

Abstract
Given the rapid increase of species with a sequenced genome, the need to identify orthologous genes between them has emerged as a central bioinformatics task. Many different methods exist for orthology detection, which makes it difficult to decide which one to choose for a particular application.
Here, we review the latest developments and issues in the orthology field, and summarize the most recent results reported at the third ‘Quest for Orthologs’ meeting. We focus on community efforts such as the adoption of reference proteomes, standard file formats and benchmarking. Progress in these areas is good, and they are already beneficial to both orthology consumers and providers. However, a major current issue is that the massive increase in complete proteomes poses computational challenges to many of the ortholog database providers, as most orthology inference algorithms scale at least quadratically with the number of proteomes.
The Quest for Orthologs consortium is an open community with a number of working groups that join efforts to enhance various aspects of orthology analysis, such as defining standard formats and datasets, documenting community resources and benchmarking.
Availability and implementation: All such materials are available at http://questfororthologs.org.
Contact: erik.sonnhammer@scilifelab.se or c.dessimoz@ucl.ac.uk
1 INTRODUCTION
Orthologs are defined as genes in different species that descend by speciation from the same gene in the last common ancestor (Fitch, 1970). Because of this, they are likely to perform equivalent functions, and even if they have diverged since the speciation event, they are more likely to be functional counterparts in different species than other types of homologs (Gabaldón and Koonin, 2013). The probable functional equivalence of orthologs has made them attractive for genome annotation, and a range of approaches have been developed to identify orthologs, which has resulted in a number of repositories for precomputed orthology relationships. In fact, there are currently at least 37 different ortholog databases (reviewed in Altenhoff and Dessimoz, 2012). Besides their importance for genome annotation and functional inference, finding orthologs is a necessary step to build species phylogenies and to perform comparative genomics analyses (e.g. anchoring chromosome alignments, reconstructing ancestral proteomes).
An unfortunate effect of the wide interest in orthology is that many different formats and datasets exist, and it is far from trivial to integrate or compare orthologs from different sources. As a forum to discuss orthology analysis, standards and ways to coordinate and compare ortholog inferences, the orthology community started a workshop series called ‘Quest for Orthologs’ which held its third event in July 2013 in Lausanne, Switzerland. We here review the latest developments and trends in the orthology field, including unpublished results presented at the latest workshop.
The rapidly increasing number of genomes sequenced creates acute computational challenges. As we discuss below, because most orthology prediction methods have at least quadratic scaling with the number of included species (e.g. owing to all-against-all sequence comparison), computation times have become a bottleneck. Computing orthologs between all complete proteomes has recently gone from typically a matter of CPU-weeks to hundreds of CPU-years, and new, faster algorithms and methods are called for.
Other areas we discuss that have received attention recently include ‘domain orthology’, i.e. orthology analysis at the protein domain level, and xenology, or horizontal gene transfer (HGT), which in some cases may instead be the result of contaminating sequences in poorly curated genomes.
Finally, we provide an update on areas of central importance to the orthology community, in particular, (i) standards for data analysis and data sharing, and (ii) the ‘orthology conjecture’, i.e. the testing of the hypothesis that orthologs are more functionally similar than paralogs. Even though this hypothesis has been considered true both from first principles and data, it has been debated (Studer and Robinson-Rechavi, 2009), and was recently challenged with counter evidence. However, a number of subsequent studies identified problems with that analysis and showed that when correcting for biases, the same type of analysis does provide support for the ortholog conjecture.
