect of salmeterol on Pseudomonas aeruginosa and Haemophilus influenzae induced epithelial damage [103,104]. In the Pseudomonas study, there was not only reduced pyocyanin-induced cytoplasmic blebbing and reduced mitochondrial damage but also a significant reduction in adherent bacteria. These data suggest that salmeterol has a cytoprotective effect on respiratory epithelial cells, most likely related to maintaining structural integrity of the epithelial cells rather than increasing antibacterial activity. Interestingly, salbutamol and isoproterenol have also been shown to increase monocyte adhesion to human airway epithelial cells in vitro, monocytes being integral to the bacterial immune response in the lung [105]. It is possible, therefore, that β2-agonists have a role to play in the prevention of ventilator associated pneumonia, which commonly complicates ALI/ARDS, by augmenting host epithelial resistance to infection.
Effects of β2-agonists on epithelial wound repair
In ARDS, histological studies have confirmed that there is a physical breach of both the alveolar endothelial and epithelial barriers. This physical damage results in pulmonary oedema that is central to the need for mechanical ventilation. Recovery of the barrier function is vital for effective alveolar epithelial repair. This process is regulated by keratinocyte growth factors (KGFs) and other related cytokines (e.g. IL-1β) that are capable of stimulating alveolar epithelial cell proliferation and migration. In a rat study, pretreatment with KGF before induction of lung injury reduced the severity of injury [106]. The protective capability of KGF is probably due to upregulation of the number of type II alveolar epithelial cells, with a corresponding increase in net alveolar fluid transport [107]. Salbutamol is a potent upregulator of human airway epithelial cells, probably via a protein kinase cascade, and isoproterenol directly increased the migration of bovine epithelial cells, speeding up the closure of mechanically and enzymatically induced wounds [108]. Currently, it is not known whether stimulating epithelial regeneration in humans improves outcome in patients with ARDS.
Effects of β2-agonists on lung mechanics
The physiological consequences of extensive alveolar–epithelial injury include a reduction in pulmonary compliance [5] and increased airway resistance [109], which are associated with an increased work of breathing and requirement for mechanical ventilation. Several studies have shown that both intravenous and nebulized salbutamol reduce peak airway and plateau pressures [109-111]] in patients with ARDS. The reduction in peak airway pressure reflects a reduction in airway resistance due to the bronchodilator effects of β2-agonists. However, the reduction in plateau pressure suggests an improvement in respiratory compliance, through as yet undetermined mechanisms. These studies suggest that β-agonists may have a beneficial role to play in improving respiratory mechanics in patients with ARDS.
Drug delivery and side effects
The optimal route for delivering β2-agonists has not beendetermined. Inhaled or nebulized therapy to mechanically ventilated patients appears attractive because it may reduce the incidence of systemic side effects compared with parenteral treatment. Initial concerns about efficacy of drug deposition into the alveolar space following nebulized or inhaled administration in mechanically ventilated patients with ALI/ARDS [112] have been superseded by a recent study that demonstrated therapeutic levels in pulmonary oedema fluid from patients with ARDS [38]. Atabai and coworkers [38] showed that nebulized salbutamol (3.5 ± 2.6 mg) in patients with ALI achieved a median concentration of 1240 ng/ml (between 10-5 mol/l and 10-6 mol/l) in pulmonary oedema fluid. No studies in patients with ARDS have yet reported the concentration of drug in plasma or BAL fluid following intravenous salbutamol administration, although preliminary studies at our institution have suggested that plasma levels of 10-6 mol/l may be achievable with a continuous infusion of salbutamol at 15 μg/kg per hour. The optimal dose remains to be identified. Higher doses of β2-agonists, used in many experimentalstudies, stimulate both β1- and β2-adrenergic receptors, and it is not possible to determine the relative roles of β1 and β2 receptor stimulation in such studies. However, the finding that β1 stimulation by high-dose terbutaline is associated withdownregulation of alveolar fluid clearance in the ex vivo rat lung [79] supports the hypothesis that β2-adrenergic stimulation is more important.
The administration of β2-agonists can lead to important car-diovascular, metabolic and renal complications. Stimulation of cardiac and vascular β1 and β2 receptors can cause tachycardia, arrhythmias, exacerbation of myocardial ischaemia, pulmonary vasodilation and loss of hypoxic–pulmonary vasoconstriction [113,114]. Metabolic sequelae include hypokalamaemia, hyperinsulinaemia and hyperglycaemia [115]. The use of intravenous β2-agonists for tocolysis duringpregnancy has been associated with the development of maternal pulmonary oedema [116,117]. Studies investigating this phenomenon in vivo in rabbits and humans found that intravenous injection of β2-agonists caused reduced sodium, potassium and water excretion, leading to a reduced haematocrit and intravascular hypervolaemia [118,119]. These adverse effects are usually more marked following intravenous than after nebulized administration. However, in general these drugs are well tolerated in the critically ill. These potentially deleterious effects may limit the potential beneficial effects of β2-agonists described in this review.
