

Eukaryotic cells license each DNA replication origin during G1 phase by assembling a prereplication complex (pre-RC) that contains a Mcm2–7 double hexamer. In this study, Sun et al. examined the helicase loading reaction in the presence of ATP, revealing the basic architecture of a number of pre-RC assembly reaction intermediates, including a newly identified ORC–Cdc6–Mcm2–7–Mcm2–7 complex. The detailed architecture of the Mcm2–7 double hexamer was also established.
Keywords: origin recognition complex, DNA replication initiation, replicative helicase, prereplication complex, electron microscopy
Abstract
Eukaryotic cells license each DNA replication origin during G1 phase by assembling a prereplication complex that contains a Mcm2–7 (minichromosome maintenance proteins 2–7) double hexamer. During S phase, each Mcm2–7 hexamer forms the core of a replicative DNA helicase. However, the mechanisms of origin licensing and helicase activation are poorly understood. The helicase loaders ORC–Cdc6 function to recruit a single Cdt1–Mcm2–7 heptamer to replication origins prior to Cdt1 release and ORC–Cdc6–Mcm2–7 complex formation, but how the second Mcm2–7 hexamer is recruited to promote double-hexamer formation is not well understood. Here, structural evidence for intermediates consisting of an ORC–Cdc6–Mcm2–7 complex and an ORC–Cdc6–Mcm2–7–Mcm2–7 complex are reported, which together provide new insights into DNA licensing. Detailed structural analysis of the loaded Mcm2–7 double-hexamer complex demonstrates that the two hexamers are interlocked and misaligned along the DNA axis and lack ATP hydrolysis activity that is essential for DNA helicase activity. Moreover, we show that the head-to-head juxtaposition of the Mcm2–7 double hexamer generates a new protein interaction surface that creates a multisubunit-binding site for an S-phase protein kinase that is known to activate DNA replication. The data suggest how the double hexamer is assembled and how helicase activity is regulated during DNA licensing, with implications for cell cycle control of DNA replication and genome stability.
DNA licensing is the focal point of several redundant cell cycle controls that guarantees that each genome segment is replicated once and only once per cell division cycle (Bell and Dutta 2002; Remus and Diffley 2009; Pospiech et al. 2010; Siddiqui et al. 2013). The molecular basis of origin licensing has been best studied using the budding yeast Saccharomyces cerevisiae system. It involves assembly of a prereplicative complex (pre-RC) by loading of the core replicative helicase minichromosome maintenance proteins 2–7 (Mcm2–7) as a double hexamer around dsDNA at each replication origin (Evrin et al. 2009; Remus et al. 2009). While in bacteria, archaea, and eukaryotic viruses the loaded helicase is active for DNA unwinding, in eukaryotic cells, the helicase is loaded in an inactive form (Yardimci and Walter 2014). This is a central feature for the temporal separation of DNA licensing in G1 phase from initiation of DNA replication in S phase that is essential for once per cell cycle DNA replication (Diffley 2001). However, what confines the Mcm2–7 double hexamer to its inactive state in G1 phase is only partially understood but is of relevance, as untimely helicase activation could result in rereplication and genomic instability.
Mcm2–7 double-hexamer formation is a multistep reaction during which two Mcm2–7 hexamers are recruited one after another to replication origins (Evrin et al. 2013; Fernandez-Cid et al. 2013; Frigola et al. 2013). In S. cerevisiae, ORC–Cdc6 recognizes the replication origin in an ATP-dependent manner (Fig. 1A, step 1; Bell and Stillman 1992; Speck et al. 2005; Speck and Stillman 2007). Chromatin-bound ORC–Cdc6 function to recruit a Cdt1–Mcm2–7 heptamer to replication origins (Fig. 1A, step 2; Speck et al. 2005; Randell et al. 2006; Speck and Stillman 2007; Tsakraklides and Bell 2010; Evrin et al. 2013; Frigola et al. 2013; Sun et al. 2013). Using an in vitro Mcm2–7 loading system that employs a purified ORC, Cdc6, Cdt1, and Mcm2–7 hexamer and an origin-containing plasmid, we recently observed formation of a loading intermediate termed OCCM in the presence of a slowly hydrolysable ATP analog, ATP-γS, which captured a complex containing ORC–Cdc6 bound to a single Mcm2–7 hexamer and one to two copies of Cdt1 (Takara and Bell 2011; Evrin et al. 2013; Sun et al. 2013). A cryoelectron microscopy (cryo-EM) structure of this pre-RC intermediate showed that the AAA+ ATPase domains of ORC/Cdc6 latch onto the C-terminal AAA+ ATPase domains of Mcm2–7, leaving the Mcm2–7 N-terminal domains (NTDs) accessible, detailing the overall architecture of this important complex. Using biochemical experiments, we observed that Orc1 and Cdc6 ATP hydrolysis promotes rapid Cdt1 release and the formation of an ORC–Cdc6–Mcm2–7 (OCM) complex (Randell et al. 2006; Fernandez-Cid et al. 2013). One question that arises from that study is whether ATP hydrolysis by ORC–Cdc6 and subsequent Cdt1 dissociation leads to a