

Abstract
Under defined conditions, mesenchymal stem cells can differentiate into unique cell types, making them attractive candidates for cell-based disease therapies. Ischemic diseases would greatly benefit from treatments that include the formation of new blood vessels from mesenchymal stem cells. However, blood vessels are complex structures composed of endothelial cells and smooth muscle cells, and their assembly and function in a diseased environment is reliant upon joining with the pre-existing vasculature. Although endothelial cell/smooth muscle cell interactions are well known, how endothelial cells may influence mesenchymal stem cells and facilitate their differentiation has not been defined. Therefore, we sought to explore how endothelial cells might drive mesenchymal stem cells toward a smooth muscle fate. Our data show that cocultured endothelial cells induce smooth muscle cell differentiation in mesenchymal stem cells. Endothelial cells can promote a contractile phenotype, reduce proliferation, and enhance collagen synthesis and secretion. Our data show that Notch signaling is essential for endothelial cell-dependent differentiation, and this differentiation pathway is largely independent of growth factor signaling mechanisms.
Introduction
Bone marrow-derived mesenchymal stem cells have the capacity to differentiate into a range of cell types, making them an attractive source of stem cells for tissue engineering and organ repair [1–3]. Of particular interest is their potential to contribute to the formation or repair of blood vessels [4]. Patients with ischemic injuries, such as stroke and myocardial infarction, would greatly benefit from newly formed vessels derived from mesenchymal stem cells [3]. Whereas neovascularization treatments to activate and recruit resident mesenchymal stem cells could be used to stave off peripheral artery disease [5]. Despite the tremendous potential that the mesenchymal stem cell precursors hold for treatment of disease, the multipotent nature of these cells offers challenges to harnessing their potential. Mesenchymal stem cells have been shown to differentiate into many cells types, including osteoblasts, chondrocytes, adipocytes, endothelial cells, and smooth muscle cells [2,3]. Several in vitro studies have identified precise techniques for their differentiation into desired cell types [2,6]. However, the in vivo environment in which they are placed likely has a substantial impact in defining the fate and function of these cells. For example, placing mesenchymal stem cells in a proangiogenic environment would presumably promote blood vessel assembly. Yet, how are mesenchymal stem cells instructed to differentiate into both endothelial cells and smooth muscle cells? A functional blood vessel is composed of two primary cells types, endothelial cells and smooth muscle cells or pericytes, and there is substantial interaction between the cells and the vasculature.
In adult blood vessels, it is well established that endothelial cells impact vascular smooth muscle cell function by governing their contractile response [7,8]. Endothelial cell-derived factors like nitric oxide and endothelin are perceived by surrounding smooth muscle cells, which alters vascular reactivity. During development, the formation of blood vessels is dependent upon the ability of endothelial cells to recruit precursor smooth muscle cells and promote their differentiation [9,10]. The recruitment and differentiation of vascular smooth muscle cells by endothelial cells is regulated by platelet-derived growth factor (PDGF), transforming growth factor-β (TGFβ), and Notch signaling [11]; all factors which have been implicated in regulating mesenchymal stem cell differentiation [12–14]. Thus, the presence of endothelial cells within the mesenchymal stem cell environment likely plays a substantial role in their differentiation decisions. Given that mesenchymal stem cells are being investigated as a source of cells for blood vessel repair and engineering, it seems valuable to understand the impact of endothelial cells on the mesenchymal stem cell population.
In this study, we examined the effect of cocultured endothelial cells on bone marrow-derived mesenchymal stem cell differentiation. The data show that endothelial cells originating from unique vascular beds can promote the differentiation of mesenchymal stem cells toward a smooth muscle fate. Endothelial cells cause an increase in contractile gene expression and function, while concomitantly decreasing stem cell markers. Further analysis of the smooth muscle cell phenotype revealed that endothelial cells promote quiescence in mesenchymal stem cells and increase a synthetic phenotype, all of which is dependent upon Notch signaling. These data highlight the importance of cellular environment on mesenchymal stem cell differentiation, and in particular demonstrate a potentially critical role of endothelial cells in mesenchymal stem cell fate decisions.
