

Abstract
Motivation: The ability to accurately model protein structures at the atomistic level underpins efforts to understand protein folding, to engineer natural proteins predictably and to design proteins de novo. Homology-based methods are well established and produce impressive results. However, these are limited to structures presented by and resolved for natural proteins. Addressing this problem more widely and deriving truly ab initio models requires mathematical descriptions for protein folds; the means to decorate these with natural, engineered or de novo sequences; and methods to score the resulting models.
Results: We present CCBuilder, a web-based application that tackles the problem for a defined but large class of protein structure, the α-helical coiled coils. CCBuilder generates coiled-coil backbones, builds side chains onto these frameworks and provides a range of metrics to measure the quality of the models. Its straightforward graphical user interface provides broad functionality that allows users to build and assess models, in which helix geometry, coiled-coil architecture and topology and protein sequence can be varied rapidly. We demonstrate the utility of CCBuilder by assembling models for 653 coiled-coil structures from the PDB, which cover >96% of the known coiled-coil types, and by generating models for rarer and de novo coiled-coil structures.
Availability and implementation: CCBuilder is freely available, without registration, at http://coiledcoils.chm.bris.ac.uk/app/cc_builder/
Contact: D.N.Woolfson@bristol.ac.uk or Chris.Wood@bristol.ac.uk
1 INTRODUCTION
The accurate prediction and modelling of protein structures remain key challenges in structural bioinformatics. Advances here enable applications in protein folding, design and engineering, where sequence information needs to be translated to give insight into protein structure, function and mutation. However, given the degrees of freedom inherent in polypeptide chains, the modelling of all potential structures that a sequence, even of modest length, could adopt is intractable computationally. Therefore, it is necessary to restrict the structural space being searched and modelled. Currently, this is done through comparative modelling (Martí-Renom et al., 2000), fold recognition (Lobley et al. 2009; Wu and Zhang, 2008), fragment-based recombination (Rohl et al., 2004) or other methods of restricting conformational space (Rooman et al., 1991; Park and Levitt, 1995; Gibbs et al., 2001).
Another route is to define the space to be searched by structural parameterization of protein folds (Bowie et al., 1991; Koga et al., 2012; Ponder and Richards, 1987). This becomes tractable (i) if there is a high probability that a sequence will fall into a broad class of protein structure; and (ii) if these 3D structures can be described mathematically to allow robust parameterization. The α-helical coiled coil is an example of such a protein fold where there are good broad-brush links between sequence and the overall structural class (Delorenzi and Speed, 2002; Lupas et al., 1991; Vincent et al., 2013). Furthermore, the backbone architectures can be described by a small number of structural parameters (Crick, 1953a; Offer and Sessions, 1995). Therefore, we have developed a tool for coiled-coil structures that uses parametric modelling.
Coiled coils are bundles of α-helices in which component strands wrap around each other to form rope-like super-helical structures (Lupas and Gruber, 2005). In the simplest cases, this is encoded by a relatively straightforward seven-residue repeating pattern of hydrophobic (h) and polar (p) amino acids, ‘hpphppp’ (Woolfson et al., 2012), often assigned abcdefg (Fig. 1a). This patterning, combined with the ∼3.6 residues per turn of an α-helix, generates a hydrophobic stripe that spirals around each helix giving the interface between the component helices (Fig. 1b). The interface is packed tightly together through so-called ‘knobs-into-holes’ (KIH) packing (Crick, 1953b) where the ‘knob’ residue on one helix projects into a ‘hole’ generated by a constellation of four residues on the partner helix (Fig. 1c).
Fig. 1.
