

Abstract
We used the allele-specific PCR-double digestion method on peripheral myelin protein 22 (PMP22) to determine duplication and deletion mutations in the proband and family members of one family with Charcot-Marie-Tooth disease type 1 and one family with hereditary neuropathy with liability to pressure palsies. The proband and one subclinical family member from the Charcot-Marie-Tooth disease type 1 family had a PMP22 gene duplication; one patient from the hereditary neuropathy with liability to pressure palsies family had a PMP22 gene deletion. Electron microscopic analysis of ultrathin sections of the superficial peroneal nerve from the two probands demonstrated demyelination and myelin sheath hyperplasia, as well as an ‘onion-like’ structure in the Charcot-Marie-Tooth disease type 1A patient. We observed an irregular thickened myelin sheath and ‘mouse-nibbled’-like changes in the patient with hereditary neuropathy with liability to pressure palsies. In the Charcot-Marie-Tooth disease type 1A patient, nerve electrophysiological examination revealed moderate-to-severe reductions in the motor and sensory conduction velocities of the bilateral median nerve, ulnar nerve, tibial nerve, and sural nerve. Moreover, the compound muscle action potential amplitude was decreased. In the patient with hereditary neuropathy with liability to pressure palsies, the nerve conduction velocity of the bilateral tibial nerve and sural nerve was moderately reduced, and the nerve conduction velocity of the median nerve and ulnar nerve of both upper extremities was slightly reduced.
Keywords: Charcot-Marie-Tooth disease, hereditary neuropathy with liability to pressure palsies, peripheral myelin protein 22, gene mutation, PCR-double digestion method, myelin sheath, action potential, neuropathology, neural regeneration
Research Highlights
(1) We used allele-specific PCR-double digestion to diagnose peripheral myelin protein 22 (PMP22) gene duplication in Charcot-Marie-Tooth disease type 1A patients and PMP22 gene deletion in hereditary neuropathy with liability to pressure palsies patients.
(2) Allele-specific PCR-double digestion is a rapid screening tool suitable for genetic diagnosis of patients with Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies.
(3) Distinctive nerve electrophysiological and pathological changes can guide the genetic diagnosis in Charcot-Marie-Tooth disease type 1A and hereditary neuropathy with liability to pressure palsies.
Abbreviations
CMT, Charcot-Marie-Tooth disease; PMP22, peripheral myelin protein 22; HNPP, hereditary neuropathy with liability to pressure palsies; REP, repeat sequence
INTRODUCTION
Charcot-Marie-Tooth disease (CMT), also called hereditary motor and sensory neuropathy, is a group of common peripheral nerve-related, single-gene-inheritance diseases with the characteristics of highly hereditary and clinical heterogeneity[1]. The incidence is 17–40/10 000[1]. In the clinic, CMT can be divided into myelin sheath type (CMT1) and axon type (CMT2) according to electrophysiological and pathological characteristics[2]. The modes of inheritance are autosomal dominant, autosomal recessive, and X-linked[3]. CMT1A, which is caused by duplication of the peripheral myelin protein 22 (PMP22) gene, is the most common subtype, accounting for about 50–70% of the total number of CMT cases. On the other hand, PMP22 gene deletion is associated with about 70% cases of hereditary neuropathy with liability to pressure palsies (HNPP)[4,5].
The reciprocal gene duplication and deletion that lead to CMT1A and HNPP, respectively, occur because of the presence of a 30-kb low-copy repeat sequence (REP) upstream and downstream of PMP22, with 98.5% homology between the two. During meiosis, a staggered arrangement between the distal and proximal REPs can result in unequal arm crossing-over of non-sister chromatids of the homologous chromosomes 17[6,7]. CMT1A and HNPP are the complementary outcomes of a large-fragment unequal arm crossing-over[6,7].
Here, we used allele-specific PCR-double digestion to screen for PMP22 gene duplications and deletions in one CMT1 family and one HNPP family. We used the method of Lin et al[8], which involves amplification with pairs of primers specific to the proximal and distal REPs, followed by EcoRI and Nsil digestion. We also analyzed the clinical, electrophysiological, and pathological characteristics of patients from these families.