Materials and Methods
Cell culture
Human adult bone marrow-derived mesenchymal stem cells (HMSC) were purchased from ScienCell and cultured in Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher Scientific) supplemented with 5% fetal bovine serum (FBS; HyClone), 2 mM glutamine, 1 mM sodium pyruvate, and 100 U/mL penicillin/streptomycin. Primary cultures of human aortic smooth muscle cells (HAoSMC) were purchased from VascuLife and maintained in DMEM with 5% FBS, insulin (4 ng/mL), EGF (5 ng/mL), ascorbic acid (50 ng/mL), and supplemented as described above. Human umbilical vein endothelial cells (HUVEC; Cascade Biologics), human microvascular endothelial cells (Lonza), and human pulmonary artery endothelial cells (Lifeline) were grown in EBM-2 supplemented with the BulletKit components (Lonza) as recommended by the supplier. Human adenocarcinoma (HeLa) cells were purchased from the American Type Culture Collection and cultured in DMEM supplemented with 5% FBS. TN-293 cells were purchased from Stratagene and cultured in DMEM as described above with 5% FBS. All cultures were maintained in humidified 5% CO2 at 37°C. For coculture, 1.25×105 HMSC or HAoSMC were plated in six-well plates, and after 24 h, equal numbers of endothelial cells were added. For blocking Notch signaling, N-[(3,5-Difluorophenyl)acetyl]-L-alanyl-2-phenylglycine 1, 1-dimethylethyl ester (DAPT; Calbiochem) was added to 10 μM of final concentration. For HMSC differentiation into smooth muscle cells, 10 ng/mL TGFβ (PeproTech) and 5 ng/mL PDGF-BB (PeproTech) were added into the culture media for 72 h. Ten micromolars SB-431542 (Reagents Direct) and 2 μM PDGFR inhibitor I (Calbiochem) were used to inhibit TGFβ and PDGF signaling. To separate endothelial cells from HMSC and HAoSMC, anti-PECAM1-conjugated Dynabeads (Invitrogen) were used following the manufacturer's instructions. All cell coculture experiments were performed in a medium consisting of EBM-2 supplemented with the BulletKit components. The separated cells were collected for RNA and protein isolation for quantitative PCR (qPCR) or western blot analysis. For adipogenic differentiation, 8×104 HMSC were cultured in a 12-well plate with 10% DMEM containing 0.5 μM dexamethasone, 0.5 μM 1-methyl-3-isobutylxanathine (IBMX; R&D Systems) and 50 μM indomethacin (Alfa Aesar) for 96 h [2]. For osteogenic differentiation, 8×104 HMSC were cultured in a 12-well plate with 10% DMEM containing 50 μg/mL ascorbic acid, 0.1 μM dexamethasone, and 100 mM β-glycerophosphate (MP Biomedicals) for 96 h [2].
Quantitative PCR
RNA from cultured cells was extracted by the RiboZol reagent (Amersco). Reverse transcription was performed using the M-MLV reverse transcriptase (Promega) to generate complement DNA. qPCR was performed using the StepOne PCR machine (Applied Biosystems) with the SYBR Green qPCR Master Mix (Applied Biosystems). Corresponding gene expression levels were normalized to GAPDH. Primer sequences were as follows: Notch3, 5′-GAG CCA ATG CCA ACT GAA GAG (forward) and 5′-GGC AGA TCA GGT CGG AGA TG (reverse); HeyL, 5′-CAT ACA ATG TCC TTG TGC AGT ACA CA (forward) and 5′-GCC AGG GCT CGG GCA TCA AAG AA (reverse); Smooth muscle α-actin, 5′-CAA GTG ATC ACC ATC GGA AAT G (forward) and 5′-GAC TCC ATC CCG ATG AAG GA (reverse); SM22α, 5′-CAA GCT GGT GAA CAG CCT GTA C (forward) and 5′-GAC CAT GGA GGG TGG GTT CT (reverse); Calponin, 5′-TGA AGC CCC ACG ACA TTT TT (forward) and 5′-GGG TGG ACT GCA CCT GTG TA (reverse); COL1A1, 5′-CAG ACA AGC AAC CCA AAC TGA A (forward) and TGA GAG ATG AAT GCA AAG GAA AAA (reverse); COL3A1, 5′-TGG TCA GTC CTA TGC GGA TAG A (forward) and 5′-CGG ATC CTG AGT CAC AGA CAC A (reverse); COL4A1, 5′-CGT AAC TAA CAC ACC CTG CTT CAT (forward) and 5′-CAC TAT TGA AAG CTT ATC GCT GTC TT (reverse); COL5A3, 5′-GAC AGA GAC TCC AGC TCC AAA TC (forward) and 5′-TCT CTA GGA TCG TGG CAT TGA G (reverse); Cyclin D1, 5′-CGT GGC CTC TAA GAT GAA GGA (forward) and 5′-CGG TGT AGA TGC ACA GCT TCT C (reverse); Cyclin D2, 5′-CCC TCT GCT GAG CGG TAC TAA (forward) and 5′-TCT TAT CCT GCC AAT TCA GTG TGA (reverse); GAPDH, 5′-ATG GAA ATC CCA TCA CCA TCT T (forward) and 5′-CGC CCC ACT TGA TTT TGG (reverse); CD73, 5′-CAC TGG GAC ATT CGG GTT TT (forward) and 5′-CGT CCA CAC CCC TCA CTT TC (reverse); CD90, 5′-CGA ACC AAC TTC ACC AGC AAA T (forward) and 5′-CCT TGC TAG TGA AGG CGG ATA (reverse); CD105, 5′-TTG TCT TGC GCA GTG CTT ACT C (forward) and 5′-CCG CCT CAT TGC TGA TCA TA (reverse); KLF4, 5′-ACC AGG CAC TAC CGT AAA CAC A (forward) and 5′-ATG CTC GGT CGC ATT TTT G (reverse); Myocardin, 5′-GAC AGT AAG AAC CGC CAC AAA AA (forward) and 5′-GGG AAT GTA CTG GTG ATA TTT AAG CTT (reverse); ALP, 5′-CCG GGC AAC TCT ATC TTT GG (forward) and 5′-GAT GGC AGT GAA GGG CTT CTT (reverse); BGLAP, 5′-TGT GAG CTC AAT CCG GAC TGT (forward) and 5′-CCG ATA GGC CTC CTG AAA GC (reverse); ADIPOQ, 5′-GCA AAA CCC ATG GAG GAA TTC (forward) and 5′-TCT TCC CTG ACC CTG TTG GT (reverse); PPARG2, 5′-TCA GGG CTG CCA GTT TCG (forward) and 5′-GCT TTT GGC ATA CTC TGT GAT CTC (reverse).