Conclusion
There is substantial evidence from in vitro and in vivo animal and human studies suggesting several mechanisms through which β2-agonists may play a potential role in the treatment of patients with ARDS. Clinical experience in the treatment of airflow obstruction in critically ill patients has demonstrated good tolerability and side-effect profiles with these drugs. They are also commercially available as intravenous, inhaled and nebulized formulations, which are relatively inexpensive. To date no randomized controlled clinical trials have yet been completed to confirm the potential benefits of this treatment. However, a double-blind, randomized and placebo-controlled trial using intravenous salbutamol (Beta Agonist Lung Injury TrIal [BALTI]) is reaching completion in the UK, and the ARDS Network in the USA is considering a large multicentre trial using nebulized salbutamol. The results of these trials will hopefully improve our understanding of the application of this treatment in patients with ALI/ARDS.
Competing interests
GDP, AR, DFM and DRT have received support in the past to attend medical conferences from manufacturers of β-agonists.
Abbreviations
ALI = acute lung injury; ARDS = acute respiratory distress syndrome; BAL = bronchoalveolar lavage; fMLP = formyl-methionyl-leucyl-phenylalanine; IL = interleukin; KGF = keratinocyte growth factor; LPS = lipopolysaccharide; SP = surfactant protein; TNF = tumour necrosis factor.
Acknowledgments
Acknowledgements
We would like to thank Stuart Hudson, Medical Illustration Department, Birmingham Heartlands Hospital for producing the illustrations that support this review.
References
- Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- Luhr OR, Antonsen K, Karlsson M, Aardal A, Thorsteinsson A, Frostell CG, Bonde J. Incidence and mortality after acute respiratory failure and acute respiratory distress syndrome in Sweden, Denmark, and Iceland. The ARF Study Group. Am J Respir Crit Care Med. 1999;159:1849–1861. doi: 10.1164/ajrccm.159.6.9808136. [DOI] [PubMed] [Google Scholar]
- Goss CH, Brower RG, Hudson LD, Rubenfeld GD. Incidence of acute lung injury in the United States. Crit Care Med. 2003;31:1607–1611. doi: 10.1097/01.CCM.0000063475.65751.1D. [DOI] [PubMed] [Google Scholar]
- Bersten AD, Davidson K, Nicholas TE, Doyle IR. Respiratory mechanics and surfactant in the acute respiratory distress syndrome. Clin Exp Pharmacol Physiol. 1998;25:955–963. doi: 10.1111/j.1440-1681.1998.tb02352.x. [DOI] [PubMed] [Google Scholar]
- Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet. 1967;2:319–323. doi: 10.1016/S0140-6736(67)90168-7. [DOI] [PubMed] [Google Scholar]
- Herridge MS, Cheung AM, Tansey CM, Matte-Martyn A, Diaz-Granados N, Al-Saidi F, Cooper AB, Guest CB, Mazer CD, Mehta S, Stewart TE, Barr A, Cook D, Slutsky AS, Canadian Critical Care Trials Group One-year outcomes in survivors of the acute respiratory distress syndrome. N Engl J Med. 2003;348:683–693. doi: 10.1056/NEJMoa022450. [DOI] [PubMed] [Google Scholar]
- Baudouin S. Improved survival in ARDS: chance, technology or experience? Thorax. 1998;53:237–238. doi: 10.1136/thx.53.4.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med. 1998;342:1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- McIntyre RC, Pulido EJ, Bensard DD, Shames BD, Abraham E. Thirty years of clinical trials in acute respiratory distress syndrome. Crit Care Med. 2000;28:3314–3331. doi: 10.1097/00003246-200009000-00034. [DOI] [PubMed] [Google Scholar]
- Ingbar DH. Mechanisms of repair and remodeling following acute lung injury. Clin Chest Med. 2000;21:589–616. doi: 10.1016/s0272-5231(05)70168-4. [DOI] [PubMed] [Google Scholar]
- Pugin J, Verghese G, Widmer MC, Matthay MA. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med. 1999;27:304–312. doi: 10.1097/00003246-199902000-00036. [DOI] [PubMed] [Google Scholar]
- Laufe MD, Simon RH, Flint A, Keller JB. Adult respiratory distress syndrome in neutropenic patients. Am J Med. 1986;80:1022–1026. doi: 10.1016/0002-9343(86)90659-5. [DOI] [PubMed] [Google Scholar]
- Chollet-Martin S. Polymorphonuclear neutrophil activation during the acute respiratory distress syndrome. Intensive Care Med. 2000;26:1575–1577. doi: 10.1007/s001340051348. [DOI] [PubMed] [Google Scholar]
- Steinberg KP, Milberg JA, Martin TR, Maunder RJ, Cockrill BA, Hudson LD. Evolution of bronchoalveolar cell populations in the adult respiratory distress syndrome. Am J Respir Crit Care Med. 1994;150:113–122. doi: 10.1164/ajrccm.150.1.8025736. [DOI] [PubMed] [Google Scholar]