resetting of ORC–Cdc6 on DNA such that it would be in a position to recruit the next Mcm2–7 hexamer (Fig. 1A, step 3, right) or whether the ORC–Cdc6 stays tightly associated with the first Mcm2–7 hexamer (Fig. 1A, step 3, left). It is clear that the OCM complex is a precursor of Mcm2–7 double-hexamer formation (Fernandez-Cid et al. 2013), but exactly how the second Mcm2–7 hexamer becomes recruited is not known (Fig. 1A, step 4; Yardimci and Walter 2014). The final product of pre-RC formation is the Mcm2–7 double hexamer (Evrin et al. 2009; Remus et al. 2009), and, in this complex, both hexamers interact via their N termini (Fig. 1A, step 5; Remus et al. 2009; On et al. 2014). While pre-RC intermediates are salt-sensitive, potentially because the Mcm2–7 ring becomes destabilized, the Mcm2–7 double hexamer remains bound to DNA in high-salt conditions, but the molecular reason for this is only partially understood. Importantly, the Mcm2–7 double hexamer serves as the platform for replisome assembly (On et al. 2014). During this process, a number of replication factors and two S-phase kinases bind to the double hexamer, which allows extrusion of one DNA strand from each Mcm2–7 ring prior to processive DNA unwinding (Ilves et al. 2010; Labib 2010, 2011; Heller et al. 2011; Siddiqui et al. 2013). The details of how these complex reactions occur are not understood (Tanaka and Araki 2013), but it is clear that ATP hydrolysis is central for Mcm2–7 helicase activity and DNA unwinding (Bochman and Schwacha 2008). The purified Mcm2–7 hexamer hydrolyzes ATP at an intermediate rate (Vijayraghavan and Schwacha 2012; Fernandez-Cid et al. 2013), but when integrated in the replisome, the ATPase activity of the helicase becomes strongly activated (Ilves et al. 2010). How Mcm2–7 ATP hydrolysis is regulated and what ATPase activity the important Mcm2–7 double hexamer has is not known. Interestingly, one of the S-phase kinases, Dbf4-dependent-kinase Cdc7 (DDK), interacts with Mcm2 and Mcm4 (Sheu and Stillman 2010; Ramer et al. 2013) and phosphorylates specifically the chromatin-bound Mcm2–7 double hexamer but not the non-chromatin-bound single hexamer (Sheu and Stillman 2006; Francis et al. 2009), suggesting that DDK recognizes a specific structure in the double hexamer.
Figure 1.

The multistep pre-RC assembly process and EM visualization of intermediates during recruitment of the first Mcm2–7 hexamer by ORC–Cdc6. (A) A sketch for loading of the Mcm2–7 double hexamer highlighting three unresolved issues as shown by three question marks: (1) Does ATP hydrolysis and Cdt1 release from OCCM lead to a large gap between ORC-Cdc6 and Mcm2–7 that may allow ORC–Cdc6 to recruit a second Mcm2–7 hexamer (step 3)? (2) How is the second Mcm2–7 hexamer recruited (step 4)? (3) How are the two Mcm2–7 hexamers arranged in the double hexamer (step 5)? (B) The OCM structure compared with the OCCM structure. (Top panel) 2D class averages of cryo-EM (top row) and negative stain images (bottom row) of OCCM formed in the presence of ATP-γS. Red arrows point to the Cdt1 density identified previously. (Bottom panel) 2D class averages of OCM in the presence of ATP at a 7-min reaction time by cryo-EM (top row) and negative stain EM (bottom row). Blue arrows point to the regions where Cdt1 is missing. In both panels, a selected 2D average is enlarged and overlaid with sketches of the proposed ORC–Cdc6 region in orange and the Mcm2–7 region in purple. Bar, 15 nm.
In this study, we examined by EM the helicase loading reaction in the presence of ATP, revealing the basic architecture of a number of pre-RC assembly reaction intermediates, including the OCM, a newly identified ORC–Cdc6–Mcm2–7-Mcm2–7 (OCMM) complex, and the final product: the Mcm2–7 double hexamer. Through a systematic mapping strategy that employs subunit fusions with the maltose-binding protein (MBP), we established the detailed architecture of the Mcm2–7 double hexamer. These structural analyses, in combination with ATP hydrolysis assays, suggest a general pathway for pre-RC assembly, Mcm2–7 double hexamer stability on DNA during G1, and mechanisms for blocking helicase activation within the Mcm2–7 double-hexamer and reveal a multihexamer-binding site in the double hexamer for recruitment of the activating kinase DDK.
Results
EM structure of the OCM complex
Reactions containing ORC–Cdc6, Cdt1–Mcm2–7, and DNA result in recruitment of the proteins to DNA, as measured by coprecipitation of the protein with DNA bound to beads after washing in low salt. The topological loading of the Mcm2–7 double hexamer around the dsDNA was determined by washing the bead-bound DNA with high salt and under conditions in which only the Mcm2–7 double hexamer remained DNA bound (Evrin et al. 2009; Remus et al. 2009). A trapped OCCM complex was obtained in the absence of ATP hydrolysis, but when ATP hydrolysis was allowed, Cdt1 was rapidly released from the OCCM, and formation of an OCM complex occurred, which serves as the platform for Mcm2–7 double-hexamer assembly (Fernandez-Cid et al. 2013).
