

Abstract
Calcineurin is a Ca2+- and calmodulin-dependent protein phosphatase that plays a key role in animal and yeast physiology. In the yeast Saccharomyces cerevisiae, calcineurin is required for survival during several environmental stresses, including high concentrations of Na+, Li+, and Mn2+ ions and alkaline pH. One role of calcineurin under these conditions is to activate gene expression through its regulation of the Crz1p transcription factor. We have identified Hph1p as a novel substrate of calcineurin. HPH1 (YOR324C) and its homolog HPH2 (YAL028W) encode tail-anchored integral membrane proteins that interact with each other in the yeast two-hybrid assay and colocalize to the endoplasmic reticulum. Hph1p and Hph2p serve redundant roles in promoting growth under conditions of high salinity, alkaline pH, and cell wall stress. Calcineurin modifies the distribution of Hph1p within the endoplasmic reticulum and is required for full Hph1p activity in vivo. Furthermore, calcineurin directly dephosphorylates Hph1p and interacts with it through a sequence motif in Hph1p, PVIAVN. This motif is related to calcineurin docking sites in other substrates, such as NFAT and Crz1p, and is required for regulation of Hph1p by calcineurin. In contrast, Hph2p neither interacts with nor is dephosphorylated by calcineurin. Ca2+-induced Crz1p-mediated transcription is unaffected in hph1Δ hph2Δ mutants, and genetic analyses indicate that HPH1/HPH2 and CRZ1 act in distinct pathways downstream of calcineurin. Thus, Hph1p and Hph2p are components of a novel Ca2+- and calcineurin-regulated response required to promote growth under conditions of high Na+, alkaline pH, and cell wall stress.
Calcineurin is a Ca2+- and calmodulin-regulated serine/threonine protein phosphatase that is highly conserved throughout evolution and exists in yeasts and metazoans (for a review see reference 3). In mammals, calcineurin plays key roles in the development of skeletal and cardiac muscle, angiogenesis, embryonic axon outgrowth, and T-cell activation (12, 19, 23, 24, 37, 40). Calcineurin is not required for growth of the budding yeast Saccharomyces cerevisiae under standard laboratory conditions. It does, however, play a critical role in enabling this yeast to survive high concentrations of Na+, Li+, and Mn2+ ions, alkaline pH, prolonged exposure to mating factor, and cell wall stress (14, 15, 20, 38). Calcineurin consists of a catalytic A subunit and regulatory B subunit; in S. cerevisiae, the A subunit is encoded by two functionally redundant genes, CNA1 and CNA2, and the regulatory subunit is encoded by CNB1 (15, 16, 30, 32). Upon exposure to environmental stresses such as high concentrations of Na+, cytosolic Ca2+ concentrations increase and activate calcineurin.
A key downstream effector of calcineurin in S. cerevisiae is the zinc finger transcription factor Crz1p (33, 35, 46). Calcineurin dephosphorylates Crz1p, enabling its translocation to the nucleus, where it transactivates a number of genes through its binding to calcineurin-dependent response elements in their promoters (46, 47). Crz1p regulates the expression of genes involved in several processes, including ion and small-molecule transport, cell wall maintenance, and vesicular transport (13, 25, 34, 36, 41, 51). One calcineurin- and Crz1p-regulated gene, ENA1, encodes a P-type ATPase that plays a critical role in the extrusion of Na+ ions under conditions of high extracellular Na+ and high pH (25).
The growth of calcineurin mutants is severely reduced at alkaline pH. Under these conditions, the activity of the vacuolar H+-ATPase and other ion pumps is critical for survival of S. cerevisiae. Also, the uptake of nutrients such as phosphate, iron, and copper ions, whose solubility decreases at high pH, becomes rate limiting for growth (1, 22, 43). Calcineurin- and Crz1p-dependent transactivation of a calcineurin-dependent response element-driven reporter gene is strongly induced in response to alkaline pH, and this signaling pathway is responsible for regulating the transcription of a subset of the genes induced under these conditions (43). Despite the central role of Crz1p in calcineurin signaling, there is evidence to suggest that it is not the sole mediator of calcineurin-dependent signaling. crz1Δ cells are less sensitive than calcineurin-null cells to high concentrations of Na+, Li+, and Mn2+ and particularly to alkaline pH (22, 46), suggesting that calcineurin regulates additional downstream targets to promote survival under these conditions. Recent microarray analyses indicate that all Ca2+- and Na+-induced calcineurin-dependent gene expression is mediated via Crz1p (51). This suggests that additional calcineurin effectors act via nontranscriptional mechanisms.
High-throughput yeast-two-hybrid studies performed by Uetz et al. identified an interaction between Cna1p and the protein encoded by uncharacterized open reading frame (ORF) YOR324C (48). Both this protein and a closely related gene product encoded by YAL028W have been shown to localize to the endoplasmic reticulum (ER), but their function has yet to be elucidated (6). In this study we showed that Yor324Cp interacts with both Cna1p and Cna2p and is a novel substrate of calcineurin. Calcineurin regulates Yor324Cp function in vivo and affects its distribution within the ER. Mutants lacking both YOR324C and its homologue YAL028W are sensitive to high concentrations of Na+ and cell wall stress and are particularly defective in their ability to grow on alkaline medium. Thus, we have identified YOR324C, designated HPH1 (high pH), and YAL028W, designated HPH2, as novel components of the yeast stress response.
MATERIALS AND METHODS
Yeast strains and media.
Yeast media and culture conditions were essentially as previously described (44) except that twice the level of amino acids and nucleotides was used in synthetic media. Yeast transformations were performed with the lithium acetate method (4). Yeast strains are listed in Table 1; strains from the yeast deletion collection were purchased from Open Biosystems. VHY60 was constructed by mating strain 1621 with 10380 (BY4739 MATα hph2Δ::KAN) (yeast deletion collection). The diploids were sporulated and tetrads were dissected to generate a strain that contained both hph1Δ::KAN and hph2Δ::KAN and was isogenic with BY4741. VHY74.1 was generated by transforming VHY60 with a cassette containing a HIS3-disrupted allele of CRZ1 from the previously described plasmid pIMG38 (35). VHY87 was generated by integrating a cassette for expressing high levels of a gene encoding Kar2 presequence-DsRed.T1-HDEL (7) into the TRP1 locus of EG123 (MATα leu2-3,112 ura3-52 trp1-1 his4 canl) (gift of D. Levin). This cassette was generated by digesting the YIplac204/TKC-DsRed-HDEL plasmid with EcoRV (gift of B. Glick).
TABLE 1.
Strains used in this study
| Strain | Relevant genotype | Source or reference |
|---|---|---|
| PJ69-4A | MATa trp1-901 leu2-3,112 ura3-52 his3-200 gal4Δ gal80Δ GAL2-ADE2 LYS2::GAL1-HIS3 met2::GAL7-lacZ | 28 |
| YPH499 | MATa ura3-52 lys2-801 ade2-101 trp1 Δ63 his3-Δ200 leu2-Δl | 45 |
| DD12 | As YPH499 except cnb1::URA3::hisG | 16 |
| BY4741 | MATa leu2Δ ura3Δ met15Δ his3Δ | Yeast deletion collection |
| 1621 | As BY4741, hph1Δ::KAN | Yeast deletion collection |
| 380 | As BY4741, hph2Δ::KAN | Yeast deletion collection |
| VHY60 | As BY4741, hph1Δ::KAN hph2Δ::KAN | This study |
| 5040 | As BY4741, cnb1Δ::KAN | Yeast deletion collection |
| 5353 | As BY4741, crz1Δ::KAN | Yeast deletion collection |
| VHY74.1 | As VHY60, crz1Δ::HIS3 | This study |
| VHY87 | MATα leu2-3,112 ura3-52 his4 can1rTRP1::DsRed-HDEL | This study |
Plasmids.
The plasmids used in this study are reported in Table 2. Recombinant DNA procedures were performed as previously described (4). In all cases, genes were cloned by amplification of genomic DNA with vent polymerase (New England Biolabs) to create fragments flanked by restriction sites for cloning into the vector of interest. Sequencing reactions with ET Terminator (Amersham) and ABI Big Dye sequencing chemistry (Amersham) were performed to ensure that all amplified DNA fragments had the correct sequence.