2 BIG DATA CHALLENGES IN ORTHOLOGY ANALYSIS
Thanks to revolutionary developments in DNA sequencing technologies, there are already many thousands of species with a sequenced genome, with the total number roughly doubling each year. In fact, the reduction in sequencing costs in the past years has overtaken the rate at which the computing capacity of processors increases, known as Moore’s law. An inevitable result of this trend is that the increase in computational demands in sequence analyses is not easily met by an increase in computational capacities but rather calls for new approaches or algorithmic implementations. Given that the number of pairwise relationships increases quadratically with the total number of species, the inference of orthology relationships across an ever-growing sequence space is severely affected. Such a computational challenge affects all methodological approaches for orthology inference, but impacts most dramatically those that include steps that scale poorly with the number of sequences considered, such as phylogenetic analysis. As a result, it is challenging to be comprehensive in terms of establishing orthology and paralogy relationships across all sequenced genomes. Some databases address such problems by implementing methodological shortcuts. For instance, the latest version of TreeFam (Schreiber et al., 2013) builds gene families based on profile-based searches that avoids all-against-all comparisons employed in graph-based approaches, while Hieranoid uses a species tree-guided approach to scale linearly (Schreiber and Sonnhammer, 2013). Sharing computations across databases also seems a promising avenue. In this direction, OMA (Altenhoff et al., 2011) and OrthoDB (Waterhouse et al., 2013) have joined forces to compute all-against-all sequence comparisons only once for the two databases, an initiative that could be extended to other databases in the future. Similarly, MetaPhOrs (Pryszcz et al., 2011) exploits gene phylogenies precomputed by other databases to infer consistency-based orthologs. The last version of EggNOG (Powell et al., 2014) reuses all-against-all comparisons from the Similarity Matrix of Protein project (SIMAP; Arnold et al., 2014). Interestingly, SIMAP itself has drawn on user-volunteered computing for nearly 10 years, using the BOINC (Berkeley Open Infrastructure for Network Computing) infrastructure (Rattei et al., 2007); they, however, have recently announced plans to move back to fully ‘in-house’ computations, which casts doubts on the effectiveness of user-volunteered computing. Finally, some databases have opted for a focused approach by limiting their analyses to predetermined sets of species; this is the case for phylome-based or collection-based inferred orthologs in PhylomeDB (Huerta-Cepas et al., 2014) and PANTHER (Mi et al., 2013). Other problems related to big data challenges relate to the need to deploy large databases on servers that include fast and efficient search and displaying tools. Thus, the limit of traditional systems such as SQL-based relational databases is being reached in many cases, calling for the need for alternative solutions. Fortunately, many of the mentioned Big Data challenges are shared by other fields, also outside the research environment, and thus, a growing number of alternative solutions for some of the problems are or will be available. This will require building the necessary expertize to adapt such solutions to the specific needs of orthology databases, and to keep up with the fast developments in the Big Data field.
Independently of the mentioned computational challenges, the growing availability of sequenced genomes poses additional challenges related to the increased resolution of the data at hand. While orthology is defined at the level of comparisons across species, the increasing availability of sequences from populations of the same species and from closely related species creates scenarios that are difficult to interpret under the canonical speciation/duplication model. This is the case, for instance, of the incomplete lineage sorting of gene alleles during speciation, which actually started diverging before the speciation event, but also of scenarios resulting from hybridization, introgression or other types of genetic exchanges. Problems appearing after the availability of genomes from populations or highly related species include not only methodological problems (e.g. resolving recent duplications and speciations when only few differing sites are present), but also operational ones (e.g. should orthology be considered between genomes of the same species?; should only a reference strain or reference species be used?). These problems notwithstanding, the availability of genomes from closely related species also provides some opportunities for improving orthology prediction such as the possibility to consider pangenomes or use synteny information (i.e. chromosomal position conservation).
3 HIERARCHICAL GROUPS
In the past few years, the concept of hierarchical orthologous groups has gained increased attention. Hierarchical orthologous groups are defined with respect to specific species clades and—barring inference errors—contain all the sequences that have evolved from a single ancestral gene in the last common ancestor of that clade (Jothi et al., 2006; Kriventseva et al., 2008; Merkeev et al., 2006; Powell et al., 2014; reviewed in Boeckmann et al., 2011). Hierarchical orthologous groups generalize the concept of orthology to more than two species at a time. Consider, for instance, the Thyroid hormone receptor family, which underwent a duplication at the base of the vertebrates, yielding the two genes TR-α and TR-β (e.g. Wu et al., 2007). At the level of vertebrate species, TR-α and TR-β belong to distinct hierarchical orthologous groups, whereas at the broader level of bilaterian species, they belong to the same group. Thus, depending on the context of investigation, the user can choose the level of granularity in a precisely defined and evolution-aware way.