- Sinclair DG, Braude S, Haslam PL, Evans TW. Pulmonary endothelial permeability in patients with severe lung injury. Clinical correlates and natural history. Chest. 1994;106:535–539. doi: 10.1378/chest.106.2.535. [DOI] [PubMed] [Google Scholar]
- Doerschuk CM. The role of CD18-mediated adhesion in neutrophil sequestration induced by infusion of activated plasma in rabbits. Am J Respir Cell Mol Biol. 1992;7:140–148. doi: 10.1165/ajrcmb/7.2.140. [DOI] [PubMed] [Google Scholar]
- Kubo H, Graham L, Doyle NA, Quinlan WM, Hogg JC, Doerschuk CM. Complement fragment-induced release of neutrophils from bone marrow and sequestration within pulmonary capillaries in rabbits. Blood. 1998;92:283–290. [PubMed] [Google Scholar]
- Skoutelis AT, Kaleridis V, Athanassiou GM, Kokkinis KI, Missirlis YF, Bassaris HP. Neutrophil deformability in patients with sepsis, septic shock, and adult respiratory distress syndrome. Crit Care Med. 2000;28:2355–2359. doi: 10.1097/00003246-200007000-00029. [DOI] [PubMed] [Google Scholar]
- Doerschuk CM, Mizgerd JP, Kubo H, Qin L, Kumasaka T. Adhesion molecules and cellular biomechanical changes in acute lung injury: Giles F. Filley Lecture. Chest. 1999;Suppl:37S–43S. doi: 10.1378/chest.116.suppl_1.37s-a. [DOI] [PubMed] [Google Scholar]
- Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination [see comments] Crit Care Med. 1999;27:1738–1744. doi: 10.1097/00003246-199909000-00007. [DOI] [PubMed] [Google Scholar]
- Carden D, Xiao F, Moak C, Willis BH, Robinson-Jackson S, Alexander S. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiol. 1998;275:H385–H392. doi: 10.1152/ajpheart.1998.275.2.H385. [DOI] [PubMed] [Google Scholar]
- Sakamaki F, Ishizaka A, Urano T, Sayama K, Nakamura H, Terashima T, Waki Y, Tasaka S, Hasegawa N, Sato K, Nakagawa N, Obata T, Kanazawa M. Effect of a specific neutrophil elastase inhibitor, ONO- on endotoxin-induced acute lung injury. Am J Respir Crit Care Med. 5046;153:391–397. doi: 10.1164/ajrccm.153.1.8542148. [DOI] [PubMed] [Google Scholar]
- Martin TR, Nakamura M, Matute-Bello G. The role of apoptosis in acute lung injury. Crit Care Med. 2003;Suppl:S184–S188. doi: 10.1097/01.CCM.0000057841.33876.B1. [DOI] [PubMed] [Google Scholar]
- Matute-Bello G, Liles WC, Radella F, Steinberg KP, Ruzinski JT, Jonas M, Chi EY, Hudson LD, Martin TR. Neutrophil apoptosis in the acute respiratory distress syndrome. Am Jof Respir Crit Care Med. 1997;156:1969–1977. doi: 10.1164/ajrccm.156.6.96-12081. [DOI] [PubMed] [Google Scholar]
- Hussain N, Wu F, Zhu L, Thrall RS, Kresch MJ. Neutrophil apoptosis during the development and resolution of oleic acid-induced acute lung injury in the rat. Am J Respir Cell Mol Biol. 1998;19:867–874. doi: 10.1165/ajrcmb.19.6.3118. [DOI] [PubMed] [Google Scholar]
- Sookhai S, Wang JJ, McCourt M, Kirwan W, Bouchier-Hayes D, Redmond P. A novel therapeutic strategy for attenuating neutrophil-mediated lung injury in vivo. Ann Surg. 2002;235:285–291. doi: 10.1097/00000658-200202000-00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhingra VK, Uusaro A, Holmes CL, Walley KR. Attenuation of lung inflammation by adrenergic agonists in murine acute lung injury. Anesthesiology. 2001;95:947–953. doi: 10.1097/00000542-200110000-00025. [DOI] [PubMed] [Google Scholar]
- Wu CC, Liao MH, Chen SJ, Chou TC, Chen A, Yen MH. Terbutaline prevents circulatory failure and mitigates mortality in rodents with endotoxemia. Shock. 2000;14:60–67. doi: 10.1097/00024382-200014010-00011. [DOI] [PubMed] [Google Scholar]
- Masclans JR, Barbera JA, MacNee W, Pavia J, Piera C, Lomena F, Chung KF, Roca J, Rodriguez-Roisin R. Salbutamol reduces pulmonary neutrophil sequestration of platelet-activating factor in humans. Am J Respir Crit Care Med. 1996;154:529–532. doi: 10.1164/ajrccm.154.2.8756833. [DOI] [PubMed] [Google Scholar]
- Bloemen PG, van den Tweel MC, Henricks PA, Engels F, Kester MH, van de Loo PG, Blomjous FJ, Nijkamp FP. Increased cAMP levels in stimulated neutrophils inhibit their adhesion to human bronchial epithelial cells. Am J Physiol. 1997;272:L580–L587. doi: 10.1152/ajplung.1997.272.4.L580. [DOI] [PubMed] [Google Scholar]
- Blease K, Burke-Gaffney A, Hellewell PG. Modulation of cell adhesion molecule expression and function on human lung microvascular endothelial cells by inhibition of phosphodiesterases 3 and 4. Br J Pharmacol. 1998;124:229–237. doi: 10.1038/sj.bjp.0701833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derian CK, Santulli RJ, Rao PE, Solomon HF, Barrett JA. Inhibition of chemotactic peptide-induced neutrophil adhesion to vascular endothelium by cAMP modulators. J Immunol. 1995;154:308–317. [PubMed] [Google Scholar]