TABLE 2.
Plasmids used in this study
| Plasmid | Vector | Insert | Reference |
|---|---|---|---|
| pVH1 | pUG36 | GFP-HPH1 | This study |
| pVH2 | pUG36 | GFP-HPH2 | This study |
| pVH3 | pACTII | HPH1 | This study |
| pVH4 | pACTII | HPH2 | This study |
| pVH5 | pACTII | HPH1-ΔPVIAVN | This study |
| pVH6 | pGBT9 | HPH1 | This study |
| pVH7 | pGBT9 | CNA2 | This study |
| pVH8 | pRS315 | HPH1 | This study |
| pVH9 | pRS315 | HPH1-ΔPVIAVN | This study |
| pVH10 | pUG36 | CFP-HPH1 | This study |
| pVH11 | pUG34 | YFP-HPH2 | This study |
| pVH12 | pUG36 | HPH1-ΔPVIAVN | This study |
| BJP2014 | pGBT9 | CNA1 | 29 |
N-terminal green fluorescent protein (GFP) fusions of HPH1 and HPH2 were created by cloning the ORF of HPH1 (YOR324C) as a BamHI-HindIII fragment and HPH2 (YAL028W) as a BamHI-XhoI fragment into pUG36 (CEN URA3 pMET25-yEGFP3; gift of U Güldener and J. H. Hegemann) to create pVH1 and pVH2, respectively. In order to create pVH10, the CFP-HPH1 fusion yEGFP3 of pUG36 was first replaced with cyan fluorescent protein (CFP) amplified from pDH3 (provided by the National Center for Research Resources Yeast Resource Center, University of Washington) and flanked by XbaI and BamHI sites; the HPH1 BamHI-HindIII fragment was then cloned into this vector. In order to create pVH11 containing the YFP-HPH2 fusion, HPH2 was first subcloned from pUG36 into pUG34 (CEN HIS3 pMET25-yEGFP3) and then yEGFP3 was replaced with yellow fluorescent protein (YFP) amplified from pDH5 (provided by the National Center for Research Resources Yeast Resource Center) flanked with XbaI and BamHI.
For yeast-two-hybrid studies, the pACTII and pGBT9 vectors were used to create GAL4 activation and binding domain fusions, respectively. A SmaI-BamHI fragment of HPH2 was cloned into pACTII to create pVH4, and a SmaI-BamHI fragment of CNA2 was cloned into pGBT9 to create pVH7. A SmaI-BamHI fragment of HPH1 was cloned into pACTII and pGBT9 to create pVH3 and pVH6, respectively. A PCR-based mutagenesis strategy was used to delete amino acids 72 to 77 of HPH1, encoding PVIAVN. Two fragments were amplified, which were then ligated together into pACTII. Fragment 1 comprised the 5′ end of HPH1 flanked by a BamHI site at the N terminus and the endogenous unique XbaI site at position 205. Fragment 2 was generated with a forward primer of nucleotides 195 to 213 and 232 to 251 of HPH1 (this contained the endogenous XbaI site and encoded the deletion of the PVIAVN motif) and a reverse primer with a flanking XhoI site. BamHI-XbaI fragment 1 and XbaI-XhoI fragment 2 were cloned into pACTII digested with BamHI and XhoI to create pVH5.
Construction of pVH12 (GFP-Hph1-ΔPVIAVN) was performed by simultaneously ligating BamHI-XbaI fragment 1 and XbaI-XhoI fragment 2 (described above) with pUG36 digested with BamHI and XhoI. In order to create pVH8, a fragment comprising the HPH1 ORF and preceding 797 nucleotides was amplified from genomic DNA, flanked with BamHI and HindIII sites, and cloned into pRS315. To construct pVH9, the BamHI-XbaI DNA fragment with the promoter and 5′ 205 bp of HPH1 from pVH8 and the XbaI-XhoI fragment from the construction of pVH5 were simultaneously ligated into pRS315 cut with BamHI and XhoI.
Immunoblot analysis.
Yeast cells expressing GFP-Hph1p or GFP-Hph2p cultures were grown to log phase in synthetic medium lacking uracil and methionine. In order to assess the mobility of GFP-Hph1p and GFP-Hph2p after treatment with CaCl2, 10 optical density units at 600 nm of cells were treated or not with CaCl2 at a final concentration of 200 mM at 30°C for 10 min. Cells were pelleted and frozen in liquid nitrogen prior to preparation of extract. Extract was prepared by a method modified from that previously described by Davis et al. (18). Cells were thawed by resuspension in 100 μl of SBS buffer (40 mM Tris-HCl [pH 6.8], 8 M urea, 0.1 mM EDTA, 1% 2-mercaptoethanol, 5% sodium dodecyl sulfate [SDS]) and added to an 80-μl volume of glass beads. Samples were then mixed by vortexing for 10 min at room temperature and incubated at 37°C for 10 min; 100 μl of 2× SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer was added, and the samples were vortexed for a further 2 min at room temperature. Finally they were centrifuged at 16,000 × g for 5 min. Samples were further diluted by adding equal volumes of supernatant with 2× SDS-PAGE sample buffer. Samples were run on 5 to 12% or 7 to 11% polyacrylamide-SDS gradient gels and transferred to nitrocellulose. Blots were incubated with JL8 monoclonal anti-GFP antibody (Becton Dickinson) and then with horseradish peroxidase-conjugated anti-mouse immunoglobulin (Amersham) and visualized with an ECL kit (Amersham).
In vitro phosphatase assay.
DD12 cells expressing GFP-Hph1p were grown to log phase in 200 ml of synthetic medium lacking uracil and methionine and harvested at an optical density at 600 nm of 1. Cells were washed and resuspended in 2 ml of TL buffer (40 mM Tris-HCl [pH 7.5], 100 mM KCl, 1 mM MgCl2, 250 mM sorbitol, 1 mM dithiothreitol, 0.1% NP-40) containing protease inhibitors (1 μg of pepstatin per ml, 2 μg of leupeptin per ml, 2 μg of aprotinin per ml, 1 mM phenylmethylsulfonyl fluoride, 1.25 mM benzamidine-HCl) and phosphatase inhibitors (5 mM sodium phosphate, 10 mM sodium molybdate, 20 mM sodium fluoride, 10 mM sodium pyrophosphate, 5 mM EGTA, and 5 mM EDTA), and extracts were prepared by glass bead lysis (50).
The resulting extract was incubated with 100 μl of a 50% slurry of washed protein A-agarose beads (Sigma) and 10 μl of polyclonal rabbit anti-GFP (Becton Dickinson) for 2 h with rotation at 4°C. Immunoprecipitates were washed twice in TL buffer containing protease inhibitors and phosphatase inhibitors, twice in TL buffer containing protease inhibitors only, and twice in CP buffer (50 mM Tris-HCl [pH 7.5], 1 mM MgCl2, 1 mM dithiothreitol) containing protease inhibitors. Washed beads were divided among five tubes prior to different phosphatase treatments. Phosphatase assays were performed in a total of 100 μl in CP buffer for calcineurin and in the supplied buffer for λ-phosphatase (New England Biolabs); 500 U of recombinant human calcineurin (Calbiochem), 2,600 U of calmodulin (Calbiochem), or 10 μl of λ-phosphatase (New England Biolabs) was used per assay. Where indicated, CaCl2 was added to a final concentration of 40 mM and EGTA to 10 mM. Phosphatase assays were incubated at 30°C for 30 min; the supernatant was then removed, and SDS-PAGE sample buffer was added to the beads. Samples were then run on a 5 to 12% gradient polyacrylamide-SDS gel and blotted as described above. Blots were visualized with SuperSignal reagent (Pierce).
Spot assays.
Growth of various mutants was assessed by spotting serial dilutions of stationary-phase yeast cultures on plates containing various ions or chemicals. Cultures were diluted to an optical density at 600 nm of 0.5, and five fivefold serial dilutions were prepared. Plates were prepared with YPD containing 50 mM Tris-HCl (pH 8.4 or 8.8), 50 mM morpholineethanesulfonic acid (MES, pH 5.5), 1 M NaCl, 2 mg of Congo Red or 0.188 mg of Calcofluor White (fluorescent brightener 28 [Sigma]) per ml. Where indicated, FK506 (Fujisawa) was added to a concentration of 2 μg/ml. Plates were incubated at room temperature for 3 days for YPD and YPD pH 5.5, 4 days for YPD pH 8.5 and pH 8.8, Calcofluor White, and Congo Red, and 5 days for NaCl.