Hierarchical orthologous groups were a recurrent theme of the latest Quest for Orthologs meeting. Evgeny Zdobnov (University of Geneva, Switzerland) presented updates in the pipeline and user interface of the OrthoDB database (Waterhouse et al., 2013). Adrian Altenhoff (ETH Zurich, Switzerland) introduced a new method to compute hierarchical orthologous groups from pairs of orthologous genes (Altenhoff et al., 2013), available in the OMA database and the OMA stand-alone software (http://omabrowser.org/standalone). Erik Sonnhammer (Stockholm University, Sweden) presented Hieranoid, an algorithm to build hierarchical orthologous groups using InParanoid (Schreiber and Sonnhammer, 2013). Hierarchical orthologous groups can be described in the OrthoXML format (Schmitt et al., 2011) discussed in Section 5.
4 ORTHOLOGY BENCHMARKING
Benchmarking continues to be a major theme for the orthology community. In the second Quest for Orthologs meeting in 2011, a working group had been formed with the goal of establishing standards in orthology benchmarking and facilitating benchmarking. Christophe Dessimoz (University College London, UK) presented its progress. The main achievements of the working group are (i) the development of a freely available Web server for orthology benchmarking and (ii) a comparison of eight orthology databases on a common set of 66 species (2011 Quest for Orthologs reference proteome dataset) on a battery of 10 phylogenetic and functional tests. Results for each test can be retrieved from the benchmarking Web server (http://orthology.benchmarkservice.org/). The Web server and the benchmark results will be presented and discussed in detail in a separate publication.
5 DATA FORMAT STANDARDS
Since the first Quest for Orthologs meeting in 2009, many ortholog databases have joined the community effort to support common data format standards. There are many advantages of using a shared format, particularly for ‘ortholog consumers’ that want to import orthology information from many different providers. Also for constructing meta-databases and for comparative analyses, it is beneficial to avoid the need to write a separate parser for each data source.
The orthology community is gradually progressing from only providing their own format (usually a text file) to adopting the OrthoXML standard (Schmitt et al., 2011). At the moment, seven databases (Ensembl Compara, InParanoid, MBGD, OMA, OrthoLuge, PhylomeDB and RoundUp) are supporting OrthoXML, and MetaPhOrs and PANTHER will support it with their next releases (See http://orthoxml.org for an updated list and Web links to the databases). The PhyloXML format can be used to store orthology information in trees, but is less general because it cannot define orthologous relationships for which a tree is not specified.
Although XML offers structured data and excellent consistency verification, it is not by itself or automatically translatable to a powerful database engine in the same way that SQL is. In recent years, Semantic Web standards like RDF (Resource Description Framework; http://www.w3.org/RDF) and SPARQL (SPARQL Protocol and RDF Query Language; http://www.w3.org/TR/rdf-sparql-query/) have attracted the attention of the bioinformatics community and, for example, UniProt (Jain et al., 2009) and EBI (Jupp et al., 2014) have data accessible using such standards. RDF provides a flexible graph-based data model that facilitates the integration of datasets by making explicit the links between the graphs of each dataset, and permits identifying any such resource in the Internet through URIs (Uniform Resource Identifiers). SPARQL permits distributed queries across RDF databases scattered around the world, which facilitates the reuse of data while reducing the maintenance effort. It also offers easy combination of different datatypes, and avoids proliferation of overlapping XML schemas.
At the Quest for Orthologs meeting in 2013, some of these benefits were practically demonstrated through the RDF versions of Roundup, OGO and MBGD (see http://questfororthologs.org/orthology_databases for Web links). The success of semantic data sharing is generally improved by the use of shared ontologies. However, the aforementioned RDF databases used different application-oriented ontologies, so our community is working on defining the set of properties and classes to be used in an RDF representation of orthology. For this purpose, ontologies like the Homology Ontology (Roux and Robinson-Rechavi, 2010) and the Comparative Data Analysis Ontology (Prosdocimi et al., 2009) will have to be studied and reused. Having orthology information available according to such ontologies would also open the door for the Quest for Orthologs consortium to exploit automated reasoning, e.g. consistency of datasets, inference based on logical properties like symmetry or transitivity, etc. The performance of RDF is likely worse than for relational databases, and it is still unclear how well RDF would work in practice for large-scale orthology applications.