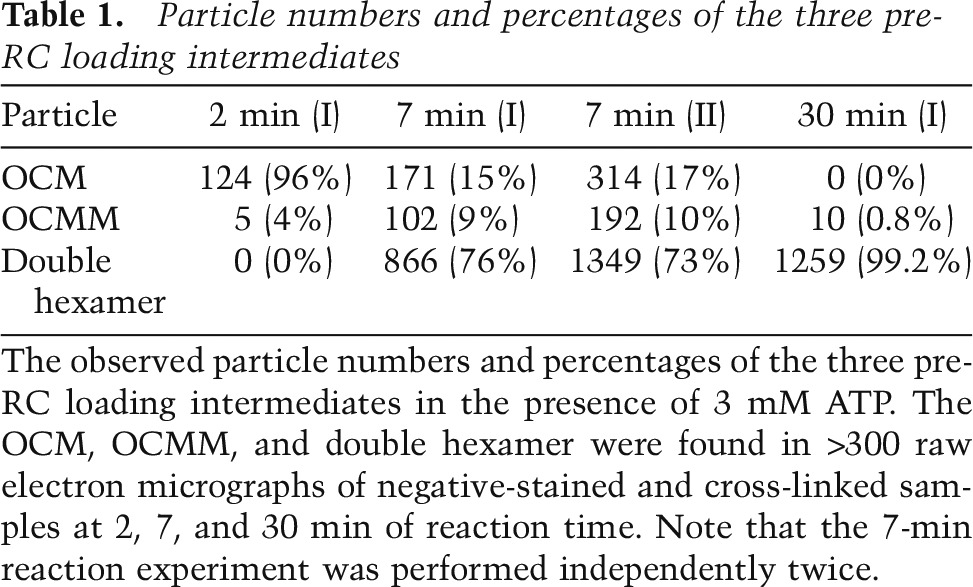
To study ATP hydrolysis-dependent intermediates in the assembly reactions while the reaction transitioned from the OCCM to the double hexamer, we harvested reaction products in the presence of 3 mM ATP at 2-, 7-, and 30-min time points; cross-linked them with glutaraldehyde; and then examined the samples by EM. Image classification of the spread complexes was used to analyze the products of the reaction. In addition to the expected fully assembled Mcm2–7 double hexamer with a distinct, four-tiered side view (see below), we found two additional complexes, one of which was the expected three-tiered OCM complex (Fig. 1B, bottom panel), and the other was a new, five-tiered OCMM that is described below. We found that in the 2-min sample, the OCM particles dominated (96%), with a small percentage of OCMM particles (4%) and without an observable double hexamer. In the 30-min sample, the double-hexamer particles dominated (99%), with an occasional OCMM particle (<1%). In the 7-min sample, all three types of particles were present in significant numbers (Table 1): Approximately 15% were OCM, ∼10% were OCMM, and ∼75% fully were assembled double hexamers. We note that the high stability of the Mcm2–7 double hexamer may have inflated its observed frequency (Remus et al. 2009); it is therefore quite likely that the OCM and OCMM are more abundant than the ratio suggests at these time points during pre-RC formation.
Table 1.
Particle numbers and percentages of the three pre-RC loading intermediates
The OCM was visualized by both negative stain EM and cryo-EM (Fig. 1B). Compared with the two-dimensional (2D) averages of the OCCM complex formed in the presence of ATP-γS (Fig. 1B, top panel), the OCM particles were missing the distinct Cdt1 density located at the side of the Mcm2–7 hexamer (Fig. 1B, bottom panel). The OCCM structure was not observed in reactions that contained ATP instead of ATP-γS, highlighting that the OCCM is extremely short-lived. Interestingly, in the OCM, ORC–Cdc6 and Mcm2–7 appear to be more closely engaged than within the OCCM, which could reflect a functional difference, as the OCM, in contrast to the OCCM, is competent for Mcm2–7 double-hexamer formation.
The OCMM—a helicase loading intermediate containing an OCM and a second Mcm2–7 single hexamer
We found by negative stain EM a five-tiered structure that we called OCMM because it is comprised of one OCM and a second Mcm2–7 single hexamer (Fig. 2A). In the four reference-free class averages of OCMM particles, the two Mcm2–7 hexamers were similar in structure to the fully assembled and isolated double hexamer (Fig. 2B). However, the left capping density is less well defined, so we were uncertain whether Cdc6 remained associated with ORC at this stage. We assumed that Cdc6 was present because ORC was capable of repeated loading of the Mcm2–7 hexamer in vitro, and the loading reaction required Cdc6 (Chen et al. 2007). The OCMM particles can also be observed in the pre-RC reaction without cross-linking, although at much lower frequency (Fig. 2C), suggesting that OCMM is a true pre-RC intermediate. The fact that OCMM nearly peaks at 7 min and disappears at the 30-min reaction time point indicates that it is a pre-RC assembly intermediate (Table 1; Fig. 2D). The OCMM is likely a late stage intermediate because the two Mcm2–7 hexamers appear to have fully assembled into a double hexamer (Tsakraklides and Bell 2010). Additional short-lived intermediates preceding the OCMM, such as a double OCM, could exist, but we did not observe them among many thousands of molecules.
