

Abstract
SGs can be visualized in cells by immunostaining of specific protein components or polyA+ mRNAs. SGs are highly dynamic and the study of their assembly and fate is important to understand the cellular response to stress. The deficiency in key factors of SGs like G3BP (RasGAP SH3 domain Binding Protein) leads to developmental defects in mice and alterations of the Central Nervous System. To study the dynamics of SGs in cells from an organism, one can culture primary cells and follow the localization of a transfected tagged component of SGs. We describe time-lapse experiment to observe G3BP1-containing SGs in Mouse Embryonic Fibroblasts (MEFs). This technique can also be used to study G3BP-containing SGs in live neurons, which is crucial as it was recently shown that these SGs are formed at the onset of neurodegenerative diseases like Alzheimer's disease. This approach can be adapted to any other cellular body and granule protein component, and performed with transgenic animals, allowing the live study of granules dynamics for example in the absence of a specific factor of these granules.
Keywords: Cellular Biology, Issue 87, Stress granule (SG), G3BP, primary cells, neurons
Introduction
Stress granules (SGs) are non-membranous cytoplasmic foci formed as a cellular protective response to environmental stress, such as elevated temperature, oxidative stress, hypoxia, osmotic shock, UV irradiation, glucose deprivation, or viral infection1. They can be induced chemically by treatment with compounds like sodium arsenite, which triggers oxidative stress. SGs accumulate stalled translation arrested messenger ribonucleoprotein (mRNPs) complexes2, sequestering mRNAs from the translational machinery, and their assembly can be triggered by the phosphorylation of eIF2α (eukaryotic initiation factor 2 α). SGs are dynamic structures which exchange components with the polysomes and other granules like the P-bodies. They constitute a "triage center" where mRNAs are sorted and processed for either translation, reinitiation, degradation, or packaging into stable non-polysomal mRNPs3. The assembly of SGs is fast but it is a gradual process with initial numerous small aggregates which coalesce into larger granules. The use of chemical inhibitors that disrupt or stabilize microtubules shows that the microtubule network is required for SG dynamics including assembly, coalescence and disassembly processes.
The dynamic assembly of SGs is also promoted by aggregation of specific RNA-binding proteins (RNA-BP) like TIA-1 (T-cell internal antigen-1) and TIAR (TIA-1-related protein), which are able to dimerize and promote polysome disassembly and the routing of mRNAs into SGs4. G3BP (RasGAP SH3 domain binding protein) is such an RNA-BP that localizes to SGs when cells are stressed with arsenite or high temperature, and overexpression of dephosphorylated G3BP can induce SGs assembly5.
G3BP is an evolutionarily conserved RNA-BP that was initially characterized through its interaction with a Ras-GTPase activating protein (RasGAP p120 6); but this interaction was recently revisited7. The G3BP family includes two members in mammals, G3BP1 (referred to as G3BP) and G3BP2 8. Both proteins colocalize in SGs, when cells are subjected to stress9. G3BPs comprise an N-terminal NTF2 domain suggested to influence their localization and oligomerization, followed by proline rich (PxxP) motifs, then C-terminal motifs associated with RNA binding: the canonical RNA-Recognition Motif (RRM) with conserved RNP1 and RNP2 motifs, followed by an arginine-glycine rich (RGG) box. Interestingly, the analysis of different parts of the protein by constructing different domains fused to EGFP showed that the NTF2-like domain and the RNA-binding domain were the most efficiently recruited to SGs, suggesting the importance of the properties of dimerization and RNA binding in the assembly of SGs. Diverse models have revealed different functions of G3BP proteins in vitro10-13. Disruption of G3BP in mice have shown the importance of this protein in developmental growth and survival14, as well as an important role for G3BP in the Central Nervous System (CNS), characterized by ataxia and defects in spatial working memory15. G3BP deficiency leads to altered neuronal plasticity and calcium homeostasis, establishing a direct link between SG formation and neurodegenerative diseases15. It is thus important to be able to study the dynamics of SGs in primary cells like neurons.