RESULTS
Patients
The family pedigrees and affected status of the individuals are shown in Figure 1.
Figure 1.
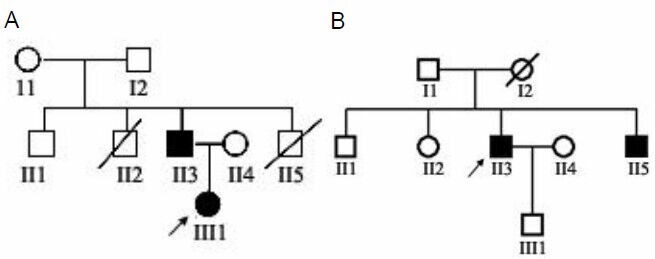
Family pedigrees.
(A) CMT1A family; (B) HNPP family.
↗: Proband; ▪: male patient; ●: female patient; □: normal male; ○: normal female;  : deceased male; ∅: deceased female.
: deceased male; ∅: deceased female.
CMT1: Charcot-Marie-Tooth disease myelin sheath type; HNPP: hereditary neuropathy with liability to pressure palsies.
Clinical manifestations of CMT1A and HNPP
The female proband of the CMT1A family, A-III1, aged 8 years, learned to walk at the age of 2. Both lower limbs were weak, especially the left lower limb. She fell often when walking, and her posture was abnormal. These symptoms worsened with time. Upon examination, the muscle force of the distal end of both lower extremities was 4+. Dorsiflexion and weakness of both feet was visible. Superficial sensation at the distal end of the lower extremities was slightly reduced. Tendon reflex was absent. There was steppage gait, ‘stork-leg’ appearance, and strephenopodia. Nerve electrophysiological examination revealed that the motor and sensory conduction velocities of the bilateral median nerve, ulnar nerve, tibial nerve, and sural nerve were moderately to severely reduced, at 11.2–23.6 m/s. Compound muscle action potential amplitude was decreased. The neuropathological characteristics were as follows: a reduced number of myelinated nerve fibers and focal irregular thickening or thinning of the myelin sheath, showing an onion-like structure. There was also an enlarged gap in the myelin sheath around the axons (Figure 2A).
Figure 2.
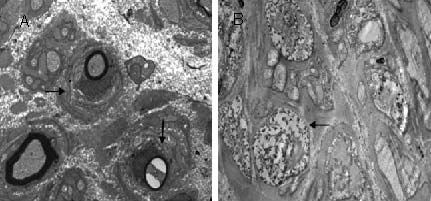
Ultrastructural nerve pathology (transmission electron microscopy, × 5 000) in CMT1A and HNPP.
(A) CMT1A: Superficial peroneal nerve pathology indicated by irregular thickening of the myelin sheath of myelinated nerve fibers, onion-like structures (→), and enlarged gaps in the myelin sheath surrounding the axons (↓).
(B) HNPP: Superficial peroneal nerve pathology indicated by a degenerated myelin sheath of myelinated nerve fibers, irregular thickening, loosing, edema, and a ‘mouse-nibbled’ appearance (←).
CMT1: Charcot-Marie-Tooth disease myelin sheath type; HNPP: hereditary neuropathy with liability to pressure palsies.
The father of the proband, A-II3, was aged 35 years. He had a normal clinical phenotype. However, nerve electrophysiological examination revealed that the motor and sensory conduction velocities of the bilateral median nerve, ulnar nerve, tibial nerve, and sural nerve were moderately reduced, at 22.2–30.4 m/s, which suggests that he was subclinically affected.
The male proband of the HNPP family, B-II3, aged 40 years, had experienced four episodes of double lower limb weakness since the age of 19, each lasting several months. During the latest attack, the muscle forces of his left lower limb and right lower limb were 4 and 5–, respectively. A weak dorsiflexion force, absent bilateral knee and ankle reflexes, and left tibial muscle atrophy were noted. Nerve electrophysiological examination revealed that the motor and sensory conduction velocities of his bilateral tibial nerve and sural nerve were moderately reduced, at 31.3–37.4 m/s. The motor and sensory conduction velocities of the median nerve and ulnar nerve of both upper limbs were slightly reduced, at 38–45.5 m/s. Pathological characteristics of the superficial peroneal nerve included a decreased density of myelinated nerve fibers, irregular thickening of the myelin sheath, a ‘mouse-nibbled’ appearance, axonal edema, and vacuolated degeneration (Figure 2B).