- Diez-Fraile A, Meyer E, Massart-Leen AM, Burvenich C. Effect of isoproterenol and dexamethasone on the lipopolysaccharide induced expression of CD11b on bovine neutrophils. Vet Immunol Immunopathol. 2000;76:151–156. doi: 10.1016/S0165-2427(00)00199-9. [DOI] [PubMed] [Google Scholar]
- Harvath L, Robbins JD, Russell AA, Seamon KB. cAMP and human neutrophil chemotaxis. Elevation of cAMP differentially affects chemotactic responsiveness. J Immunol. 1991;146:224–232. [PubMed] [Google Scholar]
- Perkins GD, Rea W, Gao F, Thickett DR. The effect of beta agonists on neutrophil adhesion molecule expression [abstract] Thorax. 2002;Suppl 3:S87. [Google Scholar]
- Silvestri M, Oddera S, Lantero S, Rossi GA. beta 2-agonist-induced inhibition of neutrophil chemotaxis is not associated with modification of LFA-1 and Mac-1 expression or with impairment of polymorphonuclear leukocyte antibacterial activity. Respir Med. 1999;93:416–423. doi: 10.1053/rmed.1999.0584. [DOI] [PubMed] [Google Scholar]
- Silvestre JS, Tamarat R, Ebrahimian TG, Le Roux A, Clergue M, Emmanuel F, Duriez M, Schwartz B, Branellec D, Levy BI. Vascular endothelial growth factor-B promotes in vivo angiogenesis. Circ Res. 2003;93:114–123. doi: 10.1161/01.RES.0000081594.21764.44. [DOI] [PubMed] [Google Scholar]
- Atabai K, Ware LB, Snider ME, Koch P, Daniel B, Nuckton TJ, Matthay MA. Aerosolized beta(2)-adrenergic agonists achieve therapeutic levels in the pulmonary edema fluid of ventilated patients with acute respiratory failure. Intensive Care Med. 2002;28:705–711. doi: 10.1007/s00134-002-1282-x. [DOI] [PubMed] [Google Scholar]
- Llewellyn-Jones CG, Stockley RA. The effects of beta 2-agonists and methylxanthines on neutrophil function in vitro. Eur Respir J. 1994;7:1460–1466. doi: 10.1183/09031936.94.07081460. [DOI] [PubMed] [Google Scholar]
- Lee E, Smigh J, Robertson P, Reynolds K, Opesan K, Kilfeather SA. Salmeterol and inhibitors of phosphodiesterase 4(PDE4) induce apoptosis in neutrophils from asthmatics: beta-adrenergic receptor-mediated salmeterol activie and additive effects with PDE4 inhibitors [abstract] Am J Respir Cell Mol Biol. 1999;159:A329. [Google Scholar]
- Dincer HE, Gangopadhyay N, Wang R, Uhal BD. Norepinephrine induces alveolar epithelial apoptosis mediated by alpha-, beta-, and angiotensin receptor activation. Am J Physiol Lung Cell Mol Physiol. 2001;281:L624–L630. doi: 10.1152/ajplung.2001.281.3.L624. [DOI] [PubMed] [Google Scholar]
- Braga PC, Mancini L, Guffanti EE, Dal S, Sala M, Reggio S. Effects of nedocromil sodium on the oxidative burst of polymorphonuclear leukocytes: comparison with salbutamol. Drugs Exp Clin Res. 1997;23:33–38. [PubMed] [Google Scholar]
- Opdahl H, Benestad HB, Nicolaysen G. Effect of beta-adrenergic agents on human neutrophil granulocyte activation with N-formyl-methionyl-leucyl-phenylalanine and phorbol myristate acetate. Pharmacol Toxicol. 1993;72:221–228. doi: 10.1111/j.1600-0773.1993.tb01640.x. [DOI] [PubMed] [Google Scholar]
- Mirza ZN, Kato M, Kimura H, Tachibana A, Fujiu T, Suzuki M, Mochizuki H, Tokuyama K, Morikawa A. Fenoterol inhibits superoxide anion generation by human polymorphonuclear leukocytes via beta-adrenoceptor-dependent and -independent mechanisms. Ann Allergy Asthma Immunol. 2002;88:494–500. doi: 10.1016/s1081-1206(10)62388-5. [DOI] [PubMed] [Google Scholar]
- Gillissen A, Wickenburg D, van Zwoll D, Schultze-Werninghaus G. Beta-2-agonists have antioxidant function in vitro. 2. The effect of beta-2-agonists on oxidant-mediated cytotoxicity and on superoxide anion generated by human polymorphonuclear leukocytes. Respiration. 1997;64:23–28. doi: 10.1159/000196638. [DOI] [PubMed] [Google Scholar]
- Park WY, Goodman RB, Steinberg KP, Ruzinski JT, Radella F, Park DR, Pugin J, Skerrett SJ, Hudson LD, Martin TR. Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;164:1896–1903. doi: 10.1164/ajrccm.164.10.2104013. [DOI] [PubMed] [Google Scholar]
- Donnelly SC, Strieter RM, Kunkel SL, Walz A, Steedman D, Grant IS, Pollok AJ, Carter DC, Haslett C. Chemotactic cytokines in the established adult respiratory distress syndrome and at-risk patients. Chest. 1994;Suppl:98S–99S. doi: 10.1378/chest.105.3_supplement.98s. [DOI] [PubMed] [Google Scholar]