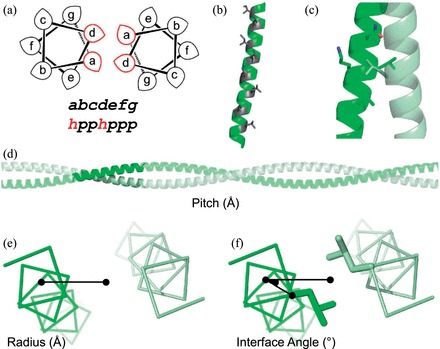
Sequence and structural features of the α-helical coiled coil. (a) Helical wheel representing a coiled-coil dimer. Hydrophobic residues are typically located at the a and d positions, with polar residues at the b, c, e, f and g positions. (b) A single helix from a coiled coil, highlighting the stripe of a/d residues (grey) that forms the assembly interface. (c) KIH packing, where the knob residue (usually a or d) from one helix (grey) projects into a hole created by four residues (black) on its partner (e.g. d’g’a’d’ or a’d’e’a’). (d) The pitch parameter describes the distance (Å) for a component helix to screw 360° around the super-helical axis. (e) The radius of assembly is measured from the super-helical axis to the centre of a component helix. (f) Interface angle, or ϕ1, is measured as the angle between the vector from the super-helical axis to the helical centre and the vector from the helical centre to the Cα carbon of an a-position residue
Despite the relatively straightforward sequence repeat and mode of packing, complexity arises because coiled coils are not limited to the common dimeric, trimeric and tetrameric oligomers: pentamers (Malashkevich et al., 1996), a decamer (Sun et al., 2014) and a dodecamer (Koronakis et al., 2000) are observed in nature, while there also exist a de novo hexamer (Zaccai et al., 2011) and an engineered heptamer (Liu et al., 2006). Furthermore, within these gross architectures, the helices can be arranged in parallel, antiparallel or mixed topologies, and the assemblies can be homo- or hetero-typic (Lupas and Gruber, 2005; Testa et al., 2009; Moutevelis and Woolfson, 2009). For the lower-order assemblies (2–4), oligomer state is largely dictated by the side chains that form the a/d interface (Harbury et al., 1993, 1994; Woolfson and Alber, 1995) and how they can pack together in space. The specification of oligomers above tetramer is less well understood, but it is clear that more complex sequence patterns than hpphppp are required (Woolfson et al., 2012). In other words, we have a good understanding of how parallel dimers, trimers and tetramers are specified by natural sequences and how these can be made de novo; however, beyond these there is more to learn.
Although methods are available to model coiled coils (Crick, 1953a; Offer et al., 2002; Grigoryan and Degrado, 2011), these only generate the α-helical backbones, which severely limits the modelling, prediction and design of coiled-coil structures. This is because of the clear link between the make up of helix–helix interfacing residues and coiled-coil oligomer state (Harbury et al., 1993; Woolfson and Alber, 1995), i.e. a side-chain packing effect, which must be captured in models.
Our aim was to improve the modelling and design of coiled-coil sequences and structures and to develop a tool for non-specialist to perform these tasks. While the large database of known coiled coils has informed and allowed us to test our approach (Lupas and Gruber, 2005; Moutevelis and Woolfson, 2009; Testa et al., 2009), coiled-coil proteins present considerable scope for generating completely de novo structures (Woolfson, 2005; Zaccai et al., 2011). This illustrates further the power and potential of this type of structural parametric modelling.
We set out to create an application that could model as many coiled-coil architectures and topologies as possible, that is, those observed already in nature plus those that are theoretically possible but currently without natural precedents. To do this, we divided the problem into two parts: first, to generate backbone models for the many coiled-coil structures possible; second, to fit and score sequences onto these frameworks.
CCBuilder is a web-based application that generates complete (backbone and side chain) atomistic models of heptad-based coiled coils in conformations specified by the user. Models are scored for feasibility with a measure of backbone strain, a test for KIH packing, and two atom-based forcefields. A basic ‘Builder’ mode is capable of modelling homo- and hetero-oligomeric coiled coils in parallel (up to 26 chains) and antiparallel (even numbered chains up to 26) conformations. This covers 96.3% of coiled coils in CC+ (Testa et al., 2009) with ≤50% sequence redundancy. Furthermore, an ‘Advanced’ builder mode allows parameters to be specified for individual α-helices around a super-helical axis, enabling some of the remaining more unusual coiled-coil conformations to be modelled. It also presents opportunities for building models for the many as yet unobserved, but theoretically possible, coiled-coil structures.
2 METHODS
2.1 Application architecture
CCBuilder has three main parts: a web-based graphical user interface (GUI), a coiled coil modelling component and a validation/scoring method. The GUI is set up as part of a Linux, Apache, NoSQL/MongoDB, Python/Django stack with a browser-based front end for collecting input parameters and returning the resultant models to the user. As models are generated server side, modelling, validation and visualization can be performed on any device with a WebGL-enabled browser, including certain mobile platforms.