This protocol provides a simple way to observe the assembly of G3BP1-containing SGs in primary cells under arsenite treatment. It can be used to study SGs assembly under different conditions, for example different kinds of stresses. It can also be adapted to other granules or other constituents of SGs. Indeed, this protocol focuses on G3BP1, but there are other stress granules markers like TIA-1/R, TTP (tristetraprolin)2, FMRP (Fragile X Mental Retardation Syndrome protein)16, TDP-43 (transactive response DNA binding protein 43)17 or Staufen18. In particular, proteins like TIA-1/R are, like G3BP, nucleating RNA binding proteins that can induce SGs assembly when overexpressed, even if the different formed SGs can differ in function, regulation and associated transcripts. Transfection of fluorophore-tagged version of any of these key components or nucleators of SGs can be performed to image particular SGs assembly and dynamics.
Protocol
All the animal procedures in this protocol are in strict adherence with the guidelines of the European Community Council Directive of 24 November 1986 (86-609/EEC). House the mice in group, allowing food and water ad libitum. Maintain them in a controlled environment (22 ±1 °C, 55 ±5% humidity) with a 12 hr:12 hr light:dark cycle (light on at 7:00 am).
1. Culture of Murine Primary Cells: Mouse Embryonic Fibroblasts (MEFs)
Autoclave thin forceps, as well as curved forceps and dissecting scissors. Store in a sterile container. Use sterile D-PBS (Dulbecco's Phosphate-Buffered Saline). Prepare complete MEFs medium: Dulbecco's modified Eagle medium (DMEM)/F12 supplemented with 10% fetal calf serum (FBS), 1 mM L-glutamine, 1 mM sodium pyruvate, 1% non-essential amino acids, and 0.5 mM 2-mercaptoethanol and warm at 37 °C.
Euthanize a pregnant mouse (at 13.5 days post-coïtum (dpc)) by cervical dislocation. Remove the uterine horns from the mouse sanitized with 90% EtOH. Under a sterile and clean hood, place the horns with the embryos in 37 °C prewarmed D-PBS. Open the horns, placethe embryos in a 100 mm sterile plastic Petri dish, clean them up from the umbilical cord material and wash them with warm D-PBS to remove blood excess. Under a binocular, remove the embryo’s limbs, the internal organs andthe upper part of the head containing the brain.
In a new Petri dish, mince theembryos into very small pieces with a sterile surgicalbladeor scissors (at least for 1 min). In the case of embryos of different genotypes, be careful to put each embryo in individual Petri dishes and keep the tail or the upper head for genotyping.
Cover the embryo with 1x trypsin-EDTA (0.25% trypsin, 1 mM EDTA) . Incubate for 30 min to 1 hr in a 37 °C incubator. Add 10 ml of complete MEFs medium to stop trypsin reaction. Pipet up and down to remove all aggregates. Let sit the cell suspension into a 50 ml tube (filled with complete medium) for 10 min, and centrifuge the supernatant for 5 min at 300 x g at room temperature. Resuspend the pellet in complete medium (6 ml for 1 embryo). Viable nucleated cells can be counted using trypanblue(around 5 x 106 cells can be expected from one embryo). Plate 3 ml per 60 mm Petri dish.
Change the medium the next day andallow the cells to growuntil the dishes are confluent. Manycell types can be seen,but only fibroblasts will survive subculture.
Split the cells and allow them to grow in 35 mm dishes with glass bottom (important for time lapse experiment), until 50-70% confluency for the transfection.Test for Mycoplasma and mouse pathogens.
2. Culture Adaptations in the Case of Neurons
The day before the culture, coat 35 mm glass bottom Petri dishes with poly-L-lysine (200 µl of 0.1 mg/ml) under a sterile and clean hood, and leave overnight.
On the next morning, rinse with sterile pure water, twice for 5 min and once for 45 min to 1 hr. Replace with 2 ml of DMEM plus 10% FBS medium and keep in a 37 °C incubator.