Figure 2.

A novel loading intermediate containing one OCM and one Mcm2–7 hexamer. (A) Four class-averaged, negative stain EM images of the novel five-tiered Mcm2–7 loading intermediate. Particles were selected from a sample that was incubated in 3 mM ATP for 7 min followed by 0.1% glutaraldehyde cross-linking. (B) Selected reference-free 2D averages of a Mcm2–7 double hexamer found in the same sample as in A. (C) The OCMM raw particle images found in the 7-min reaction sample without glutaraldehyde cross-linking. (D) The OCMM is an on-pathway intermediate during the loading of the Mcm2–7 double hexamer. The question mark denotes the possible existence of additional intermediates upstream of OCMM.
Three-dimensional (3D) reconstruction of the Mcm2–7 double hexamer
Unlike the above-described OCM and OCMM that are transient and unstable helicase loading structures, the Mcm2–7 double hexamer is highly stable, which allows a more detailed analysis of the structure. The double hexamers were visible on a 3000-base-pair (bp) circular plasmid when vitrified in the in vitro loading buffer and imaged by cryo-EM (Supplemental Fig. 1A–C). We selected several dozen of the double-hexamer particles on plasmids and subjected them to image classification. The EM images demonstrated that the protein complexes that remained on the high-salt-washed plasmid DNA were individual double hexamers and that the double hexamers did not further interact to form higher-order oligomers. The EM images further showed that dsDNA entered into the Mcm2–7 double hexamer from one end of the long axis and exited from the other, consistent with the notion that dsDNA passed through the central channel of the double hexamer (Evrin et al. 2009; Remus et al. 2009). The concentration of the plasmid-bound Mcm2–7 double hexamer was too low for efficient EM data collection. To improve the sample yield and concentration, we assembled pre-RC complexes on biotinylated origin DNA linked to streptavidin-tagged magnetic beads, washed the complexes with high salt, and finally released the protein complex from the magnetic beads with DNase I and concentrated the sample. The double-hexamer particles had good contrast in the negatively stained raw EM images and resulted in well-defined class averages, allowing a 3D EM reconstruction at 2-nm resolution (Supplemental Fig. 1D–G; Supplemental Movie 1). The 3D map reveals that the Mcm2–7 double hexamer has an approximately cylindrical structure that is 21.5 nm long and 14 nm wide, with the two hexamers in the Mcm2–7 double hexamer slightly offset from the cylindrical axis and twisted relative to each other, similar to a previous EM structure (Remus et al. 2009). However, only in the current structure were individual protein subunits resolved, which allowed for a more detailed analysis.
Mapping the architecture of the Mcm2–7 double hexamer
The identification of individual subunits in a 3D EM structure of a multiprotein complex allows for better understanding and interpretation of the data. Therefore, we employed a 3D mapping strategy employing MBP insertions into the N-terminal or C-terminal region of individual MCM protein subunits, similar to the strategy used for the OCCM (Sun et al. 2013), and assembled the MBP-inserted Mcm2–7 double hexamers one at a time (Supplemental Fig. 2, asterisks). We found that Mcm2–7 hexamers with MBP insertion into either the N-terminal region of Mcm2, Mcm3, Mcm6, or Mcm7 or the C-terminal region of Mcm2 and Mcm5 could be properly loaded by ORC–Cdc6 and Cdt1 into double hexamers, as shown by the reference-free class-averaged EM images (Fig. 3A). The 3D reconstruction of each MBP insertion double hexamer clearly showed two extra densities compared with the wild type that could be unambiguously assigned to the inserted MBP (Fig. 3B).
Figure 3.

3D reconstructions of six MBP-inserted Mcm2–7 double hexamers. (A) Two selected reference-free class averages of the double hexamer with MBP inserted to the N-terminal region of Mcm2 (MBP-M2), Mcm3 (MBP-M3), Mcm6 (MBP-M6), and Mcm7 (MBP-M7) and the C-terminal region of Mcm2 (M2-MBP) and Mcm5 (M5-MBP). Blue asterisks mark the extra and peripheral MBP densities. Box size is 36 nm. (B) Each column shows the top, front, and side views of the six MBP-fused Mcm2–7 double hexamers. The MBP density is colored blue in the 3D map. The bottom panels show horizontal sections of the 3D maps at the inserted MBP positions as indicated by dashed lines. Blue asterisks mark the MBP densities in the sections. Box size in the bottom panel is 24 nm. (C) The Mcm subunit arrangement in the lower hexamer as viewed from the N termini (left) and in the upper hexamer viewed from the C termini (right). (D) NTD and CTD assignment of each Mcm subunit in the top hexamer of the double hexamer as viewed from the side. The NTDs and the CTDs are staggered such that each Mcm subunit is tilted in the hexamer, and the double hexamer is twisted. The Mcm protein domains labeled with red numbers in C and D are positively identified in this study, and the domains in blue numbers are inferred from the known order of subunits in the hexamer.