6 REFERENCE DATASETS
The Quest for Orthologs consortium has defined a consensus dataset of proteomes and common file formats (Dessimoz et al., 2012; Gabaldón et al., 2009) to be used by diverse orthology inference methods, allowing for standardized benchmarks and to aid integration of multiple ortholog sources. The Quest for Orthologs Reference Proteomes datasets were created as a collection of data providing a representative protein for each gene in the genome of selected species. Such datasets have been generated annually from the UniProt Knowledgebase (UniProKB) database (The UniProt Consortium, 2012) for the past four years. To this end, a gene-centric pipeline has been developed and enhanced over these years at UniProt. The Quest for Orthologs Reference Proteomes are a manually compiled subset of the UniProt reference proteomes, comprising well-annotated model organisms and organisms of interest for biomedical research and phylogeny, with the intention to provide broad coverage of the tree of life.
Currently, the reference dataset provided to the Quest for Orthologs consortium comprises 66 species (40 Eukaryotes plus 26 Bacteria/Archea) that are based on the UniProtKB 2014_04 release of April 16, 2014. In total, this represents 969 707 protein sequences and 449 433 243 residues. They are all complete non-redundant reference proteome sets for the species chosen and are publicly available at http://www.ebi.ac.uk/reference_proteomes. The data are provided either as SeqXML (Schmitt et al., 2011) or as flat files composed of non-redundant FASTA files for ‘canonical’ and ‘additional’ sequence datasets, where ‘additional’ involves isoforms and/or variants of the canonical protein sequence for a given gene, including haplotypes, readthrough, pseudogenes, etc. Importantly, the last version of the reference proteomes includes the coding sequences (CDS DNA) for each protein. Finally, a gene-to-protein mapping file and an ‘id mapping’ file containing different database identifiers for those proteins are provided. One of the efforts that was initiated in the last Quest for Orthologs meeting was the construction of a reference species tree for these reference proteomes. For this, a working group has been created that is surveying the literature to establish a most supported topology for these species with information on what nodes may be less supported (http://swisstree.vital-it.ch/species_tree). Such a reference tree will serve to rationalize choices of subsets of the reference proteomes, as well as to expand ongoing efforts on benchmarking orthology prediction methods.
7 THE ORTHOLOG CONJECTURE STILL HOLDS
In 2011, the orthology field was baffled by a publication claiming that orthologs are less functionally conserved than paralogs (Nehrt et al., 2011). This would contradict one of the main motivations of the Quest for Orthologs, and came as a surprise because five recent papers (reviewed in Dessimoz et al., 2012; Gabaldón and Koonin, 2013) had provided different lines of support for the ortholog conjecture. If anything, the paper by Nehrt et al. showed that one has to be extremely careful when using Gene Ontology (GO) annotations between species and when comparing gene expression data. It was followed up by reports on how they had used GO incorrectly (Thomas et al., 2012) and showing that when controlling for confounders, the ortholog conjecture actually holds (Altenhoff et al., 2012). Furthermore, using microarrays and RNA-seq gene expression datasets, Chen and Zhang (2012); Huerta-Cepas et al. (2011) and Rogozin et al. (2014) showed that orthologs are more conserved in expression pattern than paralogs. Marc Robinson-Rechavi (University of Lausanne, Switzerland) presented further evidence that functional divergence between human and mouse orthologs is primarily owing to expression patterns and not to positive selection on protein sequences. In conclusion, analyzing functional conservation between species is challenging, and many pitfalls exist that can lead to unexpected and incorrect results.
Orthology by itself is an evolutionary concept and does not imply identical function. Conversely, non-homologous sequences may perform the same function, a situation referred to as analogy. This is well known, and an old criticism of the orthology concept is that it does not take divergence into account. For instance, it is likely that a mammal-specific paralogous gene pair is more similar in sequence and function than a human–Escherichia coli ortholog pair. It would therefore be useful to quantitatively estimate how functionally similar two genes are given their evolutionary relationship. At the Lausanne meeting, Jean-François Dufayard (CIRAD, Montpellier, France) presented a functional conservation score to this end, based on events and distances between two genes measured along a gene tree. The score needs to be empirically calibrated, which turned out to be difficult, but it is a promising approach to predict the level of functional conservation.