Dissect embryos at 18.5 dpc. Under a sterile and clean hood, place the horns with the embryos in cold sterile HBSS (Hank's Balanced Salt Solution) in 100 mm Petri dish. Neonatal pups can also be used instead of embryos in order to preserve the life of the dam and enable it to produce more offspring, especially in the case of transgenic animals which can be difficult to obtain. In individual Petri dishes, take each embryo or newborn and cut the head with scissors. Hold the head by inserting curved forceps into the eyes, cut the skin and carefully open the soft skull from the back of the head until the eyes, on each side of the head. Cut the optic nerves and the brain stem, remove the brain, and put it in a new Petri dish containing HBSS. Under a stereoscope, remove all the meninges using two thin forceps. Separate the hippocampi, the cortex or any other part of the brain depending on the structure to study.
Immerse the dissected brain structure in 4.5 ml of cold HBSS prepared previously in 15 ml tubes and keep on ice until digestion with trypsin. Add 0.5 ml of 2.5% trypsin and incubate at 37 °C for 15 min to 20 min. Rinse the trypsin 3x with HBSS, being extremely careful to not discard the digested brain parts.
Resuspend in 500 µl (hippocampi) to 1 ml (cortex) DMEM plus 10% FBS and pipet up and down several times with a 1 ml micropipette equipped with a 1 ml tip, then equipped with 1 ml plus 200 µl tips, until there is no visible aggregate.
Distribute 100-200 µl of cell suspension to each 35 mm glass bottom dish containing DMEM plus 10% FBS and let the neurons adhere at 37 °C for at least 3 hr. Replace by prewarmed neuron complete medium (Neurobasal medium supplemented with 250 µM L-glutamine and NS21, prepared as described in Chen et al.19) and leave at 37 °C to allow neuronal growth. Transfect the neurons at 5 to 14 days in vitro (div) (the efficiency of transfection is higher after a couple of div but synaptic connections are better established from 7-10 div).
3. Transfection of EGFP-G3BP1 Construct
Transfect the cells with a vector containing the cDNA of your protein of interest (any component of SGs) fused to a fluorescent marker (GFP, YFP, etc.), using 3 µg of purified plasmid per 35 mm dish.
Transfect the MEFs using a commercial method, following the manufacturer's protocol (See Table of Materials/Reagents).
- Transfect the neurons with a calcium phosphate method adapted from Xia et al.20 Briefly:
- Prepare the solutions: DMEM-wash: DMEM containing 25 mM KCl; transfection solution: DMEM-wash containing 1x DMKY (HEPES 5 mM, MgCl2 10 mM, phenol red); and shock solution: HeBS 1x, DMKY 1x, and DMSO 2% (v/v); and keep them at 37 °C.
- Remove the media from the neurons, filtrate, and keep it at 37 °C. Wash with DMEM-wash then replace with transfection medium and keep at 37 °C during the preparation of the calcium phosphate-plasmid DNA precipitates.
- In a 1.5 ml microcentrifuge tube, add (in this order) Braun water (final volume 50 µl), 5 µl of CaCl2 2.5 M, and 3 µg of plasmid DNA. Drop this mix onto 50 µl of HeBS 2x already introduced in a round bottom polypropylene tube. Mix the tube by rotation along with the dropping. Let the precipitate form at room temperature during 30 min.
- Add the precipitate dropwise onto the neurons and leave at 37 °C during 30 to 50 min.
- Replace with shock solution for 1 min, then rinse with DMEM-wash and finally reintroduce the preconditioned medium. Keep the cells at 37 °C.
4. Visualization of G3BP Containing SGs Assembly and Dynamics
Replace the medium of the cells which contains phenol red by phenol red-free medium.
Use a confocal microscope equipped with fluorescence for the acquisitions. Turn on the microscope systems: mercury lamp, computer, and lasers. Warm up the chamber to 37 °C at least 20 min before the beginning of the acquisitions.
The over expression of G3BP induces the spontaneous formation of a type of SGs. The cells may thus be observed for the assembly of SGs 24 hr right after the transfection without stress induction. Alternatively, stress can be induced to further induce the assembly of SGs. Add 0.5 mM sodium arsenite and start the acquisition right after the addition of the compound (granules will be well formed within 1 hr).
Add oil to the immersion objective (use 40X or 63X objectives) and install the Petri dish over the objective in the adapted 35 mm microscope holder, stabilized on the stage holder. Check that the dish is flat otherwise the acquisitions will be altered. Turn on the fluorescence light according to the relevant fluorophore and visualize transfected cells. Turn off fluorescence once a cell of interest is in the field in order to minimize bleaching and cytotoxicity.