The GUI has a GLmol-powered molecular viewer (http://webglmol.sourceforge.jp/index-en.html) to display the models generated (Fig. 2a). Beside this, there are six main tabs: ‘Build’, ‘Log’, ‘Models’, ‘Interface’, ‘Plot’ and ‘Information’. ‘Build’ collects input parameters and displays model scoring. It also has a list of example models based on known coiled-coil crystal structures (Table 1) as a guide for parameter selection. ‘Log’ gives a detailed list of energies per residue, which is useful for identifying clashes between side chains, as well as a full SOCKET (Walshaw and Woolfson, 2001) report to indicate if and where KIH interactions are made. ‘Models’ gives a history of the last 10 models generated and allows these to be rebuilt or downloaded by the user. ‘Interface’ allows the interface angle—a parameter that defines the rotation of helices relative to the long axis of the coiled coil (Fig. 1f)—to be set graphically, easing parameter selection. ‘Plot’ displays a line graph of the Rosetta and Bristol University Docking Engine (BUDE) scores for the last 10 models generated. The ‘Information’ tab gives a brief guide to operation and a list of useful references.
Fig. 2.
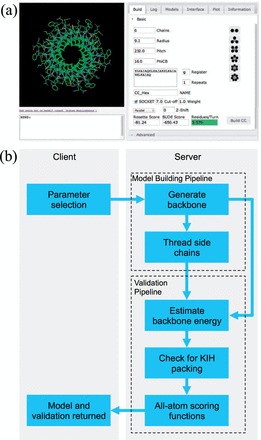
Overall architecture, appearance and workflow of CCBuilder. (a) GUI displaying the molecule viewer and the ‘Basic’ build tab, which contains fields for parameter entry and displays the returned model scoring. (b) Application architecture and workflow
Table 1.
‘Standard’ helical and coiled-coil parameters
| Oligomer state | PDB code | Sequence (gabcdef)4 | Residues per α-helical turn | Radius (Å) | Pitch (Å) | Interface angle (°) |
|---|---|---|---|---|---|---|
| 2 | 4DZM | EIAALKQ EIAALKK EIAALKW EIAALKQ | 3.62 (0.06) | 5.07 (0.26) | 225.8 (70.3) | 26.42 (2.29) |
| 3 | 4DZK | EIAAIKQ EIAAIKK EIAAIKW EIAAIKQ | 3.60 (0.03) | 6.34 (0.22) | 194.0 (36.1) | 19.98 (1.77) |
| 4 | 3R4A | ELAAIKQ ELAAIKK ELAAIKW ELAAIKQ | 3.60 (0.02) | 6.81 (0.07) | 213.2 (6.8) | 22.06 (1.27) |
| 5 | 1MZ9 | ELQETNA ALQDVRE LLRQQVK EITFLKN | 3.62 (0.04) | 8.57 (0.16) | 174.1 (12.8) | 14.27 (4.10) |
| 6 | 3R46 | ELKAIAQ ELKAIAK ELKAIAW ELKAIAQ | 3.57 (0.05) | 9.13 (0.14) | 228.4 (29.4) | 16.40 (1.13) |
Note: Values in brackets are for the standard deviations.
Building a model requires basic sequence and register information along with oligomer state and three geometric parameters: radius, pitch and interface angle/ϕ1, Figure 1d–f. Initially, a poly-glycine model of the α-helical backbone is generated using Crick’s mathematical description of a coiled coil (Crick, 1953a), as implemented in MAKECCSC (Offer and Sessions, 1995). To generate antiparallel helices, the coordinates of specified helices are rotated around the axis defined by the vector between the centre of the antiparallel helix and the centre of the whole assembly. This is performed using rotation matrices that are generated for each antiparallel chain and are applied to the atomic coordinates. If a z-shift parameter is required—which is a displacement of a helix along the length of the super-helical axis, measured in Å—helices are also rotated about, as well as translated along, the super-helical axis. This is to maintain the orientation of the helices relative to the assembly. The rotation angle (θ°) is calculated from the distance of z-shift (z) required and the pitch of the assembly (P).
| (1) |
This angle and the super-helical axis are used to generate a rotation matrix, which is applied to the shifted strands, ensuring that these remain in phase with the pitch of the assembly after translation.
After the backbone has been created, side chains are added onto this frame using SCRWL 4 (Krivov et al., 2009), and the side-chain relaxation algorithm from the Rosetta molecular modelling package as implemented in PyRosetta (Chaudhury et al., 2010). The model is then ready for validation. Three methods are applied to test model validity: a measure of backbone strain, testing for KIH packing, and two all-atom scoring functions.