Each Mcm protein has two main domains: an N-terminal zinc-binding domain (NTD) and a C-terminal domain (CTD) that contains the AAA+ ATPase motifs. The eukaryotic Mcm subunits Mcm2, Mcm4, and Mcm6 also have serine- and threonine-rich, nonstructured extensions on their very N termini (NSDs) that are targets for regulation by cyclin-dependent kinase (CDK), DDK, and checkpoint kinase Mec1 (Randell et al. 2010; Tanaka and Araki 2013). Note that archaeal MCMs have both the NTD and CTD but lack any N-terminal extensions (NTEs) like the eukaryotic NSDs. Because the order of the subunits within the Mcm2–7 hexamer has been well established as Mcm5, Mcm3, Mcm7, Mcm4, Mcm6, and Mcm2 (Bochman et al. 2008; Costa et al. 2011; Lyubimov et al. 2012; Vijayraghavan and Schwacha 2012; Sun et al. 2013), mapping one Mcm protein would have been sufficient for assigning the remaining five MCM proteins if the handedness of the EM structure was determined, and mapping two Mcm proteins was sufficient to assign both the handedness and the identity for all proteins. We experimentally mapped the NTD of four Mcm protein subunits, which is more than sufficient to identify all NTDs as well as the handedness of the structure (Fig. 3B,C). Unambiguous assignment for the identities of the six CTDs was facilitated by that fact that the CTDs should follow the same order of subunit arrangement and by our localization of the CTDs of Mcm2 and Mcm5 (Fig. 3B,D). This completes the assignment of both the NTDs and CTDs for all Mcm protein subunits in the double hexamer. If we consider the longitudinal axis of the double hexamer as the path of the DNA (i.e., vertical in the front and side views in Fig. 3B), then the Mcm5 CTD in the upper hexamer was tilted ∼30° toward the right relative to the Mcm5 NTD. Indeed, a notable feature of all of the Mcm subunits in the double hexamer is the staggered arrangement of the NTD ring relative to the CTD ring within a single Mcm2–7 complex in the double hexamer (Figs. 3D, 4). This is in stark contrast to the relative arrangement in the single Mcm2–7 hexamer and the Cdc45–Mcm2–7–GINS (CMG) helicase, where the NTD and CTD rings are vertically aligned (see the Discussion; Sun et al. 2013; Costa et al. 2014).
Figure 4.

The architecture of the Mcm2–7 double hexamer. (A) Four side views of the 3D map of the wild-type Mcm2–7 double hexamer consecutively rotated 90° around a vertical axis. The six Mcm proteins are colored differently and labeled correspondingly. A pair of blue lines trace the helical arrangement of the NTDs (red asterisks) of Mcm2, Mcm6, and Mcm4 (back side view). The black ovals in the front and back views indicate the position of the twofold axis. (B) Top and bottom views of the Mcm2–7 CTD ring. A density that is connected to the CTD of Mcm3 and located near the hexamer center is tentatively assigned as the CTE of Mcm3. (C) Top and bottom views of the Mcm2–7 NTD ring. Overlapping the two views by translation generates the interface between two hexamers of the Mcm2–7 double hexamer. Mcm2–Mcm5 in one hexamer is approximately aligned with Mcm4–Mcm6 in the other.
Segmentation and subunit assignment of the 3D EM map of the Mcm2–7 double hexamer
We carried out semiautomatic density segmentation in Chimera (Pettersen et al. 2004). First, the double hexamer was separated at the middle into two hexamers. Next, within each hexamer, six NTD densities were located in the middle of the double hexamer, and six CTD densities were located on the top and bottom of the double hexamer. Finally, the NTDs and CTDs belonging to the same proteins as described above were merged, and their subunit identities were assigned (Fig. 4A–C). The extra density near the center of the CTD ring is directly connected to the Mcm3 CTD and was therefore assigned as the Mcm3 C-terminal extension (CTE) (Fig. 4A,B). The Mcm3 CTE was shown to activate the ATPase activities of ORC–Cdc6 (Frigola et al. 2013). The Mcm3 CTE partially protrudes from the end of the MCM hexamer, providing a structural basis for its function in the initial binding to ORC–Cdc6 (Frigola et al. 2013; Sun et al. 2013). Furthermore, its prominent position suggested that it could alter Mcm2–7 subunit interactions or influence Mcm2–7 ATP hydrolysis. Indeed, for Mcm2–7-ΔC3 in comparison with wild-type Mcm2–7, we observed reduced ATP hydrolysis activity that was even more reduced in the presence of Cdt1 (Supplemental Fig. 3).