In the "acquire tab", select the lasers depending on the excitation wavelength of the tag fluorophore (488 nm in our protocol which uses EGFP-tagged G3BP) and select the adapted emission length for the PMT (photomultiplier tube). Adjust the laser power (usually not more than 10%) to minimize noise and oversaturation as well as toxicity, and set the gain and offset to modify the signal to noise ratio. Scan fast (1,000 Hz) in order to minimize the duration of laser exposition (line average 2, frame average 1). If desired, set the parameters for the Z stack to be able to reconstruct the image in 3D (a Z step of 1 µm is usually fine with cells). Determine the interval time between each acquisition: smaller intervals permit to obtain a more coherent movie but this may induce photobleaching and toxicity (an interval of 20 sec was used in the described experiments). Duration of total acquisition may vary depending on the stress type. Many G3BP-containing SGs are formed very rapidly under arsenite treatment and are well visible after 1 hr of treatment: in that case, choose rapidly the cells to film and start the acquisitions right after the stress, with a total duration of acquisitions of 15-20 min largely sufficient to observe the assembly of several SGs.
Save the pictures and reconstruct the stacks and movie using ImageJ software.
Representative Results
Stress granule formation is important in the response of cells to stress, permitting a cellular adaptation with stalled translation, until the stress has cleared, associated to prevention of apoptosis. This assay permits to study the SGs in primary cells, by following the localization of key SGs protein components (Figure 1). G3BP, a key factor of SGs assembly, is present rather diffusively in the cytoplasm of cultured MEFs or neurons (Figure 2A a and c), and is clearly present in discrete granules under arsenite treatment in MEFs as well as neurons (Figure 2A b and d). Primary cells can be obtained from transgenic mouse models, allowing for example the study of stress granules in the absence of one of their components. Interestingly, the deficiency of G3BP1 in mouse leads to severe defects of the Central Nervous System, notably an ataxic phenotype characterized by incapacity to correctly coordinate the movements (as seen in a limb clasping test, Figure 2B) and an alteration of spatial working memory. G3BP1 is present in stress granules in neurons, and this is all the more interesting that SGs have been shown to form at the onset of neurodegenerative diseases like Alzheimer disease.
This assay (Figure 1) thus permits to study SGs in primary cells, indispensable to study their composition and assembly in vivo. Furthermore, it allows the study of live cells and can thus be used to follow the dynamics of SGs, by visualizing one or several of their overexpressed components in real time. Figure 3 shows representative time-lapse images permitting to follow the assembly and dynamics of G3BP1-containing SGs in MEFs after arsenite treatment. In Figure 4A, the same experiment is performed with murine hippocampal neurons in culture. EGFP-G3BP1 localization goes from large subcellular granular structures to more defined and smaller granules, the SGs, after arsenite treatment. SGs were more and more defined if the acquisitions were continued (not shown) but the cell morphology started to change, probably due to the heat and toxicity induced by the laser excitation.
Time lapse permits the capturing of a video to directly observe the SGs dynamics. Furthermore, the use of a confocal microscope permits to reconstruct an image in Z stack and thus to follow completely the dynamics of the bodies throughout the cell in 3 dimensions (as illustrated for a neuron at a defined time in Figure 4B).