Yeast two-hybrid assays.
Yeast two-hybrid assays were performed by introducing combinations of Gal4p activation and binding domain fusions into strain PJ69-4A, which contains a GAL1 promoter-HIS3 reporter gene. To perform the yeast two-hybrid assays, these strains were grown in selective medium and spotted onto synthetic medium containing and lacking histidine. These spot assays were set up exactly as described above, and the plates were incubated for 3 days at 30°C.
Microscopy.
To examine the localization of GFP-Hph1p and GFP-Hph2p in wild-type (YPH499) cells, this strain was transformed with plasmid pVH1 and pVH2, respectively. Liquid cultures were set up in synthetic medium lacking uracil and with either low levels of methionine (3 mg/liter) for expression of GFP-Hph1p or no methionine for expression of GFP-Hph2p. In order to investigate the localization of Hph1p and Hph2p relative to the ER, strain VHY87.1, expressing HDEL-DsRed, was transformed with plasmid pVH1 or pVH2, respectively, and cultures were prepared as described above. To localize Hph1p and Hph2p in the same cell, wild-type cells (YPH499) were transformed with plasmids pVH10 and pVH11, expressing CFP-Hph1p and YFP-Hph2p, respectively. In this case, cultures were grown in synthetic medium lacking uracil, histidine, and methionine. Log-phase cultures at an optical density at 600 nm of 1.0 were concentrated by brief centrifugation and mounted on microscope slides precoated with a 25% gelatin solution made with the corresponding medium.
Cells expressing a single GFP fusion were visualized with a Nikon Eclipse E600 microscope with fluorescence optics and an HB100 mercury lamp. Fluorescein filter sets (Chroma) were used to visualize GFP. Photos were taken with a Hammamatsu digital charge-coupled device 47420-95 camera and QED software (QED Imaging). For colocalization studies, cells were imaged with a Nikon TE200 inverted microscope with a 100× 1.3NA objective lens. Independent excitation (CFP, 436 ± 10 nm; YFP; 493 ± 17 nm; and red fluorescent protein [RFP], 580 ± 20 nm) and emission (CFP, 465 ± 30 nm; YFP, 530 ± 40 nm; and RFP, 630 ± 60 nm) filters were used in conjunction with a three-pass dichroic mirror (Chroma 28311). GFP was imaged either with the YFP filter combination or with the same YFP and RFP excitation filters in combination with the Chroma 13699 three-pass emitter and Chroma 20597 three-pass beam splitter. Image acquisition, controlled via MetaMorph software (Universal Imaging Corporation), ranged from 50 to 1,500 ms with a Princeton Instruments PE1300 cooled charge-coupled device camera. Colocalization studies were performed with sequential acquisitions having 30 ms of dead time between images.
For estimation of the total number of foci in a cell, wild-type (YPH499) and cnb1Δ (DD12) cells expressing GFP-Hph1p (pVH1) were grown to log phase in synthetic medium lacking uracil and containing methionine at 3 mg/liter as described above. Cells were either imaged directly or treated with CaCl2 (200 mM) for 10 min prior to visualization with the Nikon TE200 inverted microscope in combination with MetaMorph software, as described above. A through series of 18 images at 0.5-μm steps was taken with a calibrated stepper motor to adjust focus. Images were projected into a single plane, with the maximum values at each pixel position. For each cell, fluorescent foci were counted and then divided by the area of the cell in pixels to give the value for the number of foci per unit area. Data from 30 to 40 cells were collected for each condition, and the mean was determined.
RESULTS
Hph1p but not Hph2p interacts with Cna1p and Cna2p and is a substrate of calcineurin.
Genomewide yeast two-hybrid analyses demonstrated an interaction between the calcineurin A subunit, Cna1p, and the uncharacterized ORF YOR324C (now designated HPH1) and between YOR324C and its close homolog YAL028W (designated HPH2) (48). We used a directed yeast two-hybrid approach to confirm and extend these observations. N-terminal fusions were created between the Gal4p activation domain with Hph1p and Hph2p and the Gal4p binding domain with Cna1p and Cna2p. Combinations of the constructs, as indicated in Fig. 1, were expressed in strain PJ69-4A, containing a GAL1 promoter-HIS3 reporter, and interactions were assayed by determining the extent of growth on medium lacking histidine. Hph1p but not its homolog Hph2p interacted with both calcineurin A subunits, Cna1p and Cna2p. Furthermore, using a Gal4p activation domain Hph2p fusion and a Gal4p binding domain Hph1p fusion, we reproduced the interaction seen previously between Hph1p and Hph2p (Fig. 1). We have also found by yeast two-hybrid assays that Hph2p is able to interact with itself (data not shown). In contrast, self-association of Hph1p could not be assessed due to toxicity of the Hph1p Gal4p activation domain fusion in combination with either the Hph1p or Hph2p Gal4p binding domain fusions.
FIG. 1.

Hph1p but not Hph2p interacts with Cna1p and Cna2p in yeast two-hybrid assays, and Hph1p and Hph2p interact with each other. A strain (PJ69-4A) containing a GAL1-HIS3 reporter gene was transformed with combinations of GAL4 binding domain fusions of CNA1 (BJP2014), CNA2 (pVH7) or HPH1 (pVH6) (BD) and GAL4 activation domain fusions of HPH1 (pVH3) and HPH2 (pVH4) (AD), as indicated. Cells were grown as described in Materials and Methods, plated on synthetic medium containing (+His) or lacking (−His) histidine, and incubated for 3 days at 30°C.
Since Hph1p can interact with both calcineurin A subunits, we determined whether it is a substrate of calcineurin. Functional N-terminal GFP fusions of Hph1p and Hph2p were created (see Materials and Methods), and their electrophoretic mobility was examined by SDS-PAGE. Calcium treatment of wild-type cells resulted in an increased electrophoretic mobility of GFP-Hph1p. No change in mobility was observed in CaCl2-treated calcineurin-null cells (cnb1Δ, lacking the calcineurin regulatory B subunit), and in these cells GFP-Hph1p exhibited a slower-migrating form (Fig. 2A). Similar results were seen with epitope-tagged Hph1p expressed at endogenous levels (data not shown). These data indicate that the calcium-induced mobility shift of Hph1p is calcineurin dependent, and in vitro assays demonstrated that Hph1p is a direct substrate of calcineurin (Fig. 2B). GFP-Hph1p immunoprecipitated from calcineurin-deficient cells showed an increased mobility when treated with recombinant calcineurin, calmodulin, and CaCl2, and this could be blocked by the additionof the Ca2+ chelator EGTA (Fig. 2B). Though this in vitro calcineurin-mediated increase in mobility of GFP-Hph1p is smaller than that seen in vivo, it was reproducible. Treatment with λ-phosphatase resulted in an increased mobility of GFP-Hph1p compared with the calcineurin treatment, suggesting that there are phosphorylated residues of Hph1p that are not removed by calcineurin.
FIG. 2.