As users are free to specify a broad range of values for input parameters, there is the potential to generate strained helices (i.e. helices where geometry and hydrogen bonding are far from ideal) through using either low or high pitch values. Rather than restrict the permitted values, a measure of backbone strain is used to allow users the choice of what models to accept and reject. To determine whether the backbone in the models is strained, the number of residues per α-helical turn (n) was calculated, using Helanal (Bansal et al., 2000), for a set of 32 878, non-redundant, α-helices selected from 2417 crystal structures with a resolution of better than 1.6 Å and sequence identity <30%. The value for n for the model, which is extracted from MAKECCSC when the model is generated, is returned to the user with an indication of how it compares with the reference distribution.
Models are tested for KIH packing using SOCKET (Walshaw and Woolfson, 2001). A full report is available in the ‘Log’ tab detailing which residues are involved. The cut-off distance for finding KIH packing is 7 Å as default, but this can be modified via the ‘Build’ tab. Knob residues are highlighted in the model viewer as ‘sticks’ compared with the ‘line’ representation for other residues.
Two scoring functions are applied to the final model: Rosetta (Bradley et al., 2005) using the standard force field, with the ‘Score 12’ patch; and BUDE (McIntosh-Smith et al., 2011; Mcintosh-smith et al., 2014). BUDE uses an empirical free-energy force field to predict the free energy of binding between two molecules. BUDE is a general purpose, GPU-accelerated, molecular-docking program designed to perform virtual screening, binding site identification and protein–protein docking in real space. Here, we have applied it to calculate binding energies between the component chains of coiled-coil models by making minor modifications to the original residue-based force field parameters (the resulting force field is Bude_FF-R1).
For CCBuilder, BUDE scores a single conformation specified by the user during model generation. One α-helix in the coiled coil is designated as the ‘ligand’, with the other helix/helices designated as the ‘receptor’ before energy of binding is calculated. This is repeated for each strand in the assembly and averaged to generate the final score. As BUDE is designed to minimize thousands of potential drug candidates, each in a range of different conformations, the binding energy of a single conformation can be evaluated in a fraction of a second, making it ideal for a web-based application where response time contributes to overall user experience.
2.2 Helix-building validation
To test the model-building protocol, we rebuilt a set of 594 parallel homo-oligomeric coiled coils (oligomer state between 2 and 6 and ≥40 amino acids) selected from CC+, a relational database of coiled-coil proteins (Testa et al., 2009). The parameters required to build models of these coiled coils were extracted from the crystal structures using TWISTER (Strelkov and Burkhard, 2002), which measures them in three residue frames before averaging across the length of the protein. The coordinates of the structure and the model were fitted, and the RMSD was calculated using the McLachlan algorithm (McLachlan, 1982) as implemented in the program ProFit (Martin, A.C.R., http://www.bioinf.org.uk/software/profit/). To allow for comparison between sequences of different length, the RMSD scores were normalized using the RMSD100 algorithm (Carugo and Pongor, 2001), where N = number of amino acids.
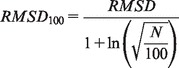 |
(2) |
CCBuilder’s antiparallel mode was tested in the same manner. A set of 59 homo-oligomeric antiparallel coiled-coil dimers, with ≥40 amino acids, was modelled in CCBuilder, and RMSD values between the model and the crystal structure calculated and normalized to RMSD100 scores.
3 RESULTS
3.1 Residues per α-helical turn as a measure of backbone strain
Helanal (Bansal et al., 2000) was used to determine the mean value of n in a set of α-helices extracted from high-resolution structures (<1.6 Å) (Fig. 3a). This was found to be 3.60, which should represent average geometry for an unstrained α-helix. However, the distribution was broader than expected, with a shoulder around n = 3.3. This closely mirrors an aforementioned distribution found in a smaller set of α-helices (Chothia et al., 1981), with the shoulder being attributed to segments of 310-helix found at the ends of α-helical regions. These non–α-helical regions were filtered out using helix definitions from DSSP (Kabsch and Sander, 1983) (Fig. 3b), generating a mean value of n = 3.65. For comparison, we determined the distribution in α-helices from coiled coils of the CC+ database (Testa et al., 2009). For these, the mean value was 3.62. As judged by Student’s t-test, the two distributions were indistinguishable, t(6768) = 27.90, P = 2.2 × 10−16. Thus, models returned with n = 3.65 ± 0.07 should be considered as models with good backbone geometry for coiled coils and helical assemblies in general.