8 DOMAIN ORTHOLOGY
Most existing ortholog databases contain orthology assignments as a property of the entire protein, i.e. they consider the whole protein as a single object. However, many proteins consist of multiple domains, and domain architectures are known to evolve over time by deletion, duplication or insertion of individual domains (Buljan and Bateman, 2009). Wu et al. (2012) reported that within the Drosophila clade, domain rearrangements occur in 35.9% of the gene families. Domains on the same protein chain may be orthologous to different genes (illustrated in Fig. 1). It is certainly true that orthologs can have different domain architectures. (Forslund et al., 2011) found that between some species, 10% of the orthologs differ in domain architecture. Likewise, Lucy Mengqi Li (Imperial College, London, UK) reported at the Quest for Orthologs meeting that, based on analyses of OMA and Pfam, up to 50% may differ.
Fig. 1.
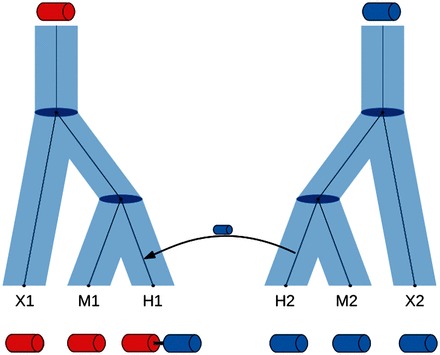
Evolutionary scenario that would give rise to inconsistent orthology relationships for different domains in a protein. The hypothetical red and blue domains are evolving by descent along the species tree of the species X, M and H, giving rise to extant proteins X1, M1, H1, H2, M2 and X2. After a duplication of protein H2 in species H, this blue domain was inserted into red-domain protein H1, such that protein H1 now has two domains. Therefore, H1’s red domain is orthologous to M1 and X1, whereas its blue domain is orthologous to M2 and X2
Other studies have analyzed independent creation of domain architectures, i.e. that the same domain architecture has been reinvented multiple times by domain rearrangements (Forslund et al., 2007; Zmasek and Godzik, 2011). Such domain architecture reinvention implies that individual domains in an architecture can have different evolutionary histories, and therefore are unlikely to be orthologous to the same genes.
Thus, as others have noted (Sjölander et al., 2011), it would make sense to apply a domain-aware approach for orthology inference. This is the case for the databases PHOG (Datta et al., 2009) and MBGD (Uchiyama, 2006; Uchiyama et al., 2012), which both consider domain architecture when making orthology assignments. Ikuo Uchiyama (National Institute for Basic Biology, Aichi, Japan) presented a new algorithm for refinement of domain boundaries using multiple alignments for the MBGD pipeline at the Quest for Orthologs meeting.
Still, many improvements can be envisaged. There is no current resource that displays both domain architecture graphics and domain-wise orthology assignments (Storm et al., 2003). The ability to distinguish between orthology supported by all domains and orthology supported by a subset of the domains only would be helpful to improve the quality of ortholog assignments and refine function inference across orthologs. Furthermore, domain orthology may in some cases reveal orthology relationships missed by full-length analyses, for instance, owing to highly divergent parts of the genes. Lucy Mengqi Li also described how domain-aware orthology inference could be used to elucidate mechanisms of architectural changes. One must keep in mind, however, that adding the domain level to orthology analysis further adds to the computational burden, and probably compromises between coverage and level of refinement have to be made.