A brother of the proband, B-II5, aged 37 years, had experienced recurrent lower limb weakness and steppage gait since adolescence. Electrophysiological studies were not conducted and no blood was collected. He was diagnosed with HNPP in the clinic.
PMP22 gene duplication and deletion analysis
We performed allele-specific PCR-double digestion analysis of PMP22 on the CMT1 and HNPP probands and family members, a normal control, and a CMT1A-positive control. The results are shown in Figure 3. A 3.57-kb fragment was amplified from the distal REP region using the primers Hot-DF + Hot-DR, and a 3.6-kb fragment was amplified from the proximal REP region using primers Hot-PF + Hot-PR. Both fragments were obtained from all individuals (Figure 3A). In addition, a 3.58-kb duplicated junction fragment could be amplified using primers Hot-DF + Hot-PR from the CMT1 patients A-III1, A-II3, and the CMT1A positive control only. Similarly, a 3.6-kb deleted junction fragment could be amplified using primers Hot-PF + Hot-DR from the HNPP patient B-II3 only (Figure 3B).
Figure 3.
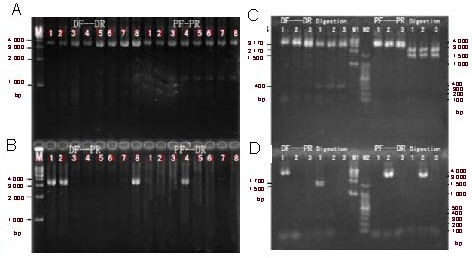
Electropherograms showing PCR and double digestion of the PMP22 allele.
(A, B) PCR amplification products with the indicated primer pairs. 1: A-III1; 2: A-II3; 3: A-II4; 4: B-II3; 5: B-II4; 6: B-III1; 7: normal control; 8: CMT1A positive control. (C, D) PCR amplification products with the indicated primer pairs (the first three lanes of each half) and EcoRI and NsiI double digestion of the PCR products (the second three lanes of each half). 1: A-III1; 2: B-II3; 3: normal control; M: DNA marker.
CMT1: Charcot-Marie-Tooth disease myelin sheath type; PMP22: peripheral myelin protein 22.
We verified the amplified products from A-III1, B-II3, and the normal control by EcoRI and NsiI double digestion. The normal distal REP fragment could be digested by EcoRI, yielding 3.17-kb and 0.4-kb fragments, but could not be digested by NsiI.
Conversely, the normal proximal REP fragment could be digested by NsiI, yielding 2.1-kb and 1.5-kb fragments, but could not be digested by EcoRI (Figure 3C). The duplicated junction fragment amplified from individual A-III1 gave 1.7-kb, 1.5-kb, and 0.4-kb fragments after digestion with both enzymes. However, the deletion junction fragment amplified from individual B-II3 could not be digested by either enzyme (Figure 3D).
DISCUSSION
PMP22 is a myelin-associated molecule associated with Schwann cells in peripheral nerves, and accounts for 2–5% of peripheral nerve myelin sheath protein[9]. PMP22 regulates the proliferation, differentiation, and apoptosis of Schwann cells[10].
Many methods have been used to diagnose duplication and deletion of PMP22. Southern blot and pulsed-field gel electrophoresis are time-consuming, laborious, and have low sensitivity. Fluorescence in situ hybridization has high sensitivity but it is subjective, and its repeatability is limited[11]. PCR-based methods, include PCR of short tandem repeats, fluorescent quantitative real-time PCR, multiplex ligation-dependent probe amplification, and allele-specific PCR-double digestion[12,13]. Latour et al[14] used three pairs of primers to amplify short tandem repeats, but the sensitivity was low. The sensitivity was improved to 99% when 15 pairs of primers were used[15], but it is impractical to perform so many assays in a clinical setting. Fluorescent quantitative real-time PCR and multiplex ligation-dependent probe amplification are relatively simple techniques, but because of the specialized equipment needed, they are generally only used in laboratories rather than in the clinic[16].