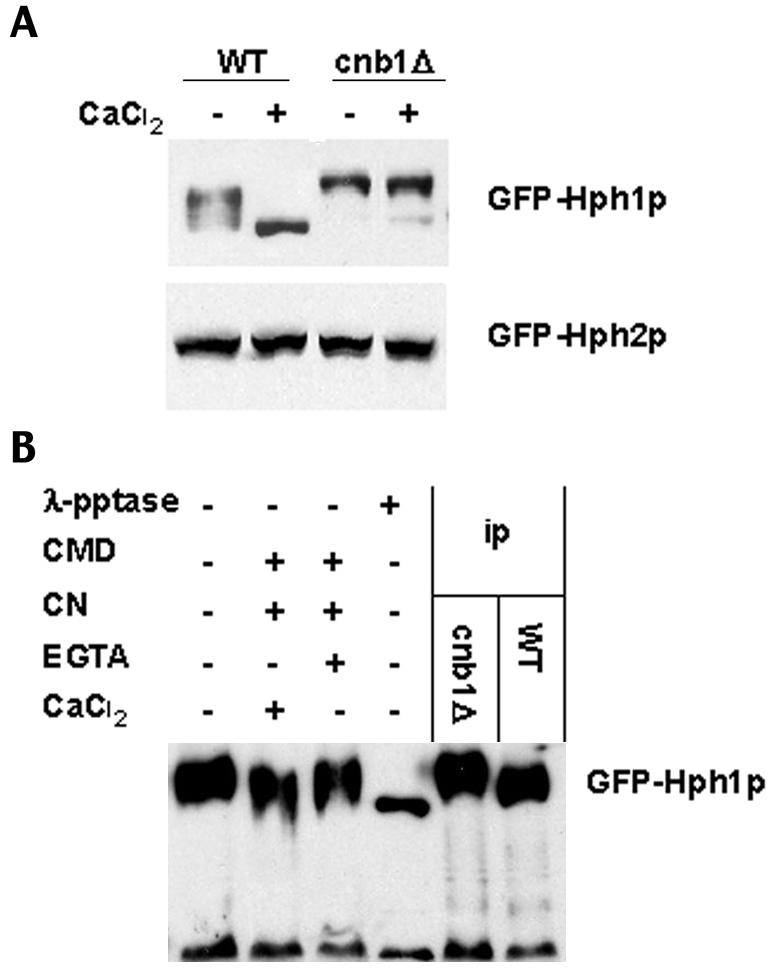
Hph1p but not Hph2p is dephosphorylated upon CaCl2 treatment in a calcineurin-dependent manner. (A) Wild-type (WT, YPH499) and cnb1Δ (DD12) cells expressing GFP-Hph1p (pVH1) or GFP-Hph2p (pVH2) were grown to log phase and either mock treated or treated with 200 mM CaCl2 for 10 min. Cells were then frozen in liquid N2 and extracts were prepared (see Materials and Methods). Extracts were subjected to SDS-PAGE and Western blotted with an anti-GFP antibody. (B) Extracts were prepared from wild-type (YPH499) and cnb1Δ (DD12) cells expressing GFP-Hph1p (pVH1). Immunoprecipitated (ip) GFP-Hph1p from cnb1Δ cells was treated with recombinant calcineurin (CN), calmodulin (CMD), CaCl2, EGTA, or λ-phosphatase (λ-pptase) as indicated in the left-hand lanes (see Materials and Methods). These samples, together with untreated immunoprecipitates of GFP-Hph1p from wild-type and cnb1Δ cells, were then subjected to SDS-PAGE and Western blotted with an anti-GFP antibody.
No change in the electrophoretic mobility of GFP-Hph2p was observed when wild-type or calcineurin-null cells were treated with CaCl2, suggesting that Hph2p is not a substrate of calcineurin. This is consistent with the lack of interaction observed between calcineurin and Hph2p in yeast two-hybrid analysis. Thus, Hph1p interacts with and is dephosphorylated by calcineurin. Hph2p binds Hph1p but does not interact with and is not dephosphorylated by calcineurin.
Hph1p and Hph2p are required for growth under high NaCl, alkaline pH, and cell wall stress.
In order to further elucidate the functions of Hph1p and Hph2p, we examined strains lacking each of these genes as well as an hph1Δ hph2Δ double mutant. Since Hph1p is a substrate of calcineurin, we examined the phenotypes of these strains in a number of conditions under which calcineurin-deficient cells exhibit growth defects. We found no growth defect for the hph1Δ and hph2Δ single mutant strains, but the hph1Δ hph2Δ strain showed reduced growth on alkaline media, media containing high levels of Na+ ions, and media containing the cell wall disruptants Calcofluor White and Congo Red. These effects were more pronounced when cells were grown at 25°C (Fig. 3). No growth defects were observed on medium containing high levels of Mn2+, Li+, or Ca2+ ions or sorbitol or at high temperature (data not shown).
FIG. 3.

hph1Δ hph2Δ cells are sensitive to growth in conditions of high pH and high NaCl and with Calcofluor White and Congo Red. Wild-type (WT, BY4741), hph1Δ (strain 1621), hph2Δ (strain 380), and hph1Δ hph2Δ (strain VHY60) strains were grown to stationary phase in YPD. Cultures were then diluted to an optical density at 600 nm of 0.5, and fivefold serial dilutions were spotted on YPD plates with no addition, 50 mM Tris-HCl at pH 8.8, 50 mM MES at pH 5.5, 1 M NaCl, 2 mg of Congo Red per ml, or 0.188 mg of Calcofluor White per ml. Plates were incubated at room temperature for 3 days for YPD and pH 5.5, 4 days for pH 8.8, Calcofluor White, and Congo Red, and 5 days for NaCl.
An uncharacterized ORF, YOR325W, is located within the coding sequence of HPH1 on the opposite strand. A DNA fragment that contained the complete sequence of YOR325W but only part of the YOR324C (HPH1) ORF failed to complement the growth defects of hph1Δ hph2Δ mutants at alkaline pH or high Na+ (data not shown). Thus, the growth defects that we observed can be attributed to elimination of the protein encoded by YOR324C (Hph1p) and not that encoded by YOR325W.
Genetic interactions of HPH1, HPH2, calcineurin, and CRZ1.
We examined the genetic interactions between HPH1, HPH2, CRZ1, and calcineurin (CNB1) to clarify the role of HPH1 and HPH2 in the calcineurin signaling pathway. CRZ1 encodes a calcineurin-dependent transcription factor, and crz1Δ mutants exhibit a growth defect at high pH that is less severe than that seen with a calcineurin mutant. hph1Δ hph2Δ crz1Δ mutants are more sensitive to high pH than crz1Δ or hph1Δ hph2Δ strains (Fig. 4), establishing that Crz1p acts independently of Hph1p and Hph2p to promote growth at high pH. Furthermore, Crz1p- and calcineurin-dependent transcription can be measured with a reporter gene (CDRE::lacZ) in which the β-galactosidase gene is controlled by the Crz1p binding site (calcineurin-dependent response element) (46). hph1Δ hph2Δ cells exhibit no defect in Ca2+- and calcineurin-dependent CDRE::lacZ expression (data not shown), confirming that Hph1p and Hph2p function independently of Crz1p. In contrast, when grown in the presence of the calcineurin inhibitor FK506, wild-type, hph1Δ hph2Δ, and crz1Δ strains exhibit identical growth defects at high pH. Thus, a defect in calcineurin is epistatic to defects in crz1 or hph1 and hph2. This observation is consistent with a role for calcineurin in controlling the functions of Crz1p, Hph1p, and Hph2p. Together with biochemical studies that establish Crz1p and Hph1p as substrates of calcineurin, these genetic observations confirm that Crz1p, Hph1p, and Hph2p act independently of each other and are downstream of (i.e., controlled by) calcineurin.
FIG. 4.

Phenotypic analysis shows that the high-pH growth defects of hph1Δ, hph2Δ, and crz1Δ mutations are additive and that loss of calcineurin function is epistatic to hph1Δ, hph2Δ, and crz1Δ. Cells were plated as described for Fig. 3 to compare growth between wild-type (WT), hph1Δ, hph2Δ, hph1Δ hph2Δ, cnb1Δ (strain 5040), crz1Δ (strain 5353) and hph1Δ hph2Δ crz1Δ (strain VHY74.1) strains on YPD at pH 5.5 (50 mM MES) and YPD at pH 8.4 (50 mM Tris) containing either solvent (−) or the calcineurin inhibitor FK506 (+) at 2 μg/ml. Plates were incubated at room temperature for 3 days for pH 5.5 and 4 days for pH 8.4.
To further examine the physiological relevance of the calcineurin-dependent dephosphorylation of Hph1p, we constructed a mutant allele of HPH1 that is defective in its regulation by calcineurin. Calcineurin has been shown to interact with substrates via a docking site that is distinct from sites of dephosphorylation. Furthermore, mutant substrates that lack this docking site are defective for calcineurin-dependent regulation (2, 11). We sought to identify the sequence in Hph1p that is required for its interaction with calcineurin as follows. Using a series of two-hybrid constructs, we established that a sequence between amino acids 50 and 100 is required for Hph1p interaction with calcineurin (data not shown). We examined this region and identified a sequence (amino acids 72 to 77), PVIAVN, that is conserved in HPH1 fungal homologs but is not present in HPH2 or its homologs. This sequence is related to the PxIxIT calcineurin-binding motif defined in NFAT and the equivalent site, PIISIQ, that mediates interaction between Crz1p and yeast calcineurin (2, 11). We therefore deleted this sequence to create the hph1-ΔPVIAVN allele.