Western blotting
Cells were homogenized in the RIPA buffer containing 50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 1% NP40, 0.25% SDS, and protease inhibitors (Sigma). Protein concentrations were determined by the Bradford assay (BioRad). After running in 10% SDS-polyacrylamide gels, proteins were transferred to nitrocellulose membranes (GE Healthcare). The membranes were incubated for 1 h at room temperature with 3% nonfat dry milk, then with primary antibodies against Notch3 (1:2,000, sc-5593, Santa Cruz Biotechnology), β-tubulin (1:20,000, T7816; Sigma), smooth muscle α-actin (1:5,000,1A4; Sigma), SM-MHC (1:2,000, BT562; Biomedical Technologies Inc.), Calponin (1:2,000, C2687; Sigma) in a blocking solution overnight at 4°C. Secondary antibodies conjugated to the horseradish peroxidase (HRP), (1:5,000; Amersham Biosciences) were incubated for 2 h at room temperature. Proteins were detected by enhanced chemiluminescence (Thermo Fisher Scientific) and films were digitally scanned for protein quantification using the NIH Image software. Relative protein expression was calculated by normalization to β-tubulin.
Gel contractile assay
For contraction assays, 70 μL of rat tail collagen I (1 mg/mL; BD Biosciences) with 2.8×104 HMSC, 2.8×104 HUVEC, or 2.8×104 HeLa cells was added to the wells of a 96-well plate. Collagen gels were allowed to polymerize for 30 min at 37°C. Cells were incubated in the EBM-2 medium for another 48 h, and replaced with fresh DMEM with 10% FBS before detaching gels from the culture plate. Images of gels were captured after 1 h following gel release, and gel areas were determined using the NIH ImageJ software. Relative gel contraction was determined as changes in gel surface area from the 0 to 1 h time point.
Immunostaining
HMSC were prestained with the cell tracker dye Green CMFDA (Invitrogen) at 10 μM in serum-free DMEM for 15 min, and then cocultured with HUVEC for 48 h. Cells were fixed with 4% paraformaldehyde for 20 min at room temperature, permeabilized with 0.3% TritonX-100, and blocked in phosphate buffered saline (PBS) containing 5% goat serum and 2% bovine serum albumin (BSA) for 1 h at room temperature, followed by the rabbit anti-Ki67 (1:500, ab66155; Abcam) incubation overnight at 4°C. Secondary antibodies were conjugated to Alexa 594 and used at a concentration of 1:500 (Invitrogen). The percentage of Ki67 positive cells was determined by costaining with the green tracker dye and calculating the ratio of fluorescently labeled cells. For co-labeling, an endothelial-specific TRITC-Lectin (L4889; Sigma) in PBS containing 1% BSA was incubated with fixed cells for 1 h.
Lentivirus expression and infection
The lentivirus plasmids containing GFP alone or GFP together with dominant-negative mastermind-like 1 (dnMAML) constructs were made as described previously [15]. The lentivirus plasmids were transfected into TN-293 cells using PolyJet (SignaGen), and the viral particles were amplified and purified as described previously [15]. For infection, HMSC were seeded in a six-well plate at a density of 8×104 cells per well, 24 h before viral infection. One milliliter of lentivirus suspension diluted in 1 mL DMEM with 10% FBS was added to each well. Polybrene was supplemented at a final concentration of 6 μg/mL. Twenty-four hours later, cells were transferred to fresh DMEM containing 10% FBS for additional 24 h incubation. After 48 h infection in each well, 105 HUVEC were cocultured with infected HMSC for another 48 h, cells were separated, and mRNA was collected for qPCR analysis.
Luciferase assay
CBF-luciferase plasmid was generated as described [15]. To measure the transcriptional activity, HMSC or HAoSMC were placed in a 12-well plate at 6×104 cells/well and transfected the pGL3-promoter-luciferase plasmid as control or with a plasmid with CBF1-binding elements upstream of the promoter (pCBF-Luc) using the PolyJet reagent (SignaGen) after 24 h. Cells were then cocultured with 5×104 HUVEC for an additional 48 h. The promoter activity was measured by luciferase assays using the Bright-Glo reagent (Promega). To normalize the transfection efficiency, Hsp68-β-galactosidase (LacZ) was cotransfected, and luciferase activities were normalized based on an equivalent amount of LacZ activity. For secreted luciferase assays, a constitutive CAG promoter sequence [16] was cloned into a NanoLuc luciferase pNL1.3 plasmid (Promega), which contains the luciferase sequence fused to an IL6 N-terminal secretion signal sequence [17]. To determine the secreted luciferase activity, HMSC were plated in a 24-well plate at a cell density of 3×104 cells/well and transfected the plasmid with the PolyJet (SignaGen). Twenty-four hours later, equal numbers of HUVEC were cocultured with HMSC for additional 48 h. The cultured medium was collected and reacted with Nano-Glo substrate to measure the secreted luciferase. Luciferase readings were performed on a LUMIstar Omega luminometer (BMG Labtech). The Hsp68-β-galactosidase (LacZ) construct was cotransfected to normalize the transfection efficiency.