The allele-specific PCR-double digestion method that we chose is fast, easy to operate, and shows good specificity. When compared with the method of Haupt et al[17], which used one pair of primers for amplification plus NsiI + EcoRI digestion, and that of Chang et al[18], which used many pairs of primers for amplification plus SacI + EcoRI digestion, the method of Lin et al[8] that we used is simple, rapid, workable, can overcome the effects of non-specific amplification, and decreases the occurrence of false positive and false negative results. However, because this method is likely to have a low sensitivity, we suggest that if the results are negative, another method such as fluorescent quantitative real-time PCR should be used.
Our CMT1A family showed autosomal dominant inheritance. Individual A-III1 had been affected since infancy-showing late walking, distal amyotrophy and myasthenia, absent tendon reflexes, peripheral hypesthesia, and talipes equinovarus. However, the lack of clinical symptoms in individual A-II3 suggests high clinical heterogeneity. This is probably associated with gene modification and environmental factors[12,19]. Interestingly, this individual did show reduced nerve conduction velocities that were consistent with the genetic diagnosis. Therefore, nerve electrophysiological examination of family members may be a good way to identify individuals with PMP22 gene duplication who are pre-symptomatic or subclinical.
A previous study demonstrated that CMT1A nerve pathology is characterized by a reduced density of myelinated nerve fibers and demyelination and/or remyelination, and that the degree of neural functional defects is strongly associated with the severity of axonal degeneration[20]. The characteristics of CMT1A nerve pathology in our patients were identical to those described in a previous study[21]. Although pathological characteristics such as myelin sheath proliferation and an onion-shaped structure may provide reference information for CMT1A genetic diagnosis, axonal lesions are helpful to predict the disease course and prognosis[21].
The average age of onset of HNPP is 22.5 years. Slightly more than half (53.6%) of patients are affected before the age of 20. The clinical manifestation is pressure palsies. Pressure on the nerves can cause tingling sensations, numbness, weakness, muscle atrophy, and even paralysis of the affected area. These symptoms tend to disappear after several weeks or months. Some patients experience neurological symptoms including reduced diffuse nerve conduction velocity and focal thickening (a ‘sausage-shaped structure’) of the myelin sheath of peripheral nerves[21]. Consistent with previous studies, our patient experienced HNPP before the age of 20, with the clinical manifestation of recurrent lower limb weakness. Nerve electrophysiology was characterized by reduced nerve conduction velocity, and neuropathology manifested as irregular thickening of the myelin sheath, and ‘mouse-nibbled’-like changes. Because HNPP displays recurrent limb numbness and weakness, it is difficult to differentiate from entrapment neuropathy, inflammatory demyelinating polyneuropathy, diabetic polyneuropathy and hereditary neuralgic amyotrophy[22,23,24]. Its distinctive nerve electrophysiological and neuropathological changes are helpful to diagnose HNPP and can guide genetic diagnosis[22,23,24].
SUBJECTS AND METHODS
Design
Genetic analysis of human neurological disease.
Time and setting
The experiments were performed at the State Key Laboratory of Medical Genetics, Third Xiangya Hospital, Central South University, China, from August 2010 to August 2011.
Subjects
One CMT1 family and one HNPP family, from the Outpatient Clinic, Department of Neurology, Third Xiangya Hospital in China, were enrolled for this study in 2010. Medical histories were taken and physical examinations performed by two physicians from the Department of Neurology on probands A-III1 and B-II3 and their family members A-II3, A-II4, B-II4, and B-III1. The Manual Muscle Test Scale (0–5) was used to evaluate muscle strength. Individual A-III1 met the diagnostic criteria for CMT1 and individuals B-II3 and B-II5 met those for HNPP[24,25]. This study was conducted in accordance with the Declaration of Helsinki and all the patients and family members signed an informed consent.
Methods
Nerve electrophysiological study
Nerve conduction velocity and compound muscle action potential were determined using a Keypoint electromyogram instrument (Alpine BioMed ApS, Skovlunde, Denmark) in probands A-III1 and B-II3 and patient A-II3.