9 HOMOLOGY RELATIONSHIPS ARISING FROM NON-VERTICAL INHERITANCE
Orthology analyses generally assume that the genetic material is propagated by vertical descent from preexisting genes either by speciation (resulting in orthologs) or duplication (resulting in paralogs). This encapsulation is however known to often be violated in prokaryotes that frequently exchange DNA with each other, a mechanism termed HGT. Although to a lesser extent, HGT is also detected in microbial groups of eukaryotes, particularly in fungi (Keeling and Palmer, 2008; Marcet-Houben and Gabaldón, 2009). To describe the homology relationship between genes related by a non-vertical transfer event, the term xenolog was introduced (Gray and Fitch, 1983). It is often difficult to assess whether a gene has been horizontally transferred or has evolved in an unusual way. This is especially true in analyses performed at large scales and using automated procedures. At the same time, a recent comparative study has shown that current orthology inference methods perform poorly in the presence of HGT (Dalquen et al., 2013). Paul Thomas (University of Southern California, Los Angeles, USA) presented a new method to reconcile trees allowing for duplication and HGT, by comparing two alternative hypotheses at each step during phylogenetic tree building. When two genes are inferred to be neighbors in the tree but are from very distant species, one hypothesis is that they were both vertically inherited from their common ancestor but were lost in the intermediate species; the alternative hypothesis is that there was a horizontal transfer and no deletions. With an increasing number of implied deletions in the vertical descent scenario, the relative likelihood of the horizontal transfer scenario becomes greater. Choosing a threshold of 15 implied deletions within a set of 82 organisms in PANTHER version 8 (Mi et al., 2013), Paul Thomas identified ∼2000 potential horizontal transfer events in 800 gene families from the PANTHER database (∼10% of all families in the database). Many of these cases are known evolutionary events, such as the acquisition of proteobacterial genes in the eukaryotic common ancestor (presumably from mitochondrial endosymbiosis), and that of cyanobacterial genes in the plant common ancestor (presumably from plastid endosymbiosis). Interestingly, however, some genomes have an excess of such cases that are not likely to reflect true evolutionary events. Examples include five apparent horizontal transfer events from a Plasmodium-like organism to platypus, and ∼40 apparent events from an alpha-proteobacterial organism to tick. This is more consistent with DNA sample contamination (Rickettsia is an alpha-proteobacterial symbiont of the tick gut) than actual inter-genome transfer. It thus seems worthwhile to annotate likely horizontally transferred genes, which for eukaryotes may well be contaminations that can be corrected in revised genome releases. Another approach was taken by Vincent Daubin (Lyon University, France), who used probabilistic modeling to simultaneously reconstruct the species tree and the gene trees, as well as all the implied gene duplication, loss and transfer events (Boussau et al., 2012). Last but not least, it is important to note that other natural processes, distinct from HGT, can cause gene tree topologies that are incongruent with the underlying species tree. These include, among others, the incomplete sorting of alleles across lineages during rapid speciation events and the recombination of paralogous genes (gene conversion).
10 OUTLOOK
The Quest for Orthologs community effort is progressing in many areas of shared interest to research groups in the field, both for method developers and consumers of orthologs. Yet many challenges remain, particularly in dealing with data growth and in extending the basic concept of orthology to hierarchical groups, to multi-domain proteins and to lateral gene transfer. Likewise, work still needs to be done to achieve the consortium’s vision of full interoperability among orthology resources and comprehensive and fair resource benchmarking.
To tackle these challenges, a roadmap has been laid out and 13 working groups have been created (see http://questfororthologs.org). The next meeting will take place in Barcelona in 2015. We invite all interested parties to join us in the QfO.
ACKNOWLEDGEMENTS
Members of the Quest for Orthologs Consortium: Adrian Altenhoff, Judith Blake, Brigitte Boeckmann, Mike Cherry, Hirokazu Chiba, Vincent Daubin, Todd DeLuca, Christophe Dessimoz, Jean-Francois Dufayard, Dannie Durand, Ingo Ebersberger, Jesualdo Tomas Fernandez Breis, Kristoffer Forslund, Toni Gabaldon, Manuel Gil, Đorđe Grbić, Javier Herrero, Ingrid Keseler, Evgenia Kriventseva, Gilles Lasalle, Suzanna Lewis, Lucy Mengqi Li, Benjamin Linard, Ari Löytynoja, Rachel Lyne, Maria Martin, Raja Mazumder, Vincent Miele, Sebastien Moretti, Matthieu Muffato, Christopher Mungall, Mateus Patricio, Simon Penel, Cecile Pereira, Malte Petersen, Rémi Planel, Marc Robinson-Rechavi, David Roos, Thomas Schmitt, Fabian Schreiber, Kimmen Sjölander, Nives Škunca, Erik Sonnhammer, Alan Wilter Sousa da Silva, Radek Szklarczyk, Paul Thomas, Ikuo Uchiyama, Asier Ullate, Michiel Van Bel, Klaas Vandepoele, Robert Waterhouse, Jeanne Wilbrandt, Ioannis Xenarios, Andy Yates, Evgeny Zdobnov.
Funding: Open access charges were covered by the UCL Library.
Conflict of Interest: none declared.