Sirius red assay
1.25×105 HMSC were plated in a six-well plate and cultured with 1.25×105 HUVEC for 48 h. 5×104 separated HMSC were plated in each well of a 12-well plate for another 12 h. Collagen concentration in a cultured medium was measured by the Sirius Red assay as described [18]. Briefly, the culture medium was collected and incubated with 25% ammonium sulfate solution overnight at 4°C. The precipitated collagen was bonded with the Sirius red F3B (Sigma) and then released by potassium hydroxide solution. Collagen concentration was determined at 540 nm absorbance using a Molecular Devices SpectraMax M5 microplate reader.
Flow cytometry
Cells recovered from culture were washed twice in PBS. Cell suspensions in PBS with 2% BSA (3×105 cells/100 μL) were incubated at 4°C for 30 min with antibodies to CD73 and CD90 according to the manufacturer's instruction. Phycoerythrin (PE) conjugated CD73 and fluorescein isothiocyanate (FITC) conjugated CD90 antibodies were purchased from BD Pharmingen (561014; 561969). Cells were then washed twice with 2 mL PBST and resuspended in 0.5 mL PBS. Cells were subjected to flow cytometry using a LSR II cytometer (BD Biosciences) and the FlowJo software for analysis.
Statistical analyses
For all quantitative analyses presented, a minimum of three independent replicates were performed in terms of individual experiment. Data are presented as mean±standard error of the mean. Data analyses were conducted using the GraphPad Prism using nonparametric Mann–Whitney test, two-tailed Student's t-test and analysis of variance for statistical significance between groups. Differences were considered significant if P<0.05.
Results
Endothelial cells activate smooth muscle-specific gene expression in mesenchymal stem cells
Although it is well established that endothelial cells can influence the phenotypes of mature vascular smooth muscle cells, how endothelial cells might regulate stem cell precursors has not been fully examined. Mesenchymal stem cells have the capacity to differentiate toward a smooth muscle lineage; therefore, we investigated if endothelial cells could promote the differentiation of these cells into smooth muscle. To do so, we utilized an established coculture system, in which endothelial cells and mesenchymal stem cells are cultured in a 1:1 ratio to permit direct cell contact and physical interaction [15]. Under coculture conditions, the mesenchymal stem cells appear to interact with the endothelial cells and together they form distinctive cellular networks (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd). Following coculture, the cells are physically separated using endothelial cell-specific anti-PECAM1-conjugated beads to allow for molecular analysis of gene and protein expression in the individual cell types. Previous analysis of the separation efficiency using magnetic beads was further confirmed here and revealed greater than 99% efficiency (Supplementary Fig. S1) [19]. Human mesenchymal stem cells (HMSC) were cultured with human endothelial cells and examined for smooth muscle-specific gene expression by qPCR. Examination of HMSC following coculture showed an increase in smooth muscle markers, SM α-actin (ACTA2), SM22α (TAGLN), and h1-calponin (CNN1) transcripts compared with the HMSC cultured alone (Fig. 1A–C). The smooth muscle transcription factor, myocardin, also exhibited an increase in RNA expression in cocultured HMSC [20]. Using three endothelial cell subtypes derived from the umbilical vein, microvascular, and pulmonary artery endothelial cells, when cocultured with HMSC showed similar expression profiles of smooth muscle genes. Analysis of smooth muscle marker protein expression in cocultured mesenchymal stem cells showed a similar result, with significant increases in expression levels after endothelial cell coculture at both 48 and 96 h (Fig. 1D–G). These results indicated that endothelial cells have the capacity to induce smooth muscle marker gene expression in naive mesenchymal stem cells. The ability of endothelial cells to induce smooth muscle markers suggested that the mesenchymal stem cells were undergoing differentiation and losing their stem cell properties. Examination of stem marker genes demonstrated a gradual decrease in CD73, CD90, CD105, and KLF4 mRNA expression (Fig. 2), indicating an ability of endothelial cells to reduce their stem cell properties. This was further confirmed by using flow cytometry with antibodies directed against CD70 and CD90, which showed a similar decrease in protein expression following coculture (Fig. 2). To further investigate the extent of this endothelial cell-dependent differentiation, we examined the contractile ability of the mesenchymal stem cells when cocultured. HMSC were cocultured with HUVEC in a collagen matrix and contractile response was measured as a percentage of contracted gel area. Mesenchymal stem cells that were cocultured with endothelial cells exhibited a greater contractile ability compared with cells cultured alone (Fig. 3). Further, coculture of HMSC with adenocarcinoma HeLa cells did not cause a similar increase in contraction, indicating that endothelial cells possess a unique capacity to impart a contractile phenotype on differentiating smooth muscle cells.
FIG. 1.

Endothelial cells derived from unique vascular beds promote smooth muscle differentiation of mesenchymal stem cells. Human adult bone marrow-derived mesenchymal stem cells (HMSC) were cocultured with human umbilical vein endothelial cells (HUVEC), human microvascular endothelial cells (HMVEC), or human pulmonary artery endothelial cells (HPAEC) for 48 h, separated using anti-PECAM1-conjugated beads, and RNA was extracted to measure gene expression by qPCR (A–C). HMSC were cultured for western blot analysis as described above and collected at 48 and 96 h time points. Smooth muscle marker genes were detected using specific antibodies and quantified (D–G). *P<0.05, relative to control without cocultured endothelial cells. qPCR, quantitative PCR.