PMP22 allele-specific PCR-double digestion analysis
10 mL of peripheral ulnar venous blood was obtained from individuals A-III1, A-II3, A-II4, B-II3, B-II4, and B-III1. Genomic DNA was extracted using a standard phenol-chloroform method. We used the primers Hot-DF, Hot-DR, Hot-PF, and Hot-PR described by Lin et al[8]. Primers were synthesized by Sangon Biotech (Shanghai, China). We amplified genomic DNA using the four primer pairs Hot-PF + Hot-PR (proximal REP fragment), Hot-DF + Hot-DR (distal REP fragment), Hot-DF + Hot-PR (CMT1A-REP fragment), and Hot-PF + Hot-DR (HNPP-REP fragment). The PCR reaction conditions were as follows: 95°C for 3 minutes, 30 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 45 seconds, followed by 72°C for 6 minutes, then a hold at 4°C. PCR products were electrophoresed on 0.8% agarose gels and stained with ethidium bromide. In addition, the PCR products were purified then digested by the restriction enzymes EcoRI and NsiI (Takara, Dalian, China) at 37°C for 6 hours, inactivated at 80°C for 20 minutes, and analyzed by agarose gel electrophoresis.
Ultrastructural analysis of the superficial peroneal nerve
A superficial peroneal nerve biopsy was conducted in the CMT1 proband and the HNPP proband. Specimens were fixed using the glutaraldehyde-osmic acid double fixation method, embedded, and sliced into 50-nm-thick sections. Following double staining with uranyl acetate and lead nitrate, sections were observed with an H-7500 transmission electron microscope (Hitachi, Tokyo, Japan) and photographed.
Footnotes
Funding: This project was funded by the National Natural Science Foundation of China, grant No. 81071001 and 30600200.
Conflicts of interest: None declared.
Ethical approval: This study was approved by the Ethics Committee, Xiangya School of Medicine, Central South University, China.
(Edited by Yu YQ, Luo W/Qiu Y/Song LP)
REFERENCES
- [1].Patzkó A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep. 2011;11(1):78–88. doi: 10.1007/s11910-010-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pareyson D, Scaioli V, Laurà M. Clinical and electrophysiological aspects of Charcot-Marie-Tooth disease. Neuromolecular Med. 2006;8(1-2):3–22. doi: 10.1385/nmm:8:1-2:3. [DOI] [PubMed] [Google Scholar]
- [3].Berger P, Young P, Suter U. Molecular cell biology of Charcot-Marie-Tooth disease. Neurogenetics. 2002;4(1):1–15. doi: 10.1007/s10048-002-0130-z. [DOI] [PubMed] [Google Scholar]
- [4].De Jonghe P, Timmerman V, Nelis E, et al. Charcot-Marie-Tooth disease and related peripheral neuropathies. J Peripher Nerv Syst. 1997;2(4):370–387. [PubMed] [Google Scholar]
- [5].Mariman EC, Gabreëls-Festen AA, van Beersum SE, et al. Prevalence of the 1. 5-Mb 17p deletion in families with hereditary neuropathy with liability to pressure palsies. Ann Neurol. 1994;36(4):650–655. doi: 10.1002/ana.410360415. [DOI] [PubMed] [Google Scholar]
- [6].Zhang F, Seeman P, Liu P, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet. 2010;86(6):892–903. doi: 10.1016/j.ajhg.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lopes J, LeGuern E, Gouider R, et al. Recombination hot spot in a 3. 2-kb region of the Charcot-Marie-Tooth type 1A repeat sequences: new tools for molecular diagnosis of hereditary neuropathy with liability to pressure palsies and of Charcot-Marie-Tooth type 1A. French CMT Collaborative Research Group. Am J Hum Genet. 1996;58(6):1223–1230. [PMC free article] [PubMed] [Google Scholar]
- [8].Lin KP, Chou CH, Lee HY, et al. Allele-specific all-or-none PCR product diagnostic strategy for Charcot-Marie-Tooth 1A and hereditary neuropathy with liability to pressure palsies. J Chin Med Assoc. 2006;69(2):68–73. doi: 10.1016/S1726-4901(09)70116-9. [DOI] [PubMed] [Google Scholar]