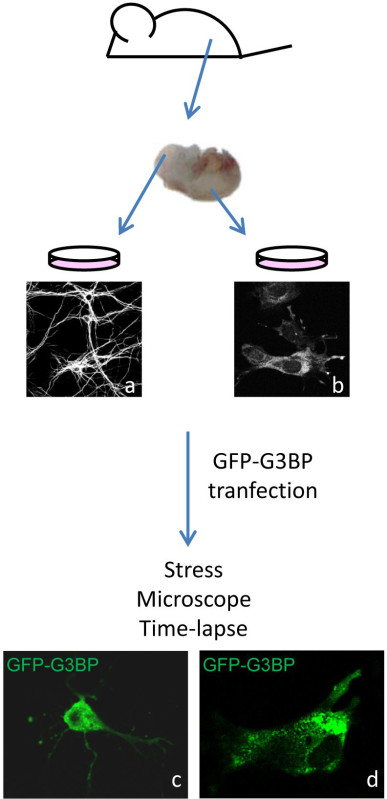 Figure 1. Descriptive of the main steps of the protocol. Primary cells are cultured from embryos (18.5 dpc (days post-coitum) (or neonatal pups) for neurons (a), 13.5 dpc for MEFs (b)). After transfection of a plasmid containing EGFP-G3BP1 cDNA, cells are stressed and imaged with a laser scanning confocal microscope. Dynamics of G3BP1-containing SGs are observed.Neurons are visualized by MAP2 (Microtubule Associated Protein 2) staining in (a), MEFs by G3BP1 staining in (b). They are given as illustrative pictures, as well as EGFP-G3BP1 transfected murine neuron (c) and fibroblast (d), which are independent pictures and do not correspond to the cells given in (a) and (b).
Figure 1. Descriptive of the main steps of the protocol. Primary cells are cultured from embryos (18.5 dpc (days post-coitum) (or neonatal pups) for neurons (a), 13.5 dpc for MEFs (b)). After transfection of a plasmid containing EGFP-G3BP1 cDNA, cells are stressed and imaged with a laser scanning confocal microscope. Dynamics of G3BP1-containing SGs are observed.Neurons are visualized by MAP2 (Microtubule Associated Protein 2) staining in (a), MEFs by G3BP1 staining in (b). They are given as illustrative pictures, as well as EGFP-G3BP1 transfected murine neuron (c) and fibroblast (d), which are independent pictures and do not correspond to the cells given in (a) and (b).
 Figure 2. (A). G3BP localization in cultured MEFs and hippocampal neurons, untreated or stressed with sodium arsenite.G3BP is mainly cytoplasmic, with a diffuse staining in MEFs (a) or in large granular structures in the soma of neurons (c) when the cells are not stressed. After 1 hr of sodium arsenite treatment, G3BP becomes localized in discrete cytoplasmic granules (MEFs, b and neurons, d). Here, cells were fixed and immunostained with an anti-G3BP1 antibody. MAP2 staining permits to identify neurons in c) and d). DNA was counter-stained in blue with Hoechst. Scale bars represent 2 µm (a and b) and 10 µm (c and d). (B). When lifted by the tail towards the floor (limb-clasping test), WT mice extend their legs (left) whereas G3BP1 KO mice show an abnormal paw-clasping reflex close to a bat-like posture. Please click here to view a larger version of this figure.
Figure 2. (A). G3BP localization in cultured MEFs and hippocampal neurons, untreated or stressed with sodium arsenite.G3BP is mainly cytoplasmic, with a diffuse staining in MEFs (a) or in large granular structures in the soma of neurons (c) when the cells are not stressed. After 1 hr of sodium arsenite treatment, G3BP becomes localized in discrete cytoplasmic granules (MEFs, b and neurons, d). Here, cells were fixed and immunostained with an anti-G3BP1 antibody. MAP2 staining permits to identify neurons in c) and d). DNA was counter-stained in blue with Hoechst. Scale bars represent 2 µm (a and b) and 10 µm (c and d). (B). When lifted by the tail towards the floor (limb-clasping test), WT mice extend their legs (left) whereas G3BP1 KO mice show an abnormal paw-clasping reflex close to a bat-like posture. Please click here to view a larger version of this figure.
 Figure 3. Successive acquisitions in time-lapse experiment obtained with MEFs transfected with EGFP-G3BP1, using a Leica SP5 laser scanning confocal microscope (excitation at a wavelength of 488 nm). Acquisitions were started 10 min after the addition of sodium arsenite, and obtained during 10 min with intervals of 20 sec. Six representative pictures are given, with t the time in sec after the addition of sodium arsenite (0.5 mM). The arrows show two areas where we can see the assembly of SGs. Scale bar = 2 µm.