- Martin TR. Lung cytokines and ARDS: Roger S. Mitchell Lecture. [review] Chest. 1999;Suppl:2S–8S. doi: 10.1378/chest.116.suppl_1.2S. [DOI] [PubMed] [Google Scholar]
- Goodman RB, Strieter RM, Martin DP, Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD, Martin TR. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:602–611. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kajikawa O, Martin TR, Sharar SR, Harlan JM, Winn RK. The role of leukocyte emigration and IL-8 on the development of lipopolysaccharide-induced lung injury in rabbits. J Immunol. 1998;161:5704–5709. [PubMed] [Google Scholar]
- Modelska K, Pittet JF, Folkesson HG, Courtney Broaddus V, Matthay MA. Acid-induced lung injury. Protective effect of anti-interleukin-8 pretreatment on alveolar epithelial barrier function in rabbits. Am J Respir Crit Care Med. 1999;160:1450–1456. doi: 10.1164/ajrccm.160.5.9901096. [DOI] [PubMed] [Google Scholar]
- Folkesson HG, Matthay MA, Hebert CA, Broaddus VC. Acid aspiration-induced lung injury in rabbits is mediated by interleukin-8-dependent mechanisms. J Clin Invest. 1995;96:107–116. doi: 10.1172/JCI118009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterlund A, Linden M, Larsson K. Effects of beta2-agonists and budesonide on interleukin-1beta and leukotriene B4 secretion: studies of human monocytes and alveolar macrophages. J Asthma. 1998;35:565–573. doi: 10.3109/02770909809048959. [DOI] [PubMed] [Google Scholar]
- Severn A, Rapson NT, Hunter CA, Liew FY. Regulation of tumor necrosis factor production by adrenaline and beta-adrenergic agonists. J Immunol. 1992;148:3441–3445. [PubMed] [Google Scholar]
- Gu Y, Seidel A. Influence of salbutamol and isoproterenol on the production of TNF and reactive oxygen species by bovine alveolar macrophages and calcitriol differentiated HL-60 cells. Immunopharmacol Immunotoxicol. 1996;18:115–128. doi: 10.3109/08923979609007114. [DOI] [PubMed] [Google Scholar]
- Monastra G, Secchi EF. Beta-adrenergic receptors mediate in vivo the adrenaline inhibition of lipopolysaccharide-induced tumor necrosis factor release. Immunol Lett. 1993;38:127–130. doi: 10.1016/0165-2478(93)90177-4. [DOI] [PubMed] [Google Scholar]
- Sekut L, Champion BR, Page K, Menius JA, Jr, Connolly KM. Anti-inflammatory activity of salmeterol: down-regulation of cytokine production. Clin Exp Immunol. 1995;99:461–466. doi: 10.1111/j.1365-2249.1995.tb05573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Poll T, Jansen J, Endert E, Sauerwein HP, van Deventer SJ. Noradrenaline inhibits lipopolysaccharide-induced tumor necrosis factor and interleukin 6 production in human whole blood. Infect Immun. 1994;62:2046–2050. doi: 10.1128/iai.62.5.2046-2050.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au BT, Teixeira MM, Collins PD, Williams TJ. Effect of PDE4 inhibitors on zymosan-induced IL-8 release from human neutrophils: synergism with prostanoids and salbutamol. Br J Pharmacol. 1998;123:1260–1266. doi: 10.1038/sj.bjp.0701723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CY, Tsai CS, Hsu PC, Wu CT, Wong CS, Ho ST. Dobutamine modulates lipopolysaccharide-induced macrophage inflammatory protein-1alpha and interleukin-8 production in human monocytes. Anesth Analg. 2003;97:210–215. doi: 10.1213/01.ane.0000066257.38180.04. [DOI] [PubMed] [Google Scholar]
- Izeboud CA, Vermeulen RM, Zwart A, Voss HP, van Miert AS, Witkamp RF. Stereoselectivity at the beta2-adrenoceptor on macrophages is a major determinant of the anti-inflammatory effects of beta2-agonists. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:184–189. doi: 10.1007/s002100000281. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kim YK, Govindarajan A, Baba A, Binnie M, Marco Ranieri V, Liu M, Slutsky AS. Effect of adrenoreceptors on endotoxin-induced cytokines and lipid peroxidation in lung explants. Am J Respir Crit Care Med. 1999;160:1703–1710. doi: 10.1164/ajrccm.160.5.9903068. [DOI] [PubMed] [Google Scholar]
- van der Poll T, Calvano SE, Kumar A, Coyle SM, Lowry SF. Epinephrine attenuates down-regulation of monocyte tumor necrosis factor receptors during human endotoxemia. J Leukoc Biol. 1997;61:156–160. doi: 10.1002/jlb.61.2.156. [DOI] [PubMed] [Google Scholar]
- Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med. 1982;3:35–56. [PubMed] [Google Scholar]
- Minnear FL, DeMichele MA, Leonhardt S, Andersen TT, Teitler M. Isoproterenol antagonizes endothelial permeability induced by thrombin and thrombin receptor peptide. J Appl Physiol. 1993;75:1171–1179. doi: 10.1152/jappl.1993.75.3.1171. [DOI] [PubMed] [Google Scholar]