Fig. 3.
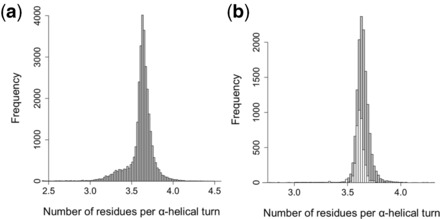
Distribution of residues per α-helical turn. (a) Residues per α-helical turn (n) in a set of 32 878 α-helices extracted from 2417 crystal structures with a resolution of <1.6 Å and sequence identity <30%, using helix definitions contained in the header of the coordinate files. Mean values of n = 3.60 (SD = 0.20). (b) Grey bars: values of n found in a subset of 13 703 helices from the distribution in Figure 3A, using helices defined by DSSP (Kabsch and Sander, 1983). Mean = 3.65 (SD = 0.07). White bars: values of n found in 4167 helices extracted from 1473 coiled-coil crystal structures. Mean = 3.62 (SD = 0.07)
3.2 Model validation
Models were constructed based on parameters derived from known crystal structures to test CCBuilder’s model-building protocol. The building process was automated to produce 653 models for comparison with the corresponding crystal structure (Table 2 and Fig. 4). We found that backbones were modelled well, with an average RMSD100 score of <0.80 Å measured over all backbone atoms, similar to previously reported values for backbone models (Grigoryan and Degrado, 2011). With side-chain atoms included, the calculated models reproduced the experimental structures with a reasonable mean RMSD100 score of 2.20 Å. This value is inflated, especially in lower order coiled coils, as five of seven residues of the heptad repeat—i.e. those other than a and d—are relatively unconstrained (Fig. 1a). It should follow that as oligomer state increases, more residues become constrained as the helix–helix interfaces broaden—i.e. to partially include those at e and g—and therefore, the averaged RMSD100 scores should decrease. This was observed with average all-atom RMSD100 scores for dimers (250), trimers (266) and tetramers (50) being 2.38 Å, 2.13 Å and 1.69 Å, respectively.
Table 2.
Average RMSD100 scores between models generated with CCBuilder and known coiled-coil structures, for both parallel and antiparallel conformations
| Orientation | Number of models | Average RMSD100 scores (Å) |
||
|---|---|---|---|---|
| All atoms | Backbone | Cα Only | ||
| Parallel | 594 | 2.17 (0.68) | 0.74 (0.45) | 0.72 (0.50) |
| Antiparallel | 59 | 2.45 (0.59) | 1.17 (0.68) | 1.11 (0.67) |
| Combined | 653 | 2.20 (0.67) | 0.77 (0.49) | 0.76 (0.53) |
Note: Values in brackets are for the standard deviations.
Fig. 4.
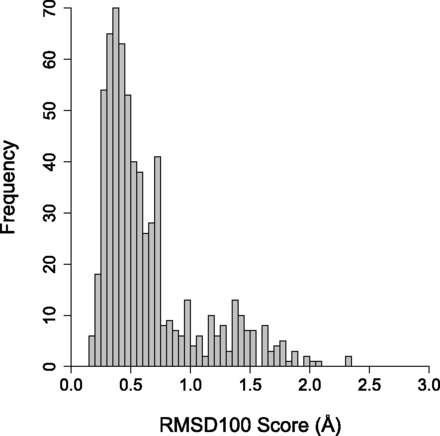
Distribution of RMSD100 scores measured between the backbone atoms of models generated with CCBuilder and crystal structures of known coiled coils. The set used for validation contained 594 parallel and 59 antiparallel coiled coils extracted from CC+ The mean RMSD100 score was 2.20 Å (SD = 0.67) for all atoms, 0.77 Å (SD = 0.49) for backbone only and 0.76 Å (SD = 0.53) for Cα carbons only
On average, the RMSD100 scores were higher for the antiparallel models than the parallel models (Table 2). This can be understood in that the latter required an extra parameter (the z-shift) to describe them, leading to a larger parameter and structure space. As TWISTER (Strelkov and Burkhard, 2002) does not measure this parameter and could not determine this a priori, z-shifts were applied to each model individually as they were constructed, through sequentially varying the value and scoring the model.