Hph1-ΔPVIAVNp failed to interact with Cna1p or Cna2p (calcineurin catalytic subunits) in two-hybrid assays, and the protein encoded by hph1-ΔPVIAVN did not display a Ca2+- and calcineurin-dependent increase in electrophoretic mobility (Fig. 5A and B). These two observations indicate that the protein encoded by hph1-ΔPVIAVN is not dephosphorylated by calcineurin. Note, however, that the mutant protein was present at levels similar to that of Hph1p (Fig. 5B). To determine the contribution of calcineurin to Hph1p function, hph1-ΔPVIAVN was introduced into hph1Δ hph2Δ cells, and the resulting strain was tested for its ability to grow at high pH. The results indicate that Hph1-ΔPVIAVNp is partially functional, i.e., the strain containing Hph1-ΔPVIAVNp grew better than the hph1Δ hph2Δ strain but not as well as a strain containing a wild-type copy of HPH1 (Fig. 5C). Similar results were observed for growth of cells on medium containing Na+ or Calcofluor White (data not shown). This suggests that calcineurin modulates Hph1p function but is not completely required for its activity.
FIG. 5.

Hph1p lacking the motif PVIAVN does not bind Cna1p or Cna2p, is not dephosphorylated upon treatment with CaCl2, and only partially complements the growth defects of the hph1Δ hph2Δ mutant at high pH. (A) A yeast two-hybrid assay was set up with PJ69-4A cells expressing CNA1 (BJP2014) or CNA2 (pVH7) GAL4 binding domain fusions (BD) in combination with GAL4 activation domain fusions (AD) of wild-type HPH1 (pVH3) or HPH1-ΔPVIAVN (Hph1*) (pVH5), as indicated. Cells were plated on medium containing (+His) and lacking (−His) histidine and incubated for 3 days at 30°C. (B) Wild-type (WT, YPH499) cells expressing GFP-Hph1p (pVH1) or GFP-Hph1-ΔPVIAVNp (GFP-Hph1*) (pVH12) were grown to log phase and then either mock treated or treated with 200 mM CaCl2 for 10 min. Samples were then frozen in liquid N2, extracts were prepared, and SDS-PAGE followed by Western blotting with an anti-GFP antibody was performed (see Materials and Methods). (C) Wild-type (WT, BY4741) and hph1Δ hph2Δ (VHY60) cells were transformed with either empty vector (−) (pRS315) or plasmids to express endogenous levels of wild-type Hph1p (pVH8) or Hph1-ΔPVIAVN (Hph1*) (pVH9). Cultures were grown in selective synthetic medium to the stationary phase, diluted as described for Fig. 3, plated on YPD at pH 5.5 (50 mM MES) and YPD at pH 8.8 (50 mM Tris), and incubated at room temperature for 3 and 4 days, respectively.
Hph1p and Hph2p are integral membrane proteins associated with the endoplasmic reticulum.
Analysis of the predicted coding sequences of Hph1p and Hph2p revealed the presence of one putative membrane-spanning domain near the C terminus of each protein. We confirmed that GFP-Hph1p and GFP-Hph2p associated with the P13 pellet during fractionation and found that they cosedimented with Dpm1p, a resident ER protein, in a sucrose gradient (data not shown). Additionally, consistent with previous studies, treatment with detergent was required to solubilize Hph1p and Hph2p (data not shown) (6). These observations suggest that Hph1p and Hph2p are both integral membrane proteins and are found in the ER.
To further investigate Hph1p and Hph2p localization, we examined cells containing functional GFP fusions of these proteins. GFP was fused to the N terminus of Hph1p and Hph2p, and expression was controlled by the MET25 promoter. In the case of GFP-Hph1p, a small amount of methionine was added to partially reduce expression of this fusion protein. It was not possible to visualize Hph1p or Hph2p expressed under their own promoters even when a fusion to three tandem copies of GFP was created. Experiments performed to determine the localization of all yeast genes and ORFs also failed to visualize Hph1p and Hph2p tagged genomically with GFP (27).
Consistent with previous observations, GFP-Hph1p localized to the cell and nuclear periphery in a pattern characteristic of proteins resident in the ER, although it exhibited a punctate localization that is not typical of ER proteins (Fig. 6A) (6). These punctate foci of GFP-Hph1p colocalized with the established HDEL-DsRed marker, which uniformly labels the ER (Fig. 6B). Characterization of GFP-Hph1p by time-lapse imaging revealed that the GFP-Hph1p foci moved rapidly in live cells and that their movements were constrained by and correlated with movements of the ER (data not shown). GFP-Hph2p also localized to the ER and, consistent with previous observations, exhibited a more diffuse distribution than that seen with GFP-Hph1p (Fig. 6A) (6). Similarly, GFP-Hph2p colocalized with the HDEL-DsRed marker, though in some cells small cytoplasmic foci were also observed exclusive of the ER marker (Fig. 6B). When coexpressed, CFP-Hph1p and YFP-Hph2p colocalized to discrete foci within the ER (Fig. 6C). We conclude, therefore, that Hph1p and Hph2p are components of the ER. Both proteins exhibit a similar localization pattern and colocalize within an individual cell. This observation, together with two-hybrid studies showing that Hph1p and Hph2p interact, suggests that the proteins can form a complex.
FIG. 6.

Hph1p and Hph2p localize to the ER. (A) Wild-type yeast cells (YPH499) expressing GFP-Hph1p (pVH1) or GFP-Hph2p (pVH2) under a methionine-repressible promoter were grown to log phase and visualized by fluorescence microscopy and differential interference microscopy (DIC) (see Materials and Methods). (B) GFP-Hph1p (pVH1) or GFP-Hph2p (pVH2) was expressed in wild-type yeast cells expressing HDEL-DsRed (VHY87) to mark the ER. In the third panel, the two images were merged, and yellow regions indicate areas of overlapping localization. (C) Wild-type cells (YPH499) were transformed with plasmids to express CFP-Hph1p (pVH10) and YFP-Hph2p (pVH11) and visualized by fluorescence microscopy. In the third panel, the two images were merged, and yellow regions indicate areas of overlapping localization.
Ca2+ modulates Hph1p distribution.
Cells expressing either GFP-Hph1p or GFP-Hph2p were exposed to 200 mM CaCl2. This treatment is known to activate calcineurin, as it rapidly stimulates calcineurin- and Crz1p-dependent transcription. Within minutes of CaCl2 addition, the number of bright punctate foci of GFP-Hph1p increased by approximately 2.5-fold (Fig. 7). Colocalization with HDEL-DsRed showed that the foci, though more numerous, were strictly colocalized to the ER (data not shown). Western blots established that the amount of GFP-Hph1p protein in cells did not change during this time (data not shown). Thus, we conclude that CaCl2 causes a redistribution of Hph1p in cells such that the number of discrete Hph1p-containing foci within the cell increases. In contrast, no change in GFP-Hph2p localization was observed in response to CaCl2 treatment, and cells expressing HDEL-DsRed to allow visualization of the ER showed no morphological changes in response to CaCl2 (data not shown).
FIG. 7.

Three-dimensional reconstruction of yeast cells expressing GFP-Hph1p in untreated and CaCl2-treated cells. (A) Wild-type (YPH499) and cnb1Δ (DD12) cells expressing GFP-Hph1p (pVH1) were grown to log phase and treated or not treated with CaCl2 (200 mM) for 10 min. They were then visualized by fluorescence microscopy by taking a through series of 18 images at 0.5-μm steps as described in Materials and Methods. Images were projected onto a single plane, with the maximum values at each pixel position. (B) The number of foci per unit area was determined from three-dimensional reconstructions of wild-type and cnb1Δ cells, which were either untreated or treated with 200 mM CaCl2 for 10 to 20 min (see Materials and Methods for details). A Student t test was performed to show that the difference in the number of foci between wild-type cells treated or not with CaCl2 was highly significant (P = 3.5 E-12).