REFERENCES
- Altenhoff AM, Dessimoz C. Inferring orthology and paralogy. In: Anisimova M, editor. Evolutionary Genomics: Statistical and Computational methods. 2012. Vol. 1, Methods in Molecular Biology, Vol. 855. Springer Humana, New York, pp. 259–279. [Google Scholar]
- Altenhoff AM, et al. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–D294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhoff AM, et al. Resolving the ortholog conjecture: orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput. Biol. 2012;8:e1002514. doi: 10.1371/journal.pcbi.1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhoff AM, et al. Inferring hierarchical orthologous groups from orthologous gene pairs. PLoS One. 2013;8:e53786. doi: 10.1371/journal.pone.0053786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold R, et al. SIMAP—the database of all-against-all protein sequence similarities and annotations with new interfaces and increased coverage. Nucleic Acids Res. 2014;42:D279–D284. doi: 10.1093/nar/gkt970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckmann B, et al. Conceptual framework and pilot study to benchmark phylogenomic databases based on reference gene trees. Brief. Bioinform. 2011;12:423–435. doi: 10.1093/bib/bbr034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boussau B, et al. Genome-scale coestimation of species and gene trees. Genome Res. 2012;23:323–330. doi: 10.1101/gr.141978.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M, Bateman A. The evolution of protein domain families. Biochem. Soc. Trans. 2009;37:751–755. doi: 10.1042/BST0370751. [DOI] [PubMed] [Google Scholar]
- Chen X, Zhang J. The ortholog conjecture is untestable by the current gene ontology but is supported by RNA sequencing data. PLoS Comput. Biol. 2012;8:e1002784. doi: 10.1371/journal.pcbi.1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalquen DA, et al. The impact of gene duplication, insertion, deletion, lateral gene transfer and sequencing error on orthology inference: a simulation study. PLoS One. 2013;8:e56925. doi: 10.1371/journal.pone.0056925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta RS, et al. Berkeley PHOG: PhyloFacts orthology group prediction web server. Nucleic Acids Res. 2009;37:W84–W89. doi: 10.1093/nar/gkp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz C, et al. Toward community standards in the quest for orthologs. Bioinformatics. 2012;28:900–904. doi: 10.1093/bioinformatics/bts050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM. Distinguishing homologous from analogous proteins. Syst. Zool. 1970;19:99–113. [PubMed] [Google Scholar]
- Forslund K, et al. Domain architecture conservation in orthologs. BMC Bioinformatics. 2011;12:326. doi: 10.1186/1471-2105-12-326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forslund K, et al. Domain tree-based analysis of protein architecture evolution. Mol. Biol. Evol. 2007;25:254–264. doi: 10.1093/molbev/msm254. [DOI] [PubMed] [Google Scholar]
- Gabaldón T, Koonin EV. Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 2013;14:360–366. doi: 10.1038/nrg3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabaldón T, et al. Joining forces in the quest for orthologs. Genome Biol. 2009;10:403. doi: 10.1186/gb-2009-10-9-403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray GS, Fitch WM. Evolution of antibiotic resistance genes: the DNA sequence of a kanamycin resistance gene from Staphylococcus aureus. Mol. Biol. Evol. 1983;1:57–66. doi: 10.1093/oxfordjournals.molbev.a040298. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. Evidence for short-time divergence and long-time conservation of tissue-specific expression after gene duplication. Brief. Bioinform. 2011;12:442–448. doi: 10.1093/bib/bbr022. [DOI] [PubMed] [Google Scholar]
- Huerta-Cepas J, et al. PhylomeDB v4: zooming into the plurality of evolutionary histories of a genome. Nucleic Acids Res. 2014;42:D897–D902. doi: 10.1093/nar/gkt1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain E, et al. Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics. 2009;10:136. doi: 10.1186/1471-2105-10-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jothi R, et al. COCO-CL: hierarchical clustering of homology relations based on evolutionary correlations. Bioinformatics. 2006;22:779–788. doi: 10.1093/bioinformatics/btl009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jupp S, et al. The EBI RDF platform: linked open data for the life sciences. Bioinformatics. 2014;30:1338–1339. doi: 10.1093/bioinformatics/btt765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat. Rev. Genet. 2008;9:605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- Kriventseva EV, et al. OrthoDB: the hierarchical catalog of eukaryotic orthologs. Nucleic Acids Res. 2008;36:D271–D275. doi: 10.1093/nar/gkm845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T. Acquisition of prokaryotic genes by fungal genomes. Trends Genet. 