Figure 3. Successive acquisitions in time-lapse experiment obtained with MEFs transfected with EGFP-G3BP1, using a Leica SP5 laser scanning confocal microscope (excitation at a wavelength of 488 nm). Acquisitions were started 10 min after the addition of sodium arsenite, and obtained during 10 min with intervals of 20 sec. Six representative pictures are given, with t the time in sec after the addition of sodium arsenite (0.5 mM). The arrows show two areas where we can see the assembly of SGs. Scale bar = 2 µm.
 Figure 4. (A). Successive acquisitions in time-lapse experiment obtained with murine hippocampal neurons transfected with EGFP-G3BP1, using a Leica SP5 laser scanning confocal microscope (excitation at a wavelength of 488 nm). Acquisitions were started 5 min after the addition of sodium arsenite, and obtained during 40 min with intervals of 20 sec. Six representative pictures are given, with t the time in sec after the addition of sodium arsenite (0.5 mM). The arrows show two areas where we can see the assembly of SGs. Scale bar represents 5 µm. d: dendrite; a: axon. (B). 3-dimension reconstruction from Z stack (5 acquisitions in Z axis over a total of 10 µm) of a neuron transfected with EGFP-G3BP1 and treated with sodium arsenite. n: neurite (dendrite or axon).
Please click here to view a larger version of this figure.
Figure 4. (A). Successive acquisitions in time-lapse experiment obtained with murine hippocampal neurons transfected with EGFP-G3BP1, using a Leica SP5 laser scanning confocal microscope (excitation at a wavelength of 488 nm). Acquisitions were started 5 min after the addition of sodium arsenite, and obtained during 40 min with intervals of 20 sec. Six representative pictures are given, with t the time in sec after the addition of sodium arsenite (0.5 mM). The arrows show two areas where we can see the assembly of SGs. Scale bar represents 5 µm. d: dendrite; a: axon. (B). 3-dimension reconstruction from Z stack (5 acquisitions in Z axis over a total of 10 µm) of a neuron transfected with EGFP-G3BP1 and treated with sodium arsenite. n: neurite (dendrite or axon).
Please click here to view a larger version of this figure.
Discussion
The most critical steps in the protocol concern the transfection and more particularly the time lapse acquisitions, which have to be carefully monitored in order to lower down the cytotoxicity.
Culture of primary cells is not a difficult part, as long as sterile conditions are maintained and caution is taken in order to prevent damage during the dissection and cell dissociation steps. MEFs can be kept frozen at early passages. Neuron transfection with calcium phosphate has been shown to work well 21 and this protocol adapted from Xia et al.20 enables good level of transfection, however it can induce a certain cell death. It is important to transfect the neurons within a few days following the culture, to check the pH of the solutions and to monitor the incubation of the neurons with the calcium phosphate-plasmid precipitate, which should not exceed 1 hr. Check under a light microscope and remove the transfection medium if aggregates are observed. The number of washes with DMEM can be increased.
The over expression of some SGs protein components leads to the spontaneous assembly of granules, which seems to involve the oligomerization and RNA binding properties of these proteins, as in the case of TIA-1/R 4 or G3BP 5. Thus, it is important to define the time at which you can start the time lapse acquisitions. A few SGs are visible as soon as 24 hr after EGFP-G3BP transfection, when the protein is expressed. However, inducing an additional stress leads to the formation of additional granules, which may differ as different kinds of SGs with different key components have been identified under different stresses1,22,23. Assembly of SGs is fast in living cells (as fast as 10-20 min), and many granules are completely formed within 1 hr of arsenite treatment, it is thus important to start the acquisitions as soon as possible after the induction of stress if the assembly has to be studied. At later stages however, the dynamics of SGs can also be studied, as small granules can coalesce into larger structures, and these structures are not fixed: their components can be exchanged with the cytoplasm or other kinds of granules like the P-bodies, sites of RNA decay3. Rapid eye-detection of transfected cells and rapid monitoring of the acquisitions parameters (especially X, Y, and Z coordinates) permit to obtain a movie including assembly of granules, as well as to limit photobleaching and laser-induced cytotoxicity, which is the other critical step in live imaging. Indeed, in some cases, we could observe important changes in cell morphology and even the death of a few cells, if the acquisitions were performed for a long time (more than 1 hr after the induction of oxydative stress with arsenite treatment). In order to prevent these phenomena, it is possible to fasten the scan and to increase the intervals duration between each acquisition. However, a shorter interval permits to better follow the dynamics of SGs containing the visualized protein, and to obtain a more complete movie. Thus, one should adapt the parameters to each experiment to obtain the best movie with limited photobleaching and toxicity, depending on the primary cell type, on the granule component which is followed, and on the associated fluorophore molecule.