To investigate whether the Ca2+-dependent change in GFP-Hph1p distribution correlated with calcineurin-dependent dephosphorylation of Hph1p, we quantified the number of GFP-Hph1p foci in calcineurin-deficient cells (cnb1Δ) before and after Ca2+ treatment. Under these conditions, we did not observe a significant change in the number of GFP-Hph1p-containing foci (Fig. 7). We also treated cells containing GFP-Hph1-ΔPVIAVNp with CaCl2 and did not observe a significant change in the pattern of GFP-Hph1p localization or number of foci (data not shown). Therefore, we conclude that the observed increase in GFP-Hph1p foci is Ca2+- and calcineurin-dependent and reflects a change in Hph1p distribution in the cell rather than a change in protein level.
DISCUSSION
Identification of a novel calcineurin substrate.
In this study, we identified Hph1p as a novel substrate of the Ca2+- and calmodulin-dependent phosphatase calcineurin and characterize the role of Hph1p and the closely related Hph2p in the response of S. cerevisiae to environmental stress. In addition to Hph1p, only three other yeast proteins, Crz1p, Rcn1p, and Cch1p, have been shown to be direct substrates of calcineurin (9, 26, 47). Studies of mammalian and yeast calcineurin have established that calcineurin interacts with a docking site in its substrates that is distinct from dephosphorylated residues and that this interaction is required for calcineurin to act on the substrate. We have made use of this paradigm to identify Hph1p as a novel calcineurin substrate based solely on its ability to interact with the phosphatase. Furthermore, by identifying and deleting the site within Hph1p that mediates its interaction with calcineurin, we were able to create a special allele of HPH1, hph1-ΔPVIAVN, which is specifically defective in its regulation by calcineurin. Characterization of this allele allowed us to assess the contribution of calcineurin to Hph1p function in vivo and establish that calcineurin is required for full Hph1p activity. Both of these approaches should be generally applicable to the future identification and characterization of additional calcineurin substrates.
Calcineurin docking sites have been identified in several substrates. The consensus binding motif for mammalian calcineurin defined in NFAT family members is PxIxIT, and direct selection of a peptide optimized for binding to mammalian calcineurin identified the related sequence PVIVIT (2, 3a) Docking sequences for yeast calcineurin, i.e., PVIAVN in Hph1p and PIISIQ in Crz1p, are closely related to those for mammalian calcineurin, indicating that the substrate interaction domain in calcineurin is highly conserved (11). Comparison of these sequences allows the consensus calcineurin binding motif to be further refined and suggests in particular that the last residue in the docking site can be any of several hydrophilic amino acids (i.e., T, Q, or N). Ultimately, a detailed understanding of the interaction between calcineurin and its substrates could lead to the development of compounds that inhibit specific downstream pathways of calcineurin. Such compounds would minimize the unwanted side effects caused by cyclosporine A and FK506, two immunosuppressants currently in wide clinical use that act through general inhibition of calcineurin.
Role of Hph1p and Hph2p in calcineurin signaling.
Like calcineurin mutants, cells lacking both Hph1p and Hph2p display growth defects under conditions of high salinity, high pH, and cell wall stress. A major function of calcineurin in promoting survival during environmental stress is to activate the Crz1p transcription factor. Genetic analyses indicate that Hph1p and Hph2p act independently of Crz1p, and Crz1p-dependent gene expression is unaffected in hph1Δ hph2Δ mutants. Thus, Hph1p and Hph2p define a novel branch of the calcineurin-regulated stress response. Although Hph1p is functionally redundant with Hph2p, we provide evidence that the two proteins differ in their regulation: Hph1p is a calcineurin substrate, whereas Hph2p does not interact with and is not dephosphorylated by the phosphatase. Since Hph2p is not a substrate of calcineurin but can substitute for Hph1p, it must either constitutively perform a function that is calcineurin regulated in Hph1p or be regulated by an alternative process that we have yet to identify. We also found that loss of calcineurin is epistatic to the hph1Δ hph2Δ mutations, suggesting that calcineurin is required in some way for the function of both Hph1p and Hph2p. Since calcineurin does not regulate Hph2p directly, this observation suggests that calcineurin acts through an additional, indirect mechanism to regulate Hph1p and Hph2p activity in vivo. In the future, as we learn more about the molecular functions of Hph1p and Hph2p, we should gain further insight into the relationship between calcineurin and Hph1p and Hph2p.
Currently, we know little about the role of calcineurin in regulating events associated with the ER. Calcineurin signaling is required for the long-term survival of cells undergoing ER stress. Calcineurin-null mutants but not crz1Δ mutants show decreased viability when treated with tunicamycin, a compound that blocks N-glycosylation in the ER, causing accumulation of misfolded proteins and activation of the unfolded protein response. The downstream effector(s) of calcineurin involved in this pathway has yet to be identified (10). Since Hph1p and Hph2p localize to the ER, we investigated a possible role for these proteins in cells undergoing the unfolded protein response. We found, however, no difference between the survival of wild-type cells and hph1Δ hph2Δ mutants during exposure to tunicamycin (data not shown). Thus, we conclude that Hph1p and Hph2p do not play a role in the calcineurin-mediated response to ER stress.
Hph1p and Hph2p localization.
Hph1p and Hph2p belong to a family of tail-anchored proteins which comprise a cytosolic N-terminal domain tethered to internal membranes via a single C-terminal transmembrane segment (6). These proteins fold cotranslationally and are inserted posttranslationally into the membrane by their C-terminal hydrophobic domain (21, 31). In accordance with previous studies, we found that both Hph1p and Hph2p localize to the ER (6). Hph1p displays a punctate pattern of localization, whereas Hph2p is more evenly distributed within this organelle. Beilharz et al. (6) showed that the C-terminal hydrophobic transmembrane domain is sufficient to target Hph1p to the ER but that additional determinants in the N-terminal region drive its punctate distribution within this organelle. Since it has not been possible to visualize either Hph1p or Hph2p expressed at endogenous levels, we cannot be certain that Hph1p would form foci when expressed at its natural levels. However, we did find that activating calcineurin potently increased the number of Hph1p foci in the ER and that calcineurin-null (cnb1Δ) cells had fewer foci than wild-type cells. Thus, the punctate distribution of Hph1p may reflect a physiologically relevant property of the protein that is regulated by phosphorylation. Hph1p and Hph2p interacted in a yeast two-hybrid assay; this interaction may be mediated by predicted coiled-coil domains found just N-terminal to the transmembrane domain in each protein. Addition of the calcineurin inhibitor FK506 in the yeast two-hybrid assays did not affect the interaction between Hph1p and Hph2p, suggesting that the interaction between these two proteins is not modified by calcineurin (data not shown). When coexpressed, Hph1p and Hph2p colocalized within the ER in foci, suggesting that Hph1p is able to bind Hph2p in vivo and cause its localization into foci.
Function of Hph1p and Hph2p.
Future characterization of Hph1p and Hph2p is required to understand their physiological roles at a molecular level. Other tail-anchored transmembrane proteins include the SNARE proteins of the Vamp/synaptobrevin and syntaxin families, which are involved in mediating vesicle trafficking and small components of the translocon, which mediates protein translocation into the ER. Thus, Hph1p and Hph2p may function during the stress response to facilitate protein trafficking. Alternatively, Hph1p and Hph2p may be involved in recruiting enzymes to the ER in response to stress, as other tail-anchored proteins are known to serve a similar function (for a review, see reference 49). Also, Hph1p and Hph2p have been reported to interact with Tcp1p, a subunit of the tailless complex polypeptide (TCP1) ring complex/chaperonin (TriC/CCT), via yeast two-hybrid studies (K. Dolinski, R. Balakrishnan, K. R. Christie, M. C. Costanzo, S. S. Dwight, S. R. Engel, D. G. Fisk, J. E. Hirschman, E. L. Hong, L. Issel-Tarver, A. Sethuraman, C. L. Theesfeld, G. Binkley, C. Lane, M. Schroeder, S. Dong, S. Weng, R. Andrada, D. Botstein, and J. M. Cherry, 2004, Saccharomyces genome database, http://www.yeastgenome.org; Doris Ursic, unpublished data). The TRiC/CCT chaperone promotes protein folding and interacts with proteins that undergo posttranslational translocation into the ER (39). Thus, Hph1p and Hph2p may interact with CCT/TriC components during their insertion into the ER membrane or may function together with TriC/CCT as part of the stress response. Although Hph1p and Hph2p show no significant sequence similarity to any proteins of known function, we have identified several regions of high sequence conservation by comparing Hph1p and Hph2p with closely related proteins from other fungi (data not shown). In the future, functional characterization of these regions and identification of Hph1p- and Hph2p-interacting proteins should provide insight into the mechanisms by which Hph1p and Hph2p act.