2009;26:5–8. doi: 10.1016/j.tig.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Merkeev IV, et al. PHOG: a database of supergenomes built from proteome complements. BMC Evol. Biol. 2006;6:52. doi: 10.1186/1471-2148-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, et al. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–D386. doi: 10.1093/nar/gks1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehrt NL, et al. Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput. Biol. 2011;7:e1002073. doi: 10.1371/journal.pcbi.1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell S, et al. eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res. 2014;42:D231–D239. doi: 10.1093/nar/gkt1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosdocimi F, et al. Initial implementation of a comparative data analysis ontology. Evol. Bioinform. Online. 2009;5:47–66. doi: 10.4137/ebo.s2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz LP, et al. MetaPhOrs: orthology and paralogy predictions from multiple phylogenetic evidence using a consistency-based confidence score. Nucleic Acids Res. 2011;39:e32. doi: 10.1093/nar/gkq953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattei T, et al. Distributed, High-Performance and Grid Computing in Computational Biology. 2007. Lecture Notes in Computer Science. Using public resource computing and systematic pre-calculation for large scale sequence analysis. Springer, Berlin Heidelberg, pp. 11–18. [Google Scholar]
- Rogozin IB, et al. Gene family level comparative analysis of gene expression in mammals validates the ortholog conjecture. Genome Biol. Evol. 2014;6:754–762. doi: 10.1093/gbe/evu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux J, Robinson-Rechavi M. An ontology to clarify homology-related concepts. Trends Genet. 2010;26:99–102. doi: 10.1016/j.tig.2009.12.012. [DOI] [PubMed] [Google Scholar]
- Schmitt T, et al. Letter to the editor: SeqXML and OrthoXML: standards for sequence and orthology information. Brief. Bioinform. 2011;12:485–488. doi: 10.1093/bib/bbr025. [DOI] [PubMed] [Google Scholar]
- Schreiber F, Sonnhammer ELL. Hieranoid: hierarchical orthology inference. J. Mol. Biol. 2013;425:2072–2081. doi: 10.1016/j.jmb.2013.02.018. [DOI] [PubMed] [Google Scholar]
- Schreiber F, et al. TreeFam v9: a new website, more species and orthology-on-the-fly. Nucleic Acids Res. 2013;42:D922–D925. doi: 10.1093/nar/gkt1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjölander K, et al. Ortholog identification in the presence of domain architecture rearrangement. Brief. Bioinform. 2011;12:413–422. doi: 10.1093/bib/bbr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm CEV, Sonnhammer ELL. Comprehensive analysis of orthologous protein domains using the HOPS database. Genome Res. 2003;13:2353–2362. doi: 10.1101/gr1305203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer RA, Robinson-Rechavi M. How confident can we be that orthologs are similar, but paralogs differ? Trends Genet. 2009;25:210–216. doi: 10.1016/j.tig.2009.03.004. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium. Reorganizing the protein space at the universal protein resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, et al. On the use of gene ontology annotations to assess functional similarity among orthologs and paralogs: a short report. PLoS Comput. Biol. 2012;8:e1002386. doi: 10.1371/journal.pcbi.1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama I. MBGD: a platform for microbial comparative genomics based on the automated construction of orthologous groups. Nucleic Acids Res. 2006;35:D343–D346. doi: 10.1093/nar/gkl978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama I, et al. MBGD update 2013: the microbial genome database for exploring the diversity of microbial world. Nucleic Acids Res. 2012;41:D631–D635. doi: 10.1093/nar/gks1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse RM, et al. OrthoDB: a hierarchical catalog of animal, fungal and bacterial orthologs. Nucleic Acids Res. 2013;41:D358–D365. doi: 10.1093/nar/gks1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, et al. Thyroid hormone receptor orthologues from invertebrate species with emphasis on Schistosoma mansoni. BMC Evol. Biol. 2007;7:150. doi: 10.1186/1471-2148-7-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YC, et al. Evolution at the subgene level: domain rearrangements in the Drosophila phylogeny. Mol. Biol. Evol. 2012;29:689–705. doi: 10.1093/molbev/msr222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmasek CM, Godzik A. Strong functional patterns in the evolution of eukaryotic genomes revealed by the reconstruction of ancestral protein domain repertoires. Genome Biol. 2011;12:R4. doi: 10.1186/gb-2011-12-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]