FIG. 2.

Coculture of mesenchymal stem cells with endothelial cells causes a reduction in stem cell markers. HMSC were cocultured with endothelial cells (HUVEC) for 48 and 96 h, separated, and RNA was isolated to detect stem cell markers by qPCR. Transcripts detected were, mesenchymal stem cell markers CD73, CD90, CD105 (A–C), and stem cell marker KLF4 (D). *P<0.05, compared with HMSC cultured alone. After coculturing for 96 h, separated HMSC were stained with PE-CD73 and FITC-CD90 antibodies and analyzed by flow cytometry. Filled histogram represents HMSC cultured alone and dashed histogram displays cocultured HMSC, following separation (E, F). The median florescence intensities of PE-CD73 and FITC-CD90 florescence signal were quantified (G, H). *P<0.05, compared with HMSC cultured alone. FITC, fluorescein isothiocyanate; PE, phycoerythrin.
FIG. 3.
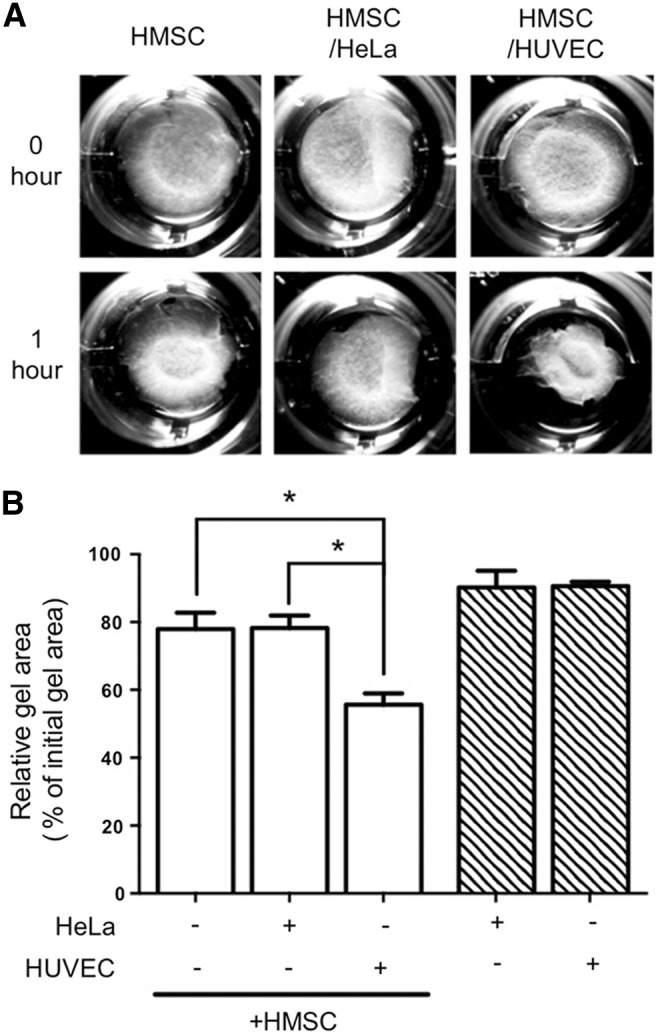
Contractile ability is increased in mesenchymal stem cells when cocultured with endothelial cells. HMSC were cocultured with HUVEC or HeLa adenocarcinoma cells (as a control) in a collagen gel matrix. (A) Gel images were captured immediately following and 1 h after gel release. (B) Contraction area was quantified by the NIH ImageJ software. HUVEC and HeLa cells cultured alone exhibited negligible contraction. *P<0.05.
Endothelial cells decrease proliferation while activating a synthetic phenotype
Differentiated smooth muscle cells are classically characterized as being highly contractile with reduced proliferative and synthetic abilities [21]. To determine if endothelial cells were promoting a quiescent phenotype in mesenchymal stem cells, we measured indices of proliferation by immunostaining to detect Ki67-expressing cells, and by examining expression of Cyclin D1 (CCND1) and Cyclin D2 (CCND2) genes. Consistent with a contractile and quiescent smooth muscle cell phenotype, endothelial cells caused a decrease in Ki67-positive mesenchymal stem cells and a corresponding decrease in Cyclin D1 and D2 mRNA expression (Fig. 4A–C). For determining the synthetic state of cocultured mesenchymal stem cells, we evaluated collagen gene expression, and measured the levels of secreted collagen in the media. Both assays showed a significant increase in the collagen content in cocultured mesenchymal stem cells compared with cells cultured alone (Fig. 4D, E). Additionally, we utilized a constitutively expressed secreted luciferase plasmid transfected into HMSC to measure general cell secretion rates [22]. Consistent with the upregulation of collagen gene expression and collagen content in the media, there was a significant increase in the secretion rate of cocultured mesenchymal stem cells (Fig. 4F). Overall, these data support the notion that endothelial cells can induce both a contractile and synthetic phenotype in mesenchymal stem cells, while suppressing a proliferative phenotype.