S. cerevisiae response to alkaline stress.
Many yeasts and fungi pose significant threats to human health, especially the health of immunocompromised persons. Fungal pathogens typically colonize a variety of host environments, and many of these environments exist at neutral or alkaline pH. Several studies of Candida albicans demonstrate an important correlation between growth at high pH and pathogenesis; for example, mutants lacking a transcriptional regulator, rim101, are defective for growth in alkaline conditions and display attenuated virulence (17). Additionally, calcineurin is required for the pathogenicity of C. albicans, since it is necessary for growth at high pH and survival in the presence of serum (5, 8, 42). Understanding the role that Hph1p and Hph2p play in promoting the survival S. cerevisiae at high pH will likely shed light on the pH-adaptive responses of other fungi and may ultimately lead to the development of novel antifungal therapies.
Acknowledgments
We thank U Güldener, J. H. Hegemann, B Glick, and I. F. de Larrinoa for generously providing plasmids. We also thank Kim Kafadar, Valérie Denis, Kim Williams, and Leila Boustany for technical help and useful discussion.
Funding for this work was provided by NIH research grant GM-48729.
REFERENCES
- 1.Anraku, Y., N. Umemoto, R. Hirata, and Y. Ohya. 1992. Genetic and cell biological aspects of the yeast vacuolar H(+)-ATPase. J. Bioenerg. Biomembr. 24:395-405. [DOI] [PubMed] [Google Scholar]
- 2.Aramburu, J., F. Garcia-Cozar, A. Raghavan, H. Okamura, A. Rao, and P. G. Hogan. 1998. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell 1:627-637. [DOI] [PubMed] [Google Scholar]
- 3.Aramburu, J., A. Rao, and C. B. Klee. 2000. Calcineurin: from structure to function. Curr. Top. Cell. Regul. 36:237-295. [DOI] [PubMed] [Google Scholar]
- 3a.Aramburu, J., M. B. Yaffe, C. Lopez-Rodriquez, L. C. Cantley, P. G. Hogan, and A. Rao. 1999. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 285:2129-2133. [DOI] [PubMed]
- 4.Ausubel, F., R. Brent, R. E. Kingston, R. E. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 1987. Current protocols in molecular biology. Wiley, New York, N.Y.
- 5.Bader, T., B. Bodendorfer, K. Schroppel, and J. Morschhauser. 2003. Calcineurin is essential for virulence in Candida albicans. Infect Immun. 71:5344-5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beilharz, T., B. Egan, P. A. Silver, K. Hofmann, and T. Lithgow. 2003. Bipartite signals mediate subcellular targeting of tail-anchored membrane proteins in Saccharomyces cerevisiae. J. Biol. Chem. 278:8219-8223. [DOI] [PubMed] [Google Scholar]
- 7.Bevis, B. J., A. T. Hammond, C. A. Reinke, and B. S. Glick. 2002. De novo formation of transitional ER sites and Golgi structures in Pichia pastoris. Nat. Cell Biol. 4:750-756. [DOI] [PubMed] [Google Scholar]
- 8.Blankenship, J. R., F. L. Wormley, M. K. Boyce, W. A. Schell, S. G. Filler, J. R. Perfect, and J. Heitman. 2003. Calcineurin is essential for Candida albicans survival in serum and virulence. Eukaryot. Cell 2:422-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bonilla, M., and K. W. Cunningham. 2003. Mitogen-activated protein kinase stimulation of Ca2+ signaling is required for survival of endoplasmic reticulum stress in yeast. Mol. Biol. Cell 14:4296-4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonilla, M., K. K. Nastase, and K. W. Cunningham. 2002. Essential role of calcineurin in response to endoplasmic reticulum stress. EMBO J. 21:2343-2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boustany, L. M., and M. S. Cyert. 2002. Calcineurin-dependent regulation of Crz1p nuclear export requires Msn5p and a conserved calcineurin docking site. Genes Dev. 16:608-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crabtree, G. R., and E. N. Olson. 2002. NFAT signaling: choreographing the social lives of cells. Cell 109(Suppl.):S67-S79. [DOI] [PubMed] [Google Scholar]
- 13.Cunningham, K. W., and G. R. Fink. 1994. Calcineurin-dependent growth control in Saccharomyces cerevisiae mutants lacking PMC1, a homolog of plasma membrane Ca2+ ATPases. J. Cell Biol. 124:351-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cyert, M. S. 2003. Calcineurin signaling in Saccharomyces cerevisiae: how yeast go crazy in response to stress. Biochem. Biophys. Res. Commun. 311:1143-1150. [DOI] [PubMed] [Google Scholar]
- 15.Cyert, M. S., R. Kunisawa, D. Kaim, and J. Thorner. 1991. Yeast has homologs (CNA1 and CNA2 gene products) of mammalian calcineurin, a calmodulin-regulated phosphoprotein phosphatase. Proc. Natl. Acad. Sci. USA 88:7376-7380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cyert, M. S., and J. Thorner. 1992. Regulatory subunit (CNB1 gene product) of yeast Ca2+/calmodulin-dependent phosphoprotein phosphatases is required for adaptation to pheromone. Mol. Cell. Biol. 12:3460-3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Davis, D. 2003. Adaptation to environmental pH in Candida albicans and its relation to pathogenesis. Curr. Genet. 44:1-7. [DOI] [PubMed] [Google Scholar]
- 18.Davis, N. G., J. L. Horecka, and G. F. Sprague, Jr. 1993. Cis- and trans-acting functions required for endocytosis of the yeast pheromone receptors. J. Cell Biol. 122:53-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de la Pompa, J. L., L. A. Timmerman, H. Takimoto, H. Yoshida, A. J. Elia, E. Samper, J. Potter, A. Wakeham, L. Marengere, B. L. Langille, G. R. Crabtree, and T. W. Mak. 1998. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 392:182-186. [DOI] [PubMed] [Google Scholar]
- 20.Farcasanu, I. C., D. Hirata, E. Tsuchiya, F. Nishiyama, and T. Miyakawa. 1995. Protein phosphatase 2B of Saccharomyces cerevisiae is required for tolerance to manganese, in blocking the entry of ions into the cells. Eur. J. Biochem. 232:712-717. [PubMed] [Google Scholar]
- 21.Fedorov, A. N., and T. O. Baldwin. 1997. Cotranslational protein folding. J. Biol. Chem. 272:32715-32718. [DOI] [PubMed] [Google Scholar]
- 22.Giaever, G., A. M. Chu, L. Ni, C. Connelly, L. Riles, S. Veronneau, S. Dow, A. Lucau-Danila, K. Anderson, B. Andre, A. P. Arkin, A. Astromoff, M. El-Bakkoury, R. Bangham, R. Benito, S. Brachat, S. Campanaro, M. Curtiss, K. Davis, A. Deutschbauer, K. D. Entian, P. Flaherty, F. Foury, D. J. Garfinkel, M. Gerstein, D. Gotte, U. Guldener, J. H. Hegemann, S. Hempel, Z. Herman, D. F. Jaramillo, D. E. Kelly, S. L. Kelly, P. Kotter, D. LaBonte, D. C. Lamb, N. Lan, H. Liang, H. Liao, L. Liu, C. Luo, M. Lussier, R. Mao, P. Menard, S. L. Ooi, J. L. Revuelta, C. J. Roberts, M. Rose, P. Ross-Macdonald, B. Scherens, G. Schimmack, B. Shafer, D. D. Shoemaker, S. Sookhai-Mahadeo, R. K. Storms, J. N. Strathern, G. Valle, M. Voet, G. Volckaert, C. Y. Wang, T. R. Ward, J. Wilhelmy, E. A. Winzeler, Y. Yang, G. Yen, E. Youngman, K. Yu, H. Bussey, J. D. Boeke, M. Snyder, P. Philippsen, R. W. Davis, and M. Johnston. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418:387-391. [DOI] [PubMed] [Google Scholar]