From the segmented and subunit-assigned structure, it is clear that the twofold axis of the double hexamer relating the two hexamers is a horizontal line running between subunits Mcm3 and Mcm7 at one side and Mcm2 and Mcm6 on the other side (Fig. 4A; Supplemental Movie 2). Therefore, in the Mcm2–7 double hexamer, the NTDs of Mcm5, Mcm3, Mcm7, Mcm4, Mcm6, and Mcm2 of one hexamer come into close proximity to the NTDs of Mcm7, Mcm3, Mcm5, Mcm2, Mcm6, and Mcm4 of the opposing hexamer, respectively. This arrangement places the NTDs of both Mcm3 subunits adjacent to each other, but because the hexamers are in a head-to-head arrangement, the remaining subunits are opposite nonidentical NTDs. For example, Mcm4 and Mcm6 that are adjacent on one hexamer associate with Mcm2 on the other, forming a composite heterotrimer of Mcm2, Mcm4, and Mcm6. One prominent feature of the double hexamer is that the NTDs of several Mcm subunits protrude toward the CTDs (Fig. 4A, front view). This is most obvious in the case of Mcm4 and Mcm6, which have, in contrast to Mcm3 and Mcm5, long NSDs. Indeed the NSD of Mcm4 contains sequences that block the initiation of DNA replication until it is hyperphosphorylated by the DDK kinase (Supplemental Fig. 2; Sheu and Stillman 2006, 2010).
The two Mcm2 NTDs in the double hexamer are related by the twofold axis and are in close proximity to each other, whereas the neighboring Mcm6 NTD is situated up toward the CTD by 15 Å, and the next subunit, the Mcm4 NTD, is situated further upward of the Mcm6 NTD by yet another 15 Å, making the NTDs of the Mcm2, Mcm6, and Mcm4 subunits form a staircase-like structure (as outlined by two blue lines in Fig. 4A, back view, NTDs marked by red asterisks). Such an NTD arrangement occurs only at the backside of the double hexamer. On the front side, the NTDs of 3, 5, 7 are approximately coplanar (Fig. 4A, front view). Thus, our analysis details how the individual Mcm2–7 hexamers are twisted relative to each other.
The NTD of Methanobacterium thermoautotrophicum archaeal MCM that corresponds to the NTD of the eukaryotic MCM subunit had been crystallized in the double-hexameric form (Fletcher et al. 2003). This structure can be docked into the NTD rings of the 3D reconstruction of the Mcm2–7 double hexamer (Supplemental Fig. 4A,C). If we align the NTD of the nearly full length archaeon Sulfolobus solfataricus monomeric MCM crystal structure to an NTD of the docked archaeal NTD hexamer (Brewster et al. 2008), the CTD of the monomeric archaeal MCM falls into the CTD density of the segmented MCM subunits in the double-hexamer EM structure. The six Mcm proteins segmented from the double hexamer are of a similar size and shape (Supplemental Fig. 4D). They all have a bifurcated NTD that is connected to a globular CTD via a constricted linker region and approximately fit with the rigid body-docked monomeric S. solfataricus MCM protein structure (Brewster et al. 2008). Notable differences exist, mainly in the top C-terminal region and the bottom N-terminal region, where some of the eukaryotic Mcm subunits have evolved CTEs and NTEs of variable lengths (Supplemental Fig. 2; Vijayraghavan and Schwacha 2012). Another source of difference arises from the single rigid body docking of the archaeal crystal structure because the relative angles and positions of the NTD and CTD may be slightly different in the eukaryotic Mcm proteins. A movie of the double-hexamer structure docked with the S. solfataricus MCM crystal structure (in the top hexamer) and the M. thermoautotrophicum NTD hexamer crystal structure (in the bottom hexamer) is shown in Supplemental Movie 3.
The ATP hydrolysis activity of Mcm2–7 is inhibited in the double-hexamer structure
The purified Mcm2–7 hexamer can hydrolyze ATP in vitro (Schwacha and Bell 2001; Ilves et al. 2010; Fernandez-Cid et al. 2013), but the active form of the helicase, when Mcm2–7 is part of a CMG complex, has a significantly higher ATPase activity (Ilves et al. 2010). On the other hand, the ATPase rate of the Mcm2–7 double hexamer is not known. The ∼2-nm staggered and ∼5° tilted stacking of the two hexamers and the ∼30° tilted arrangement of the individual Mcm proteins in the double hexamer, as revealed by our EM study, suggested that Mcm2–7 ATP hydrolysis could be altered (Fig. 5A). We measured the ATP hydrolysis rate of the hexameric and double-hexameric Mcm2–7 employing radioactive-labeled ATP and found that the ATP hydrolysis rate of the purified Mcm2–7 hexamer was 10.04 ± 0.66 pmol/min, similar to that previously observed (Schwacha and Bell 2001; Fernandez-Cid et al. 2013). In contrast, the in vitro assembled and purified Mcm2–7 double hexamer had markedly reduced ATPase activity of 1.41 ± 0.02 pmol/min (Fig. 5B). It is known that ATP is bound at the interface between the Mcm subunits and that ATPase activity relies on accurate positioning of the catalytic residues in the Walker A and B motifs and the arginine finger of the adjacent AAA+ subunit (Davey et al. 2003; Bochman and Schwacha 2009; Wendler et al. 2012). We therefore suggest that the inhibition of the ATPase activity in the double hexamer is due to the tilted conformation of Mcm subunits, resulting in uncoupling of the ATP hydrolysis motifs (Fig. 5A,C). The observed ATPase rate of the double hexamer is two orders of magnitude lower than the rate of the active Drosophila melanogaster CMG helicase (Ilves et al. 2010). Another possibility could be that the double-hexamer conformation per se is not compatible with ATP hydrolysis. However, an MCM double hexamer of the thermophilic archaeon M. thermoautotrophicum displays robust ATPase and helicase activity (Chong et al. 2000). Therefore, we suggest that the tilted subunit arrangement in the eukaryotic double hexamer is a powerful mechanism to restrict Mcm2–7 helicase activity in G1 phase and S phase prior to helicase activation (Diffley 2001).