- [9].Snipes GJ, Suter U, Welcher AA, et al. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13) J Cell Biol. 1992;117(1):225–238. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Niemann S, Sereda MW, Suter U, et al. Uncoupling of myelin assembly and schwann cell differentiation by transgenic overexpression of peripheral myelin protein 22. J Neurosci. 2000;20(1):4120–4128. doi: 10.1523/JNEUROSCI.20-11-04120.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Choi JR, Lee WH, Sunwoo IN, et al. Effectiveness of real-time quantitative PCR compare to repeat PCR for the diagnosis of Charcot-Marie-Tooth Type 1A and hereditary neuropathy with liability to pressure palsies. Yonsei Med J. 2005;46(3):347–352. doi: 10.3349/ymj.2005.46.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Finsterer J. Hypertelorism in Charcot-Marie-Tooth disease 1A from the common PMP22 duplication: A case report. Oman Med J. 2012;27(2):164–167. doi: 10.5001/omj.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Baets J, Deconinck T, De Vriendt E, et al. Genetic spectrum of hereditary neuropathies with onset in the first year of life. Brain. 2011;134(Pt 9):2664–2676. doi: 10.1093/brain/awr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Latour P, Boutrand L, Levy N, et al. Polymorphic short tandem repeats for diagnosis of the Charcot-Marie-Tooth 1A duplication. Clin Chem. 2001;47(5):829–837. [PubMed] [Google Scholar]
- [15].Badano JL, Inoue K, Katsanis N, et al. New polymorphic short tandem repeats for PCR-based Charcot-Marie-Tooth disease type 1A duplication diagnosis. Clin Chem. 2001;47(1):838–843. [PubMed] [Google Scholar]
- [16].Stangler Herodez S, Zagradisnik B, Erjavec Skerget A, et al. Molecular diagnosis of PMP22 gene duplications and deletions: comparison of different methods. J Int Med Res. 2009;37(5):1626–1631. doi: 10.1177/147323000903700542. [DOI] [PubMed] [Google Scholar]
- [17].Haupt A, Schöls L, Przuntek H, et al. Polymorphisms in the PMP-22 gene region (17p11.2-12) are crucial for simplified diagnosis of duplications/deletions. Hum Genet. 1997;99(5):688–691. doi: 10.1007/s004390050431. [DOI] [PubMed] [Google Scholar]
- [18].Chang JG, Jong YJ, Wang WP, et al. Rapid detection of a recombinant hotspot associated with Charcot-Marie-Tooth disease type IA duplication by a PCR-based DNA test. Clin Chem. 1998;44(2):270–274. [PubMed] [Google Scholar]
- [19].Birouk N, Gouider R, Le Guern E. Charcot-Marie-Tooth disease type 1A with 17p11.2 duplication Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain. 1997;120(pt5):813–823. doi: 10.1093/brain/120.5.813. [DOI] [PubMed] [Google Scholar]
- [20].Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie- Tooth disease type 1A. Brain. 2000;123(Pt 7):1516–1527. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- [21].Li J, Krajewski K, Lewis RA, et al. Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2004;29(2):205–210. doi: 10.1002/mus.10521. [DOI] [PubMed] [Google Scholar]
- [22].Sander MD, Abbasi D, Ferguson AL. The prevalence of hereditary neuropathy with liability to pressure palsies in patients with multiple surgically treated entrapment neuropathies. J Hand Surg (Am) 2005;30(6):1236–1241. doi: 10.1016/j.jhsa.2005.06.020. [DOI] [PubMed] [Google Scholar]
- [23].Rana AQ, Masroor MS. Hereditary neuropathy with liability to pressure palsy: a brief review with a case report. Int J Neurosci. 2012;122(3):119–123. doi: 10.3109/00207454.2011.633719. [DOI] [PubMed] [Google Scholar]
- [24].Grossman MJ, Feinberg J, DiCarlo EF, et al. Hereditary neuropathy with liability to pressure palsies: case report and discussion. HSS J. 2007;3(2):208–212. doi: 10.1007/s11420-007-9056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shy ME. Charcot-Marie-Tooth disease: an update. Curr Opin Neurol. 2004;17(5):579–585. doi: 10.1097/00019052-200410000-00008. [DOI] [PubMed] [Google Scholar]