- 23.Graef, I. A., F. Chen, L. Chen, A. Kuo, and G. R. Crabtree. 2001. Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105:863-875. [DOI] [PubMed] [Google Scholar]
- 24.Graef, I. A., F. Wang, F. Charron, L. Chen, J. Neilson, M. Tessier-Lavigne, and G. R. Crabtree. 2003. Neurotrophins and netrins require calcineurin/NFAT signaling to stimulate outgrowth of embryonic axons. Cell 113:657-670. [DOI] [PubMed] [Google Scholar]
- 25.Haro, R., B. Garciadeblas, and A. Rodriguez-Navarro. 1991. A novel P-type ATPase from yeast involved in sodium transport. FEBS Lett. 291:189-191. [DOI] [PubMed] [Google Scholar]
- 26.Hilioti, Z., D. A. Gallagher, S. T. Low-Nam, P. Ramaswamy, P. Gajer, T. J. Kingsbury, C. J. Birchwood, A. Levchenko, and K. W. Cunningham. 2004. GSK-3 kinases enhance calcineurin signaling by phosphorylation of RCNs. Genes Dev. 18:35-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huh, W. K., J. V. Falvo, L. C. Gerke, A. S. Carroll, R. W. Howson, J. S. Weissman, and E. K. O'Shea. 2003. Global analysis of protein localization in budding yeast. Nature 425:686-691. [DOI] [PubMed] [Google Scholar]
- 28.James, P., J. Halladay, and E. A. Craig. 1996. Genomic libraries and a host strain designed for highly efficient two-hybrid selection in yeast. Genetics 144:1425-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang, B., and M. S. Cyert. 1999. Identification of a novel region critical for calcineurin function in vivo and in vitro. J. Biol. Chem. 274:18543-18551. [DOI] [PubMed] [Google Scholar]
- 30.Kuno, T., H. Tanaka, H. Mukai, C. D. Chang, K. Hiraga, T. Miyakawa, and C. Tanaka. 1991. cDNA cloning of a calcineurin B homolog in Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 180:1159-1163. [DOI] [PubMed] [Google Scholar]
- 31.Kutay, U., G. Ahnert-Hilger, E. Hartmann, B. Wiedenmann, and T. A. Rapoport. 1995. Transport route for synaptobrevin via a novel pathway of insertion into the endoplasmic reticulum membrane. EMBO J. 14:217-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, Y., S. Ishii, M. Tokai, H. Tsutsumi, O. Ohki, R. Akada, K. Tanaka, E. Tsuchiya, S. Fukui, and T. Miyakawa. 1991. The Saccharomyces cerevisiae genes (CMP1 and CMP2) encoding calmodulin-binding proteins homologous to the catalytic subunit of mammalian protein phosphatase 2B. Mol. Gen. Genet. 227:52-59. [DOI] [PubMed] [Google Scholar]
- 33.Matheos, D. P., T. J. Kingsbury, U. S. Ahsan, and K. W. Cunningham. 1997. Tcn1p/Crz1p, a calcineurin-dependent transcription factor that differentially regulates gene expression in Saccharomyces cerevisiae. Genes Dev. 11:3445-3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazur, P., N. Morin, W. Baginsky, M. el-Sherbeini, J. A. Clemas, J. B. Nielsen, and F. Foor. 1995. Differential expression and function of two homologous subunits of yeast 1,3-β-D-glucan synthase. Mol. Cell. Biol. 15:5671-5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mendizabal, I., G. Rios, J. M. Mulet, R. Serrano, and I. F. de Larrinoa. 1998. Yeast putative transcription factors involved in salt tolerance. FEBS Lett. 425:323-328. [DOI] [PubMed] [Google Scholar]
- 36.Mendoza, I., F. Rubio, A. Rodriguez-Navarro, and J. M. Pardo. 1994. The protein phosphatase calcineurin is essential for NaCl tolerance of Saccharomyces cerevisiae. J. Biol. Chem. 269:8792-8796. [PubMed] [Google Scholar]
- 37.Molkentin, J. D., J. R. Lu, C. L. Antos, B. Markham, J. Richardson, J. Robbins, S. R. Grant, and E. N. Olson. 1998. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakamura, T., Y. Liu, D. Hirata, H. Namba, S. Harada, T. Hirokawa, and T. Miyakawa. 1993. Protein phosphatase type 2B (calcineurin)-mediated, FK506-sensitive regulation of intracellular ions in yeast is an important determinant for adaptation to high salt stress conditions. EMBO J. 12:4063-4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Plath, K., and T. A. Rapoport. 2000. Spontaneous release of cytosolic proteins from posttranslational substrates before their transport into the endoplasmic reticulum. J. Cell Biol. 151:167-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ranger, A. M., M. J. Grusby, M. R. Hodge, E. M. Gravallese, F. C. de la Brousse, T. Hoey, C. Mickanin, H. S. Baldwin, and L. H. Glimcher. 1998. The transcription factor NF-ATc is essential for cardiac valve formation. Nature 392:186-190. [DOI] [PubMed] [Google Scholar]
- 41.Rudolph, H. K., A. Antebi, G. R. Fink, C. M. Buckley, T. E. Dorman, J. LeVitre, L. S. Davidow, J. I. Mao, and D. T. Moir. 1989. The yeast secretory pathway is perturbed by mutations in PMR1, a member of a Ca2+ ATPase family. Cell 58:133-145. [DOI] [PubMed] [Google Scholar]
- 42.Sanglard, D., F. Ischer, O. Marchetti, J. Entenza, and J. Bille. 2003. Calcineurin A of Candida albicans: involvement in antifungal tolerance, cell morphogenesis and virulence. Mol. Microbiol. 48:959-976. [DOI] [PubMed] [Google Scholar]
- 43.Serrano, R., A. Ruiz, D. Bernal, J. R. Chambers, and J. Arino. 2002. The transcriptional response to alkaline pH in Saccharomyces cerevisiae: evidence for calcium-mediated signalling. Mol. Microbiol. 46:1319-1333. [DOI] [PubMed] [Google Scholar]
- 44.Sherman, F. 1991. Getting started with yeast, p. 3-22. In C. Guthrie and G. R. Fink (ed.), Guide to yeast genetics and molecular biology. Academic Press, San Diego, Calif.
- 45.Silorski, R. S., and P. Heiter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stathopoulos, A. M., and M. S. Cyert. 1997. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 11:3432-3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stathopoulos-Gerontides, A., J. J. Guo, and M. S. Cyert. 1999. Yeast calcineurin regulates nuclear localization of the Crz1p transcription factor through dephosphorylation. Genes Dev. 13:798-803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uetz, P., L. Giot, G. Cagney, T. A. Mansfield, R. S. Judson, J. R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, A. Qureshi-Emili, Y. Li, B. Godwin, D. Conover, T. Kalbfleisch, G. Vijayadamodar, M. Yang, M. Johnston, S. Fields, and J. M. Rothberg. 2000. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403:623-627. [DOI] [PubMed] [Google Scholar]
- 49.Wattenberg, B., and T. Lithgow. 2001. Targeting of C-terminal (tail)-anchored proteins: understanding how cytoplasmic activities are anchored to intracellular membranes. Traffic 2:66-71. [DOI] [PubMed] [Google Scholar]
- 50.Withee, J. L., J. Mulholland, R. Jeng, and M. S. Cyert. 1997. An essential role of the yeast pheromone-induced Ca2+ signal is to activate calcineurin. Mol. Biol. Cell 8:263-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoshimoto, H., K. Saltsman, A. P. Gasch, H. X. Li, N. Ogawa, D. Botstein, P. O. Brown, and M. S. Cyert. 2002. Genome-wide analysis of gene expression regulated by the calcineurin/Crz1p signaling pathway in Saccharomyces cerevisiae. J. Biol. Chem. 277:31079-31088. [DOI] [PubMed] [Google Scholar]