- Minnear FL, DeMichele MA, Moon DG, Rieder CL, Fenton JW. Isoproterenol reduces thrombin-induced pulmonary endothelial permeability in vitro. Am J Physiol. 1989;257:H1613–H1623. doi: 10.1152/ajpheart.1989.257.5.H1613. [DOI] [PubMed] [Google Scholar]
- Sigurdsson GH, Christenson JT. Influence of terbutaline on endotoxin-induced lung injury. Circ Shock. 1988;25:153–163. [PubMed] [Google Scholar]
- Ding Z, Jiang M, Li S, Zhang Y. Vascular barrier-enhancing effect of an endogenous beta-adrenergic agonist. Inflammation. 1995;19:1–8. doi: 10.1007/BF01534375. [DOI] [PubMed] [Google Scholar]
- Basran GS, Hardy JG, Woo SP, Ramasubramanian R, Byrne AJ. Beta-2-adrenoceptor agonists as inhibitors of lung vascular permeability to radiolabelled transferrin in the adult respiratory distress syndrome in man. Eur J Nucl Med. 1986;12:381–384. doi: 10.1007/BF00252194. [DOI] [PubMed] [Google Scholar]
- Abraham E. Coagulation abnormalities in acute lung injury and sepsis. Am J Respir Cell Mol Biol. 2000;22:401–404. doi: 10.1165/ajrcmb.22.4.f184. [DOI] [PubMed] [Google Scholar]
- Prabhakaran P, Ware LB, White KE, Cross MT, Matthay MA, Olman MA. Elevated levels of plasminogen activator inhibitor-1 in pulmonary edema fluid are associated with mortality in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L20–L28. doi: 10.1152/ajplung.00312.2002. [DOI] [PubMed] [Google Scholar]
- Ware LB, Fang X, Matthay MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2003;285:L514–L521. doi: 10.1152/ajplung.00442.2002. [DOI] [PubMed] [Google Scholar]
- Stein CM, Brown N, Vaughan DE, Lang CC, Wood AJ. Regulation of local tissue-type plasminogen activator release by endothelium-dependent and endothelium-independent agonists in human vasculature. J Am Coll Cardiol. 1998;32:117–122. doi: 10.1016/S0735-1097(98)00210-1. [DOI] [PubMed] [Google Scholar]
- Chandler WL, Levy WC, Stratton JR. The circulatory regulation of TPA and UPA secretion, clearance, and inhibition during exercise and during the infusion of isoproterenol and phenylephrine. Circulation. 1995;92:2984–2994. doi: 10.1161/01.cir.92.10.2984. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev. 2002;82:569–600. doi: 10.1152/physrev.00003.2002. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Okaniwa G, Nakada T, Nishimura T, Fujimura S, Matthay MA. Alveolar fluid clearance in the resected human lung. Am J Respir Crit Care Med. 1994;150:305–310. doi: 10.1164/ajrccm.150.2.8049807. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Suzuki S, Usuda K, Handa M, Okaniwa G, Nakada T, Fujimura S, Matthay MA. Preservation of alveolar epithelial fluid transport mechanisms in rewarmed human lung after severe hypothermia. J Appl Physiol. 1996;80:1681–1686. doi: 10.1152/jappl.1996.80.5.1681. [DOI] [PubMed] [Google Scholar]
- Tibayan FA, Chesnutt AN, Folkesson HG, Eandi J, Matthay MA. Dobutamine increases alveolar liquid clearance in ventilated rats by beta-2 receptor stimulation. Am J Respir Crit Care Med. 1997;156:438–444. doi: 10.1164/ajrccm.156.2.9609141. [DOI] [PubMed] [Google Scholar]
- Sakuma T, Tuchihara C, Ishigaki M, Osanai K, Nambu Y, Toga H, Takahashi K, Ohya N, Kurihara T, Matthay MA. Denopamine, a beta1-adrenergic agonist, increases alveolar fluid clearance in ex vivo rat and guinea pig lungs. J Appl Physiol. 2001;90:10–16. doi: 10.1152/jappl.2001.90.1.10. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Wiener-Kronish JP. Intact epithelial barrier function is critical for the resolution of alveolar edema in humans. Am Rev Respir Dis. 1990;142:1250–1257. doi: 10.1164/ajrccm/142.6_Pt_1.1250. [DOI] [PubMed] [Google Scholar]
- Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376–1383. doi: 10.1164/ajrccm.163.6.2004035. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Folkesson HG, Verkman AS. Salt and water transport across alveolar and distal airway epithelia in the adult lung. Am J Physiol. 1996;270:L487–L503. doi: 10.1152/ajplung.1996.270.4.L487. [DOI] [PubMed] [Google Scholar]
- Garat C, Meignan M, Matthay MA, Luo DF, Jayr C. Alveolar epithelial fluid clearance mechanisms are intact after moderate hyperoxic lung injury in rats. Chest. 1997;111:1381–1388. doi: 10.1378/chest.111.5.1381. [DOI] [PubMed] [Google Scholar]
- Pittet JF, Lu LN, Morris DG, Modelska K, Welch WJ, Carey HV, Roux J, Matthay MA. Reactive nitrogen species inhibit alveolar epithelial fluid transport after hemorrhagic shock in rats. J Immunol. 2001;166:6301–6310. doi: 10.4049/jimmunol.166.10.6301. [DOI] [PubMed] [Google Scholar]