Fluorescent protein tags are usually sufficiently photostable to be imaged for the duration of the experiment. Different fluorescent proteins can work well, as long as they produce a strong signal, are slow to bleach and nontoxic. Importantly, they should be bright enough in order to give signal above the autofluorescence of the cells and be thus be reliably detected. Indeed, primary cells can induce a background signal due to the autofluorescence of organelles (like mitochondria and lysosomes). However in our hands, autofluorescence was very low compared to the brightness of EGFP-G3BP, and it was easy to distinguish the EGFP signal of transfected cells versus not transfected cells. Overall, the choice of optimal filters and laser light intensity levels will be critical to obtain the right signal-to-noise ratio, associated to low toxicity. Finally, if images have to be quantitatively studied, it is important to be sure that the fluorescence intensity of the protein is not sensitive to environmental factors like components of the culture medium.
SGs dynamics can indeed be compared (speed of assembly, size and number of granules) between different conditions (cells from wild-type and knock-out animals for example). In order to get statistically significant results, one should culture cells from at least three different animals of each genotype or condition (from three different litters), and image between 10 and 50 cells in each independent experiment. It is possible to mark several positions in the acquisitions software and thus to image several transfected cells at the same time, permitting to increase the data obtained in a relatively short time. Imaging several cells simultaneously is interesting as it will be difficult to use further the same Petri dish once the cells are stressed and imaged (in cases where the assembly of SGs is studied, or if the experimental conditions in long acquisitions induce cytotoxicity). However, when quantifying the parameters "number" and "size" of SGs - and in order to increase the number of cells imaged to obtain more statistical relevance (more than 50 cells of each individual) - cells can be fixed after arsenite treatment, so that more cells on the same Petri dish can be studied. Videos will still be used as qualitative results in this case.
The protocol described here permits to study G3BP-containing SGs in primary live cells, and can be adapted to other cellular bodies and other protein components of these bodies (like TIA-1/R for SGs, FRMP and Staufen for SGs and neuronal transport granules). The use of primary cells is particularly interesting in the context of physiological studies, involving for instance transgenic animals. Indeed, G3BP1 KO mice reveal for example the essential function of a SGs factor in survival and development of an organism, as well as in the CNS functioning14,15.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge the Montpellier Rio Imaging (MRI) platform where the acquisitions were performed. They thank Isabel Cristina Lopez Mejia, Alexandra Metz, Irina Lassot, Solange Desagher, Fabien Loustalot, and Virginie Georget for their help in different parts of the protocol. This work was supported by the Fondation pour la Recherche Médicale (FRM) (Equipe FRM 2011 -n°DEQ20111223745).