The overlays of Figure 5 are used to illustrate the fits between the model and experimental structures in more detail. These are for the well-characterized coiled-coil dimer from the yeast transcriptional activator GCN4 (2ZTA) (Gonzalez et al., 1996), an antiparallel dimeric coiled coil from bovine IF1 (1GMJ) (Cabezón et al., 2001), the de novo parallel hexameric coiled coil CC-Hex (3R3K) (Zaccai et al., 2011) and a slipped engineered heptameric mutant of the aforementioned GCN4 peptide (2HY6) (Liu et al., 2006). These were selected to demonstrate a range of different model types that CCBuilder can generate. From these overlays, it is apparent that CCBuilder captures the gross structural properties of the assemblies, with good alignment of the backbones.
Fig. 5.
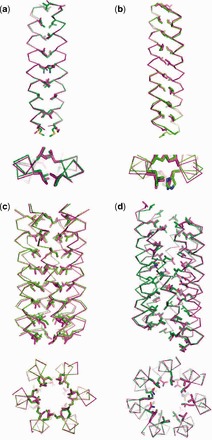
Overlays of crystal structures and models generated by CCBuilder. (a) Parallel dimer (2ZTA) RMSD 0.59 Å all atoms, 0.30 Å backbone and 0.30 Å Cα only. (b) Antiparallel dimer, with distinct z-shifted, (1GMJ) RMSD 0.98 Å all atoms, 0.63 Å backbone and 0.64 Å Cα only. (c) Parallel hexamer (3R3K) RMSD 0.71 Å all atoms, 0.34 Å backbone and 0.34 Å Cα only. (d) Slipped parallel heptamer (2HY6) 0.35 Å all atoms, 0.30 Å backbone and 0.29 Å Cα only. Key: Magenta = Crystal structure, Green = CCBuilder model
As mentioned above, a possible consequence of coiled-coil folding is that residues outside the a and d sites could be relatively unconstrained (Fig. 1). That said, along with residues at the e and g sites, it is those at a and d that contribute most to stability and oligomer-state definition for the vast majority of coiled-coil structures. Thus, we felt it important that these residues were modelled accurately. Of the core residues in the examined structures, the experimentally observed rotamers were consistently selected in our models for valine and isoleucine, and the correct leucine rotamers were reproduced well in both dimers and the slipped heptamer. However, for CC-Hex, the rotamers of the leucine side chains were almost all incorrectly assigned, possibly indicating that these residues are less constrained in this particular structure. Interestingly, and consistent with this, we find that these leucine residues, which fall at a sites, are highly mutable in CC-Hex where we have made and solved X-ray crystal structures for Leu→Asp, His and Cys mutants (Burton et al., 2013; Zaccai et al., 2011). We will explore elsewhere if rotamer selection can be improved using a coiled-coil–specific rotamer library.
4 DISCUSSION
We have described CCBuilder, a web-based interactive tool for generating models of coiled-coil structures. The input sequences and parameters can be taken from known examples, or can be completely novel, allowing modelling of natural and de novo coiled coils. For the former, CCBuilder produces models that accurately match existing crystal structures, not just for the common parallel dimers, trimers, tetramers and antiparallel dimers but also for more unusual coiled-coil geometries. For de novo coiled coils, unrestricted selection of parameters allows the user to freely explore potential coiled-coil conformation space, but this is only useful if there is robust model validation to guide the users design. In CCBuilder, this is provided through the combination of a measure of backbone strain, checking for KIH packing and the choice of two all-atom scoring functions.
Using the number of residues per α-helical turn as a measure of backbone strain exploits a parameter that is generated during backbone construction at little computational cost. It also ensures good α-helical backbone geometry, which we find to be tightly defined in general protein structures and coiled coils. The KIH check, which is done rapidly on the fly with SOCKET (Walshaw and Woolfson, 2001), is important to ensure that this signature feature of coiled coils that gives intimate helix–helix packing is captured in the models.
For the final stage of model validation, both the Rosetta and BUDE force fields are included to offer the user a choice of scoring method. For this application of constructing coiled-coil bundles, however, we find that BUDE is better suited to scoring interactions between the component helices than the standard Rosetta force field. We believe that this is because of (i) the relative hardness of atoms in the two force fields, with BUDE being the ‘softer’, allowing better accommodation of minor geometrical inaccuracies; and (ii) the BUDE hydrophobicity function giving a better representation of desolvation.