- Modelska K, Matthay MA, Brown LA, Deutch E, Lu LN, Pittet JF. Inhibition of beta-adrenergic-dependent alveolar epithelial clearance by oxidant mechanisms after hemorrhagic shock. Am J Physiol. 1999;276:L844–L857. doi: 10.1152/ajplung.1999.276.5.L844. [DOI] [PubMed] [Google Scholar]
- Laffon M, Lu LN, Modelska K, Matthay MA, Pittet JF. alpha-adrenergic blockade restores normal fluid transport capacity of alveolar epithelium after hemorrhagic shock. Am J Physiol. 1999;277:L760–L768. doi: 10.1152/ajplung.1999.277.4.L760. [DOI] [PubMed] [Google Scholar]
- Frank JA, Wang Y, Osorio O, Matthay MA. Beta-adrenergic agonist therapy accelerates the resolution of hydrostatic pulmonary edema in sheep and rats. J Appl Physiol. 2000;89:1255–1265. doi: 10.1152/jappl.2000.89.4.1255. [DOI] [PubMed] [Google Scholar]
- Campbell AR, Folkesson HG, Berthiaume Y, Gutkowska J, Suzuki S, Matthay MA. Alveolar epithelial fluid clearance persists in the presence of moderate left atrial hypertension in sheep. J Appl Physiol. 1999;86:139–151. doi: 10.1152/jappl.1999.86.1.139. [DOI] [PubMed] [Google Scholar]
- Lane SM, Maender KC, Awender NE, Maron MB. Adrenal epinephrine increases alveolar liquid clearance in a canine model of neurogenic pulmonary edema. Am J Respir Crit Care Med. 1998;158:760–768. doi: 10.1164/ajrccm.158.3.9802031. [DOI] [PubMed] [Google Scholar]
- Lasnier JM, Wangensteen OD, Schmitz LS, Gross CR, Ingbar DH. Terbutaline stimulates alveolar fluid resorption in hyperoxic lung injury. J Appl Physiol. 1996;81:1723–1729. doi: 10.1152/jappl.1996.81.4.1723. [DOI] [PubMed] [Google Scholar]
- Saldias FJ, Comellas A, Ridge KM, Lecuona E, Sznajder JI. Iso-proterenol improves ability of lung to clear edema in rats exposed to hyperoxia. J Appl Physiol. 1999;87:30–35. doi: 10.1152/jappl.1999.87.1.30. [DOI] [PubMed] [Google Scholar]
- Saldias FJ, Lecuona E, Comellas AP, Ridge KM, Rutschman DH, Sznajder JI. beta-adrenergic stimulation restores rat lung ability to clear edema in ventilator-associated lung injury. Am J Respir Crit Care Med. 2000;162:282–287. doi: 10.1164/ajrccm.162.1.9809058. [DOI] [PubMed] [Google Scholar]
- Vivona ML, Matthay M, Chabaud MB, Friedlander G, Clerici C. Hypoxia reduces alveolar epithelial sodium and fluid transport in rats: reversal by beta-adrenergic agonist treatment. Am J Respir Cell Mol Biol. 2001;25:554–561. doi: 10.1165/ajrcmb.25.5.4420. [DOI] [PubMed] [Google Scholar]
- Planes C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem. 2002;277:47318–47324. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- Sartori C, Allemann Y, Duplain H, Lepori M, Egli M, Lipp E, Hutter D, Turini P, Hugli O, Cook S, Nicod P, Scherrer U. Salmeterol for the prevention of high-altitude pulmonary edema. N Engl J Med. 2002;346:1631–1636. doi: 10.1056/NEJMoa013183. [DOI] [PubMed] [Google Scholar]
- Gunther A, Ruppert C, Schmidt R, Markart P, Grimminger F, Walmrath D, Seeger W. Surfactant alteration and replacement in acute respiratory distress syndrome. Respir Res. 2001;2:353–364. doi: 10.1186/rr86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd VL, Lopez JP. The role of surfactant-associated protein A in pulmonary host defense. Immunol Res. 2001;23:111–120. doi: 10.1385/IR:23:2-3:111. [DOI] [PubMed] [Google Scholar]
- Kumar VH, Christian C, Kresch MJ. Effects of salmeterol on secretion of phosphatidylcholine by alveolar type II cells. Life Sci. 2000;66:1639–1646. doi: 10.1016/S0024-3205(00)00483-5. [DOI] [PubMed] [Google Scholar]
- von Wichert P, Muller B, Meyer-Ingold W. Influence of a beta-adrenergic agonist on septic shock-induced alterations of phosphatidylcholine metabolism in rat lung. Lung. 1988;166:257–267. doi: 10.1007/BF02714056. [DOI] [PubMed] [Google Scholar]
- Chen Q, Bates SR, Fisher AB. Secretagogues increase the expression of surfactant protein A receptors on lung type II cells. J Biol Chem. 1996;271:25277–25283. doi: 10.1074/jbc.271.41.25277. [DOI] [PubMed] [Google Scholar]
- Gobran LI, Rooney SA. Regulation of SP-B and SP-C secretion in rat type II cells in primary culture. Am J Physiol Lung Cell Mol Physiol. 2001;281:L1413–L1419. doi: 10.1152/ajplung.2001.281.6.L1413. [DOI] [PubMed] [Google Scholar]
- Fagon JY, Chastre J, Vuagnat A, Trouillet JL, Novara A, Gibert C. Nosocomial pneumonia and mortality among patients in intensive care units. JAMA. 1996;275:866–869. doi: 10.1001/jama.275.11.866. [DOI] [PubMed] [Google Scholar]