References
- Thomas MG, Loschi M, Desbats MA, Boccaccio GL. RNA granules: The good, the bad and the ugly. Cellular Signalling. 2011;23(2):324–334. doi: 10.1016/j.cellsig.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochemical Society transactions. 2002;30(6):963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends in Biochemical Sciences. 2008;33(3):141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Gilks N, et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Molecular biology of the cell. 2004;15(12):5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourrière H, et al. The RasGAP-associated endoribonuclease G3BP assembles stress granules. The Journal of Cell Biology. 2003;160(6):823–831. doi: 10.1083/jcb.200212128. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Parker F, et al. A Ras-GTPase-activating protein SH3-domain-binding protein. Molecular and Cellular Biology. 1996;16(6):2561–2569. doi: 10.1128/mcb.16.6.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annibaldi A, Dousse A, Martin S, Tazi J, Widmann C. Revisiting G3BP1 as a RasGAP binding protein: sensitization of tumor cells to chemotherapy by the RasGAP 317-326 sequence does not involve G3BP1. PLOS ONE. 2011;6(12) doi: 10.1371/journal.pone.0029024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy D, French J, Guitard E, Ru K, Tocque B, Mattick J. Characterization of G3BPs: tissue specific expression, chromosomal localisation and rasGAP(120) binding studies. Journal of Cellular Biochemistry. 2001;84(1):173–187. doi: 10.1002/jcb.1277. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Winslow S, Sunesson L, Hellman U, Larsson C. PKCα Binds G3BP2 and Regulates Stress Granule Formation Following Cellular Stress. PLOS ONE. 2012;7(4) doi: 10.1371/journal.pone.0035820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourrière H, et al. RasGAP-associated endoribonuclease G3Bp: selective RNA degradation and phosphorylation-dependent localization. Molecular and Cellular Biology. 2001;21(22):7747–7760. doi: 10.1128/MCB.21.22.7747-7760.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Ochem A, Staub A, Falaschi A. Human DNA helicase VIII: a DNA and RNA helicase corresponding to the G3BP protein, an element of the ras transduction pathway. Nucleic acids research. 1999;27(3):817–821. doi: 10.1093/nar/27.3.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon S, et al. Distinct structural features of caprin-1 mediate its interaction with G3BP-1 and its induction of phosphorylation of eukaryotic translation initiation factor 2alpha, entry to cytoplasmic stress granules, and selective interaction with a subset of mRNAs. Molecular and Cellular Biology. 2007;27(6):2324–2342. doi: 10.1128/MCB.02300-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soncini C, Berdo I, Draetta G. Ras-GAP SH3 domain binding protein (G3BP) is a modulator of USP10, a novel human ubiquitin specific protease. Oncogene. 2001;20(29):3869–3879. doi: 10.1038/sj.onc.1204553. [DOI] [PubMed] [Google Scholar]
- Zekri L, et al. Control of fetal growth and neonatal survival by the RasGAP-associated endoribonuclease G3BP. Molecular and Cellular Biology. 2005;25(19):8703–8716. doi: 10.1128/MCB.25.19.8703-8716.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S, et al. Deficiency of G3BP1, the stress granules assembly factor, results in abnormal synaptic plasticity and calcium homeostasis in neurons. Journal of neurochemistry. 2013. [DOI] [PubMed]
- Mazroui R, Huot M-E, Tremblay S, Filion C, Labelle Y, Khandjian EW. Trapping of messenger RNA by Fragile X Mental Retardation protein into cytoplasmic granules induces translation repression. Human molecular genetics. 2002;11(24):3007–3017. doi: 10.1093/hmg/11.24.3007. [DOI] [PubMed] [Google Scholar]
- Colombrita C, et al. TDP-43 is recruited to stress granules in conditions of oxidative insult. Journal of neurochemistry. 2009;111(4):1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Thomas MG, Tosar LJM, Desbats MA, Leishman CC, Boccaccio GL. Mammalian Staufen 1 is recruited to stress granules and impairs their assembly. Journal of Cell Science. 2009;122(4):563–573. doi: 10.1242/jcs.038208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: re-defined and modified supplement B27 for neuronal cultures. Journal of neuroscience. 2008;171(2):239–247. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dudek H, Miranti CK, Greenberg ME. Calcium Influx via the NMDA Receptor Induces Immediate Early Gene Transcription by a MAP Kinase/ERK-Dependent Mechanism. The Journal of Neuroscience. 1996;16(17):5425–5436. doi: 10.1523/JNEUROSCI.16-17-05425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Ghosh A, Greenberg ME. Calcium phosphate transfection of DNA into neurons in primary culture. Current protocols in neuroscience. 2001;3 doi: 10.1002/0471142301.ns0311s03. [DOI] [PubMed] [Google Scholar]
- Reineke LC, Dougherty JD, Pierre P, Lloyd RE. Large G3BP-induced granules trigger eIF2α phosphorylation. Molecular biology of the cell. 2012;23(18) doi: 10.1091/mbc.E12-05-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedersha N, Tisdale S, Hickman T, Anderson P. Real-time and quantitative imaging of mammalian stress granules and processing bodies. Methods in enzymology. 2008;448:521–552. doi: 10.1016/S0076-6879(08)02626-8. [DOI] [PubMed] [Google Scholar]