FIG. 4.

Proliferation is reduced, but synthetic phenotype is increased with endothelial cells. (A) Ki67 immunostaining of HMSC cultured alone or cocultured with endothelial cells. The HMSC were prestained with a tracker dye before plating for identification. Scale bar=100 μm (B) Percentage of Ki67 positive mesenchymal stem cells was determined by costaining with a preloaded tracker dye and Ki67 antibody to measure the ratio of fluorescently labeled cells. (C) Cyclin D1 and Cyclin D2 mRNA expression was analyzed by qPCR in mesenchymal stem cells (HMSC) following separation from endothelial cells. (D) Expression of collagen genes was measured by qPCR after coculture and separation from endothelial cells (HUVEC). (E) Secreted collagen from HMSC culture medium was measured by Sirius Red assay after coculture and separation from HUVEC. (F) HMSC were transfected with the secreted luciferase plasmid and cocultured with HUVEC. The culture medium was collected and luciferase activity was measured. *P<0.05, relative to control without HUVEC.
To determine if endothelial cell-dependent conversion of mesenchymal stem cells to smooth muscle cells was stable following the removal of cocultured endothelial cells, we replated the mesenchymal stem cells following coculture for 48 h and separation. Interestingly, the mesenchymal stem cells rapidly lost their smooth muscle properties and regained a more proliferative, stem cell-like phenotype (Fig. 5A–D). We then asked if these cocultured mesenchymal stem cells could differentiate into other cell types following coculture. Incubation of mesenchymal stem cells with osteogenic differentiation media and adipogenic differentiation media indicated that these cells retained the ability to differentiate toward nonsmooth muscle cell lineages (Fig. 5E, F). These data indicate that endothelial cells are actively required to promote and maintain differentiation of smooth muscle cells, and that they may not have the capacity to cause terminal differentiation of the smooth muscle cells.
FIG. 5.

Endothelial cells do not cause terminal differentiation of HSMC to smooth muscle cells. After coculturing with HUVEC for 48 h, HMSC were separated, replated, and then cultured for indicated time points. (A) mRNA expression of Notch3, HeyL, and smooth muscle markers, (B) collagen genes, (C) proliferation genes Cyclin D1 and Cyclin D2, and (D) mesenchymal stem cell markers were analyzed by qPCR. (E, F) Following separation from HUVEC, HMSC were cultured in osteogenic and adipogenic differentiation media for 96 h. mRNA expression of osteogenic and adipogenic markers were measured by qPCR. *P<0.05, relative to control without HUVEC. n.s., not significant.
Endothelial cells promote Notch signaling in mesenchymal stem cells
Previous analysis of endothelial cell communication with smooth muscle cells demonstrated that endothelial cells activate Notch signaling and this is required for endothelial cell-induced smooth muscle gene expression and differentiation [15,23]. Examination of Notch signaling components in HMSC after coculture revealed activation similar to that observed in HAoSMC (Fig. 6). Notch3 and Jagged1 are established autoregulated targets of Notch signaling [15,24], and both showed an increase of RNA and protein expression in cocultured HMSC that was similar to smooth muscle cells under identical conditions (Fig. 6A–C). Moreover, expression of downstream target gene HeyL was robustly induced in mesenchymal stem cells as it was in smooth muscle cells. Using a Notch sensor luciferase plasmid containing five CBF1/RBPJ binding repeats [25] showed that endothelial cells activate Notch signaling in HMSC to a similar level as smooth muscle cells (Fig. 6D, E). To further evaluate the role of Notch activity in regulating endothelial cell-induced smooth muscle differentiation, we blocked Notch signaling and evaluated the phenotypic changes. Using a dominant-negative mastermind-like-1-expressing plasmid [26] (Fig. 7) and chemical inhibitor DAPT (Supplementary Fig. S2) to suppress Notch signaling, the data show that Notch inhibition causes a decrease in contractile protein gene expression, collagen expression, and cell secretion, while attenuating the decrease in proliferation. These results indicate that Notch signaling plays a significant role in the regulation of smooth muscle differentiation from stem cells and suggest it is a critical mediator of phenotypic modulation.
FIG. 6.

Notch signaling is activated by endothelial cells in mesenchymal stem cells. (A) HMSC and (B) HAoSMC cultured with or without HUVEC. mRNA expression of Notch3 and Notch downstream genes was measured by qPCR following separation from HUVEC. (C) Western blot shows Notch3 and Jagged1 protein expression following coculture with endothelial cells. (D, E) HMSC and HAoSMC were transiently transfected with 5XCBF1-luciferase plasmid (pCBF-Luc) to assess Notch signaling activity. Twenty-four hours after transfection, cells were cocultured with HUVEC, and luciferase activity was measured after 48 h. *P<0.05, compared with cells cultured alone. HAoSMC, human aortic smooth muscle cells.
FIG. 7.

Notch signaling confers endothelial cell-induced differentiation. HMSC were infected with dominant-negative Mastermind-like 1 (dnMAML) lentivirus to block Notch signaling before culturing with HUVEC. (A) mRNA expression of Notch3, HeyL, and smooth muscle markers, (B) proliferation genes Cyclin D1 and Cyclin D2, and (C) collagen genes were analyzed by qPCR. (D) Cell secretory activity was determined by quantifying secreted luciferase in the media. *P<0.05.