- Dowling RB, Johnson M, Cole PJ, Wilson R. Effect of salmeterol on Haemophilus influenzae infection of respiratory mucosa in vitro. Eur Respir J. 1998;11:86–90. doi: 10.1183/09031936.98.11010086. [DOI] [PubMed] [Google Scholar]
- Dowling RB, Johnson M, Cole PJ, Wilson R. Effect of fluticasone propionate and salmeterol on Pseudomonas aeruginosa infection of the respiratory mucosa in vitro. Eur Respir J. 1999;14:363–369. doi: 10.1034/j.1399-3003.1999.14b21.x. [DOI] [PubMed] [Google Scholar]
- Romberger DJ, Heires P, Rennard SI, Wyatt TA. beta-Adrenergic agonist modulation of monocyte adhesion to airway epithelial cells in vitro. Am J Physiol Lung Cell Mol Physiol. 2000;278:L139–L147. doi: 10.1152/ajplung.2000.278.1.L139. [DOI] [PubMed] [Google Scholar]
- Just N, Tillie-Leblond I, Guery BP, Fourneau C, Tonnel AB, Gosset P. Keratinocyte growth factor (KGF) decreases ICAM-1 and VCAM-1 cell expression on bronchial epithelial cells. Clin Exp Immunol. 2003;132:61–69. doi: 10.1046/j.1365-2249.2003.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Folkesson HG, Jayr C, Ware LB, Matthay MA. Alveolar epithelial fluid transport can be simultaneously upregulated by both KGF and beta-agonist therapy. J Appl Physiol. 1999;87:1852–1860. doi: 10.1152/jappl.1999.87.5.1852. [DOI] [PubMed] [Google Scholar]
- Spurzem JR, Gupta J, Veys T, Kneifl KR, Rennard SI, Wyatt TA. Activation of protein kinase A accelerates bovine bronchial epithelial cell migration. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1108–L1116. doi: 10.1152/ajplung.00148.2001. [DOI] [PubMed] [Google Scholar]
- Wright PE, Carmichael LC, Bernard GR. Effect of bronchodilators on lung mechanics in the acute respiratory distress syndrome (ARDS) Chest. 1994;106:1517–1523. doi: 10.1378/chest.106.5.1517. [DOI] [PubMed] [Google Scholar]
- Morina P, Herrera M, Venegas J, Mora D, Rodriguez M, Pino E. Effects of nebulized salbutamol on respiratory mechanics in adult respiratory distress syndrome. Intensive Care Med. 1997;23:58–64. doi: 10.1007/s001340050291. [DOI] [PubMed] [Google Scholar]
- Pesenti A, Pelosi P, Rossi N, Aprigliano M, Brazzi L, Fumagalli R. Respiratory mechanics and bronchodilator responsiveness in patients with the adult respiratory distress syndrome. Crit Care Med. 1993;21:78–83. doi: 10.1097/00003246-199301000-00016. [DOI] [PubMed] [Google Scholar]
- Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, Raventos AA, Lemaire F, Long W, Zaccardelli DS, Pattishall EN. Aerosolized surfactant in adults with sepsis-induced acute respiratory distress syndrome. Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group. N Engl J Med. 1996;334:1417–1421. doi: 10.1056/NEJM199605303342201. [DOI] [PubMed] [Google Scholar]
- Conover WB, Benumof JL, Key TC. Ritodrine inhibition of hypoxic pulmonary vasoconstriction. Am J Obstet Gynecol. 1983;146:652–656. doi: 10.1016/0002-9378(83)91007-4. [DOI] [PubMed] [Google Scholar]
- Harris L. Comparison of the effect on blood gases, ventilation, and perfusion of isoproterenol-phenylephrine and salbutamol aerosols in chronic bronchitis with asthma. J Allergy Clin Immunol. 1972;49:63–71. doi: 10.1016/0091-6749(72)90057-7. [DOI] [PubMed] [Google Scholar]
- Neville A, Palmer JB, Gaddie J, May CS, Palmer KN, Murchison LE. Metabolic effects of salbutamol: comparison of aerosol and intravenous administration. BMJ. 1977;1:413–414. doi: 10.1136/bmj.1.6058.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JF, Grant A, Keirse MJ, Chalmers I. Beta-mimetics in preterm labour: an overview of the randomized controlled trials. Br J Obstet Gynaecol. 1988;95:211–222. doi: 10.1111/j.1471-0528.1988.tb06860.x. [DOI] [PubMed] [Google Scholar]
- Bader AM, Boudier E, Martinez C, Langer B, Sacrez J, Cherif Y, Messier M, Schlaeder G. Etiology and prevention of pulmonary complications following beta-mimetic mediated tocolysis. Eur J Obstet Gynecol Reprod Biol. 1998;80:133–137. doi: 10.1016/S0301-2115(98)00105-5. [DOI] [PubMed] [Google Scholar]
- Grospietsch G, Fenske M, Girndt J, Uhlich E, Kuhn W. The renin–angiotensin–aldosterone system, antidiuretic hormone levels and water balance under tocolytic therapy with Fenoterol and Verapamil. Int J Gynaecol Obstet. 1980;17:590–595. doi: 10.1002/j.1879-3479.1980.tb00214.x. [DOI] [PubMed] [Google Scholar]
- Grospietsch G, Ulbrich R, Saul U, Fenske M, Ensink FB, Kuhn W. Urinary excretion, osmolarity and electrolytes after bolus-injection of fenoterol in female rabbits. Gynecol Obstet Invest. 1984;17:317–325. doi: 10.1159/000299169. [DOI] [PubMed] [Google Scholar]