Figure 5.

Functional implications of the Mcm2–7 double-hexamer structure. (A) There are two types of tilt found in the double-hexamer EM structure. The first is the end-to-end stacking of the two hexamers that is staggered by ∼2 nm and tilted by ∼5° off the vertical cylinder axis. The second is a ∼30° tilt of individual Mcm subunits in each hexamer with respect to the vertical axis. (B) The ATP hydrolysis rates were determined for the Mcm2–7 helicase in single-hexamer (H) and double-hexamer (DH) conformations. The single hexamer exhibits a robust ATPase activity that is strongly reduced in the double-hexamer configuration. (C, left) A speculative sketch illustrating that the arginine finger (R) of one Mcm subunit in the hexamer is properly positioned toward the Walker A and B motifs (A/B) of a neighbor protein for ATPase activity. (Right) Subunit tilt in the double hexamer misaligns the arginine finger (R) with respect to the A/B motifs and abolishes the ATPase activity. (D) The backside view of the double-hexamer structure shows that the Mcm2/5 interface in the upper hexamer is nearly opposite to the Mcm2/5 interface in the lower hexamer. The NTD of Mcm4 (M4N) wedges against the potential Mcm2/5 gate. (E) A sketch illustrating that the potential Mcm2/5 is unconstrained in the single hexamer (shown at left) but is constrained or locked by structural features such as the Mcm4 NTD of the opposing hexamer in the double hexamer. (F) A model for DDK interaction with the Mcm2–7 double hexamer but not with the single hexamer. The heterodimeric DDK requires a bipartite binding site (NTDs of Mcm2 and Mcm4) that is not available in the single hexamer but is available in the double hexamer. The double hexamer is in the right side view as in Figure 4A. (G) DDK activity on the purified single versus double Mcm2–7 hexamers. The top panels are by silver staining, and the bottom panel is by 32P autoradiography.
Function of the Mcm2/5 gate in the Mcm2–7 double hexamer
The opening of the Mcm2–7 ring is central to helicase loading onto DNA. Indeed, a DNA entry gate between Mcm2 and Mcm5 has been identified that is essential for pre-RC formation (Bochman and Schwacha 2008; Samel et al. 2014). If Mcm2–7 double-hexamer loading is a concerted process, as suggested (Remus et al. 2009; Yardimci and Walter 2014), then DNA insertion into both Mcm2–7 hexamers needs to be coordinated, requiring the Mcm2/5 gate of each hexamer to be aligned to promote simultaneous ring opening. However, the Mcm2–7 double-hexamer structure clearly shows that both Mcm2–7 rings are misaligned relative to the Mcm2/5 gate (Fig. 5D). Thus, our data show that Mcm2–7 loading must occur prior to double-hexamer formation, consistent with the finding that a single Mcm2–7 hexamer is loaded onto DNA during OCCM formation, although the first hexamer requires ORC–Cdc6 for stability (Sun et al. 2013; Samel et al. 2014). The Mcm2–7 double hexamer encircles dsDNA (Evrin et al. 2009; Remus et al. 2009), but the active helicase, as part of the CMG, encircles ssDNA (Fu et al. 2011), indicating that CMG formation in S phase involves remodeling of Mcm2–7 from a dsDNA-binding mode to an ssDNA-binding mode. This remodeling may involve opening of a gate between Mcm2 and Mcm5 (Bochman and Schwacha 2010; Costa et al. 2011; Samel et al. 2014) . However, interactions between Mcm4 of one hexamer and Mcm2 and Mcm5 of the other hexamer may lock the complex in the closed conformation (Fig. 5D,E). Thus, separation of both hexamers would have to precede or coincide with Mcm2–7 ring opening during helicase activation. In summary, we propose that the locked Mcm2/5 gate in the double hexamer represents a fundamental mechanism to restrict Mcm2–7 ring opening and premature helicase activation, causing stability of the loaded double hexamer during G1 phase and until the initiation of DNA replication at that origin (Diffley 2001).