Previously, mesenchymal stem cells were shown to differentiate into smooth muscle cells using a combination of PDGF-B and TGFβ growth factors [6,14]. Our data indicate that endothelial cells drive differentiation through utilization of the Notch signaling pathway. Therefore, we wondered if these two differentiation mechanisms were independent or shared some overlap. To test this, we promoted smooth muscle differentiation with PDGF-B and TGFβ in the presence of the Notch inhibitor, DAPT. The data indicate that PDGF-B and TGFβ are sufficient to drive smooth muscle-specific gene expression and do not require activated Notch signaling to promote smooth muscle differentiation from mesenchymal stem cells (Fig. 8A). Furthermore, there was an additive increase in differentiation when PDGF-B and TGFβ were added in coculture conditions, which was only partially blocked by DAPT (Fig. 8A). We next utilized a combination of inhibitors to the PDGFβ and TGFβ signaling pathways in coculture conditions to evaluate endothelial cell-dependent smooth muscle differentiation. These experiments showed that while there was no significant effect on Notch signaling genes, expression of smooth muscle contractile markers were only slightly attenuated (Fig. 8B). Overall, these data demonstrate that mesenchymal stem cells can be driven toward a smooth muscle cell fate by two independent pathways with similar sufficiency.
FIG. 8.

TGFβ/PDGF-B induced differentiation of mesenchymal stem cells into smooth muscle cells is Notch independent. (A) HMSC cultured alone or cocultured were treated with TGFβ and PDGF-B to induce smooth muscle differentiation in the presence or absence of DAPT to block Notch signaling. Gene expression of smooth muscle-specific markers was measured by qPCR. (B) TGFβ and PDGFβ receptor inhibitors were added together to HMSC cocultured with endothelial cells (HUVEC). Cocultured HMSC were separated from HUVEC before gene expression analysis. Expression of Notch3, HeyL, and smooth muscle-specific genes was analyzed by qPCR. *P<0.05, compared with HMSCs cultured alone or control. PDGF, platelet-derived growth factor; TGFβ, transforming growth factor-β.
Discussion
These data show that endothelial cells have a profound effect on mesenchymal stem cell phenotypes, which highlights the importance of considering the endothelial cell environment when manipulating mesenchymal stem cell fate. Our findings demonstrate that mature endothelial cells derived from arteries, veins, and microvascular beds can drive mesenchymal stem cells toward a smooth muscle cell fate in a similar fashion. Although these different endothelial cell subtypes may have unique physiological properties that can alter mature smooth muscle cell function in unique ways, they appear to share a common ability to promote differentiation. The results presented here demonstrate that endothelial cells cause a decrease in proliferation, consistent with a differentiated phenotype, but promote a synthetic phenotype that is consistent with the expression of collagen genes. Smooth muscle cell phenotypic modulation is a well-established phenomenon, where smooth muscle cells can exist in a range of phenotypes [21]. Our data indicate that endothelial cells do not drive smooth muscle cells toward a completely contractile phenotype, but establish a phenotype in which they are both contractile and synthetic. This particular phenotype may be beneficial for the establishment of the basement membrane and might represent an important transition phenotype that exists before a more classically differentiated contractile phenotype. Interestingly, endothelial cells are required to maintain this phenotype, and do not cause these cells to terminally differentiate. This is consistent with smooth muscle cells being highly plastic cells that are greatly influenced by environmental stimuli to dictate their phenotype. Additionally, after removal of endothelial cells, the mesenchymal stem cells retain the ability to differentiate toward other nonsmooth muscle cell phenotypes, indicating that the endothelial cell-dependent differentiation is tightly coupled to their presence.
Our findings show that Notch signaling is critical for endothelial cell-dependent differentiation. This result is consistent with the previous findings from our laboratory and others that demonstrated the importance of Jagged1 on endothelial cells to activate Notch signaling in smooth muscle cells [15,23]. Notch activation in mesenchymal stem cells is similar to that observed in smooth muscle cells, showing a robust increase in Notch3 and Jagged1 expression, and strong induction of downstream targets. Interestingly, the ability of endothelial cells to induce mesenchymal stem cell differentiation toward a smooth muscle cell fate appears separate and distinct from previously described differentiation cues. Previous studies showed that the combination of PDGF-B and TGFβ promotes smooth muscle cell differentiation from mesenchymal stem cells [6,14]. Our results reveal that endothelial cell-dependent differentiation is independent of these two growth factors, while Notch signaling is dispensable for PDGF-B and TGFβ-dependent differentiation. Thus, these data imply that multiple independent mechanisms can be utilized to differentiate mesenchymal stem cells toward a smooth muscle cell fate. Overall, our data demonstrate a role of endothelial cells in modulating mesenchymal stem cell differentiation. The results indicate that the vascular environment may have a substantial effect on mesenchymal stem cell fate decisions, and the presence of endothelial cells could serve as a method to direct their differentiation. Additionally, the data demonstrate a novel mechanism by which mesenchymal stem cells can differentiate into smooth muscle cells through Notch signaling, and this pathway appears largely independent of TGFβ and PDGF-B signaling.