Both the models created for the large rebuild of known coiled-coil structures (Table 2 and Fig. 4) and the dimeric and hexameric models shown in Figure 5a–c used only the parameters included in the ‘Basic’ build mode. With this functionality alone, it is possible to model >96% of known coiled-coil structures (Moutevelis and Woolfson, 2009; Testa et al., 2009), which should account for the vast majority of biologically relevant coiled coils. It should be possible to model most of the remaining coiled coils using the features included on the ‘Advanced’ mode, which allows parameters to be specified for each chain individually relative to a central helical axis. The advanced mode was used to recreate the heptameric model (Fig. 5d), where one of the interfaces between component helices is slipped by a heptad (∼10 Å), leaving a layer of hydrophobic ‘core’ residue unsatisfied on two strands. Modelling this required systematic variation of z-shift for each component chain. Nonetheless, the model returned closely matched the experimental structure.
CCBuilder also allows the construction of models for coiled-coil sequences where structural data are not available. The resulting coordinate sets have potential as search models for molecular-replacement solutions for X-ray diffraction data or as initial structures in molecular modelling and dynamics simulations. Furthermore, CCBuilder could be used predictively, allowing the rational design of mutants of existing coiled coils, or for completely de novo sequences and structures. Thus, CCBuilder should expedite the generation and visualization of models and allow real-time refinement of these via the input parameters by the user, above and beyond what is available through any other software currently available. The combination of measuring backbone geometry, checking for KIH packing and all-atom scoring functions allows the robust assessment of the feasibility of models. Thus, we suggest that CCBuilder is the best currently available tool for the design of coiled-coil proteins and assemblies that are theoretically possible, but hav1e yet to be observed in nature (Woolfson et al., 2012).
Funding: C.W.W. thanks the BBSRC for a PhD studentship through the South West Doctoral Training Partnership. D.N.W. thanks the EPSRC (EP/J001430/1), BBSRC (BB/J008990/1; grant with RLB) and the ERC (340764) for financial support.
Conflict of Interest: none declared.
REFERENCES
- Bansal M, et al. HELANAL: a program to characterize helix geometry in proteins. J. Biomol. Struct. Dyn. 2000;17:811–819. doi: 10.1080/07391102.2000.10506570. [DOI] [PubMed] [Google Scholar]
- Bowie JU, et al. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- Bradley P, et al. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- Burton AJ, et al. Accessibility, reactivity, and selectivity of side chains within a channel of de novo peptide assembly. J. Am. Chem. Soc. 2013;135:12524–12527. doi: 10.1021/ja4053027. [DOI] [PubMed] [Google Scholar]
- Cabezón E, et al. The structure of bovine IF(1), the regulatory subunit of mitochondrial F-ATPase. EMBO J. 2001;20:6990–6996. doi: 10.1093/emboj/20.24.6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carugo O, Pongor S. A normalized root-mean-square distance for comparing protein three-dimensional structures. Protein Sci. 2001;10:1470–1473. doi: 10.1110/ps.690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury S, et al. PyRosetta: a script-based interface for implementing molecular modeling algorithms using Rosetta. Bioinformatics. 2010;26:689–691. doi: 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C, et al. Helix to helix packing in proteins. J. Mol. Biol. 1981;145:215–250. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- Crick FHC. The Fourier transform of a coiled-coil. Acta Crystallogr. 1953a;6:685–689. [Google Scholar]
- Crick FHC. The packing of α-helices: simple coiled-coils. Acta Crystallogr. 1953b;6:689–697. [Google Scholar]
- Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18:617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- Gibbs N, et al. Ab initio protein structure prediction using physicochemical potentials and a simplified off-lattice model. Proteins. 2001;43:186–202. doi: 10.1002/1097-0134(20010501)43:2<186::aid-prot1030>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Gonzalez L, et al. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat. Struct. Mol. Biol. 1996;3:1011–1018. doi: 10.1038/nsb1296-1011. [DOI] [PubMed] [Google Scholar]
- Grigoryan G, DeGrado WF. Probing designability via a generalized model of helical bundle geometry. J. Mol. Biol. 2011;405:1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury PB, et al. A switch between, two- , three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Harbury PB, et al. Crystal structure of an isoleucine-zipper trimer. Nature. 1994;371:80–83. doi: 10.1038/371080a0. [DOI] [PubMed] [Google Scholar]
- Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- Koga N, et al. Principles for designing ideal protein structures. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koronakis V, et al. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–919. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- Krivov GG, et al. Improved prediction of protein side-chain conformations with SCWRL4. Proteins. 2009;77:778–795. doi: 10.1002/prot.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, et al. A seven-helix coiled coil. Proc. Natl Acad. Sci. USA. 2006;103:15457–15462. doi: 10.1073/pnas.0604871103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobley A, et al. pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics. 2009;25:1761–1767. doi: 10.1093/bioinformatics/btp302. [DOI] [PubMed] [Google Scholar]
- Lupas A, et al. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Lupas AN, Gruber M. The structure of α-helical coiled coils. Adv. Protein Chem. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- Malashkevich VN, et al. The crystal structure of a five-stranded coiled coil in COMP: a prototype ion channel? Science. 1996;274:761–765. doi: 10.1126/science.274.5288.761. [DOI] [PubMed] [Google Scholar]
- Martí-Renom MA, et al. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- McIntosh-Smith S, et al. Benchmarking energy efficiency, power costs and carbon emissions on heterogeneous systems. Comput. J. 2011;55:192–205. [Google Scholar]
- Mcintosh-smith S, et al. High performance in silico virtual drug screening on many-core processors. Int. J. High Perform. Comput. Appl. 2014 doi: 10.1177/1094342014528252. doi:10.1177/1094342014528252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan A. Rapid comparison of protein structures. Acta Crystallogr. Sect. A. 1982;38:871–873. [Google Scholar]
- Moutevelis E, Woolfson DN. A periodic table of coiled-coil protein structures. J. Mol. Biol. 2009;385:726–732. doi: 10.1016/j.jmb.2008.11.028. [DOI] [PubMed] [Google Scholar]
- Offer G, et al. Generalized Crick equations for modeling noncanonical coiled coils. J. Struct. Biol. 2002;137:41–53. doi: 10.1006/jsbi.2002.4448. [DOI] [PubMed] [Google Scholar]
- Offer G, Sessions RB. Computer modelling of the a α-helical coiled coil: packing of side-chains in the inner core. J. Mol. Biol. 1995;249:967–987. doi: 10.1006/jmbi.1995.0352. [DOI] [PubMed] [Google Scholar]
- Park BH, Levitt M. The Complexity and Accuracy of Discrete State Models of Protein Structure. J. Mol. Biol. 1995;249:493–507. doi: 10.1006/jmbi.1995.0311. [DOI] [PubMed] [Google Scholar]
- Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J. Mol. Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- Rohl CA, et al. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Rooman MJ, et al. Prediction of protein backbone conformation based on seven structure assignments. Influence of local interactions. J. Mol. Biol. 1991;221:961–979. doi: 10.1016/0022-2836(91)80186-x. [DOI] [PubMed] [Google Scholar]
- Strelkov SV, Burkhard P. Analysis of alpha-helical coiled coils with the program TWISTER reveals a structural mechanism for stutter compensation. J. Struct. Biol. 2002;137:54–64. doi: 10.1006/jsbi.2002.4454. [DOI] [PubMed] [Google Scholar]
- Sun L, et al. Icosahedral bacteriophage ΦX174 forms a tail for DNA transport during infection. Nature. 2014;505:432–435. doi: 10.1038/nature12816. [DOI] [PubMed] [Google Scholar]
- Testa OD, et al. CC+: a relational database of coiled-coil structures. Nucleic Acids Res. 2009;37:D315–D322. doi: 10.1093/nar/gkn675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent TL, et al. LOGICOIL—multi-state prediction of coiled-coil oligomeric state. Bioinformatics. 2013;29:69–76. doi: 10.1093/bioinformatics/bts648. [DOI] [PubMed] [Google Scholar]
- Walshaw J, Woolfson DN. Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol. 2001;307:1427–1450. doi: 10.1006/jmbi.2001.4545. [DOI] [PubMed] [Google Scholar]
- Woolfson DN, Alber T. Predicting oligomerization states of coiled coils. Protein Sci. 1995;4:1596–1607. doi: 10.1002/pro.5560040818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfson DN. The design of coiled-coil structures and assemblies. Adv. Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- Woolfson DN, et al. New currency for old rope: from coiled-coil assemblies to α-helical barrels. Curr. Opin. Struct. Biol. 2012;22:432–441. doi: 10.1016/j.sbi.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhang Y. MUSTER: improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547–556. doi: 10.1002/prot.21945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaccai NR, et al. A de novo peptide hexamer with a mutable channel. Nat. Chem. Biol. 2011;7:935–941. doi: 10.1038/nchembio.692. [DOI] [PMC free article] [PubMed] [Google Scholar]