Supplementary Material
Acknowledgments
This work was supported by the American Heart Association Grant to BL and the Research Institute at Nationwide Children's Hospital.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Choi YH, Kurtz A. and Stamm C. (2011). Mesenchymal stem cells for cardiac cell therapy. Hum Gene Ther 22:3–17 [DOI] [PubMed] [Google Scholar]
- 2.Marion NW. and Mao JJ. (2006). Mesenchymal stem cells and tissue engineering. Methods Enzymol 420:339–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Williams AR. and Hare JM. (2011). Mesenchymal stem cells: biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res 109:923–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Krawiec JT. and Vorp DA. (2012). Adult stem cell-based tissue engineered blood vessels: a review. Biomaterials 33:3388–3400 [DOI] [PubMed] [Google Scholar]
- 5.Yan J, Tie G, Xu TY, Cecchini K. and Messina LM. (2013). Mesenchymal stem cells as a treatment for peripheral arterial disease: current status and potential impact of type II diabetes on their therapeutic efficacy. Stem Cell Rev 9:360–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ross JJ, Hong Z, Willenbring B, Zeng L, Isenberg B, Lee EH, Reyes M, Keirstead SA, Weir EK, Tranquillo RT. and Verfaillie CM. (2006). Cytokine-induced differentiation of multipotent adult progenitor cells into functional smooth muscle cells. J Clin Invest 116:3139–3149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dora KA. (2001). Cell-cell communication in the vessel wall. Vasc Med 6:43–50 [PubMed] [Google Scholar]
- 8.Triggle CR, Samuel SM, Ravishankar S, Marei I, Arunachalam G. and Ding H. (2012). The endothelium: influencing vascular smooth muscle in many ways. Can J Physiol Pharmacol 90:713–738 [DOI] [PubMed] [Google Scholar]
- 9.Udan RS, Culver JC. and Dickinson ME. (2013). Understanding vascular development. Wiley Interdiscip Rev Dev Biol 2:327–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majesky MW, Dong XR, Regan JN. and Hoglund VJ. (2011). Vascular smooth muscle progenitor cells: building and repairing blood vessels. Circ Res 108:365–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaengel K, Genove G, Armulik A. and Betsholtz C. (2009). Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler Thromb Vasc Biol 29:630–638 [DOI] [PubMed] [Google Scholar]
- 12.Kurpinski K, Lam H, Chu J, Wang A, Kim A, Tsay E, Agrawal S, Schaffer DV. and Li S. (2010). Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 28:734–742 [DOI] [PubMed] [Google Scholar]
- 13.Guo X, Stice SL, Boyd NL. and Chen SY. (2013). A novel in vitro model system for smooth muscle differentiation from human embryonic stem cell-derived mesenchymal cells. Am J Physiol Cell Physiol 304:C289–C298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheung C, Bernardo AS, Trotter MW, Pedersen RA. and Sinha S. (2012). Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat Biotechnol 30:165–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Kennard S. and Lilly B. (2009). NOTCH3 expression is induced in mural cells through an autoregulatory loop that requires endothelial-expressed JAGGED1. Circ Res 104:466–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niwa H, Yamamura K. and Miyazaki J. (1991). Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199 [DOI] [PubMed] [Google Scholar]
- 17.Rose-John S, Schooltink H, Schmitz-Van de Leur H, Mullberg J, Heinrich PC. and Graeve L. (1993). Intracellular retention of interleukin-6 abrogates signaling. J Biol Chem 268:22084–22091 [PubMed] [Google Scholar]
- 18.Keira SM, Ferreira LM, Gragnani A, Duarte IdS. and Barbosa J. (2004). Experimental model for collagen estimation in cell culture. Acta Cir Bras 19:17–22 [Google Scholar]
- 19.Lilly B. and Kennard S. (2009). Differential gene expression in a coculture model of angiogenesis reveals modulation of select pathways and a role for Notch signaling. Physiol Genomics 36:69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Wang DZ, Hockemeyer D, McAnally J, Nordheim A. and Olson EN. (2004). Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 428:185–189 [DOI] [PubMed] [Google Scholar]
- 21.Owens GK, Kumar MS. and Wamhoff BR. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 84:767–801 [DOI] [PubMed] [Google Scholar]
- 22.Lippincott-Schwartz J, Roberts TH. and Hirschberg K. (2000). Secretory protein trafficking and organelle dynamics in living cells. Annu Rev Cell Dev Biol 16:557–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.High FA, Lu MM, Pear WS, Loomes KM, Kaestner KH. and Epstein JA. (2008). Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc Natl Acad Sci U S A 105:1955–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boucher J, Gridley T. and Liaw L. (2012). Molecular pathways of notch signaling in vascular smooth muscle cells. Front Physiol 3:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennard S, Liu H. and Lilly B. (2008). Transforming growth factor-beta (TGF-1) down-regulates Notch3 in fibroblasts to promote smooth muscle gene expression. J Biol Chem 283:1324–1333 [DOI] [PubMed] [Google Scholar]
- 26.Weng AP, Nam Y, Wolfe MS, Pear WS, Griffin JD, Blacklow SC. and Aster JC. (2003). Growth suppression of pre-T acute lymphoblastic leukemia cells by inhibition of notch signaling. Mol Cell Biol 23:655–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.