The Mcm2–7 double-hexamer architecture provides a structural basis for it being a DDK target
DDK acts on the Mcm2–7 double hexamer but not the single Mcm2–7 hexamer in solution and in vivo (Sheu and Stillman 2006; Randell et al. 2010; Heller et al. 2011; Ramer et al. 2013; Tanaka and Araki 2013), and the DDK action precedes the S-CDK action to activate the steps toward the actual initiation of DNA synthesis at each origin (Heller et al. 2011). However, it was unclear how DDK distinguishes a double hexamer from the single hexamer, as they both contain the same protein components. Recent studies found that DDK interacts with the NTDs of Mcm2 and Mcm4 (Sheu and Stillman 2010; Ramer et al. 2013). The architecture of the Mcm2–7 double hexamer, as determined in the present study, now provides a physical explanation for the timed DDK action. Mcm2 and Mcm4 are far apart in the Mcm2–7 hexamer, separated by Mcm6. In the double hexamer, however, the NTDs of Mcm2 and Mcm4 are in direct proximity, forming an integrated docking platform for the DDK (Fig. 5F). Consistent with that, we found that the purified double hexamer is a far better substrate of DDK than the single hexamer in vitro (Fig. 5G). Adjacent to the DDK docking domains in the Mcm2 and Mcm4 NTDs are the respective NSDs of each subunit that are the targets for DDK phosphorylation (Lei et al. 1997; Sheu and Stillman 2006, 2010). Phosphorylation of the Mcm4 NSD is most critical, since deletion of this intrinsic inhibitor of the initiation of DNA replication in Mcm2–7 bypasses the requirement for DDK (Sheu and Stillman 2010). Another mutation, mcm5-bob1 (P83L), in the N terminus of the Mcm5 subunit also bypasses the requirement for DDK (Hardy et al. 1997). Interestingly, this amino acid lies adjacent to the Mcm4 and Mcm2 DDK docking domains (Fig. 5F). The purified bob-1-containing mutant double hexamer was unstable, but the DDK-modified wild-type double hexamer (Supplemental Fig. 5A) was stable and amenable to structural analysis. EM revealed that DDK modification neither separates the two hexamers nor drastically alters the overall structure of the double hexamer (Supplemental Fig. 5B; On et al. 2014). Therefore, we suggest that phosphorylation mainly affects the surface properties of the double hexamer to promote recruitment of additional helicase subunits such as GINS and Cdc45 and eventually Mcm2/5 gate opening and DNA unwinding (Labib 2010; On et al. 2014).
Discussion
For a long time, eukaryotic replication licensing and pre-RC formation were merely abstract notions. Recent establishment of an in vitro helicase loading reaction with purified proteins and origin DNA has transformed these concepts into tangible biochemical processes that are amenable to mechanistic dissections (Evrin et al. 2009; Remus et al. 2009; Mehanna and Diffley 2012). By taking advantage of the established in vitro pre-RC assembly process and the fact that the proteins form large complexes on DNA, we succeeded in capturing and visualizing a number of pre-RC assembly steps by EM, which, combined with image classification and detailed subunit mapping, provided mechanistic insights into key eukaryotic replication initiation events (Fig. 6, steps 1–7).
Figure 6.

EM of pre-RC intermediates suggests possible helicase loading and initial DNA-melting mechanisms. Steps 1–5 describe the proposed model for the eukaryotic pre-RC formation. Each of the five steps as drawn is based on an experimentally captured Mcm2–7 loading intermediate. ORC–Cdc6 on origin DNA in step 1 is the activated platform for loading of Cdt1–Mcm2–7. Formation of OCCM as shown in step 2 represents the recruitment of the first hexamer. ATP hydrolysis in step 3 leads to the release of Cdt1 and formation of OCM. Formation of the OCMM in step 4 completes the recruitment of the second hexamer. The question mark in step 4 indicates a possible short-lived intermediate not captured in the present study. In step 5, ATP hydrolysis by ORC–Cdc6 leads to maturation of the double-hexamer structure on DNA and its separation from ORC–Cdc6. Mcm subunits are colored purple in the single hexamer but are colored orange in the double hexamer to highlight the subunit tilt. The two hexamers in the OCMM are also colored orange (step 4) because their structure is highly similar to the final double hexamer. The OCM, OCMM, and double hexamer in steps 3, 4, and 5 are characterized in this study. During the transition from step 5 to step 6, a possible untwisting of the right-hand-tilted Mcm proteins in the double hexamer may locally unwind the dsDNA. Further modification by DDK and CDK and binding to Cdc45 and GINS lead to the separation of the double hexamer and formation of two CMG complexes, each encircling ssDNA (step 6).
The role of the OCM in recruiting the second Mcm2–7 hexamer
The wild-type OCM is a transient and salt-sensitive pre-RC intermediate (Fernandez-Cid et al. 2013). It is therefore not feasible to obtain homogeneous OCM preparations for 3D reconstruction. Because we know the 3D EM structures of ORC–Cdc6 and the OCCM complexes (Sun et al. 2012, 2013), we are able to interpret the 2D structure of the OCM with confidence.
We know that the OCM is competent to recruit a second Mcm2–7 hexamer (Fernandez-Cid et al. 2013). The key question has been which part of the OCM structure is responsible for recruiting the next Cdt1-bound Mcm2–7 (Yardimci and Walter 2014). Our discovery that the interfaces between ORC–Cdc6 and Mcm2–7 in OCM and OCCM are similar is significant, as it indicates that the N-terminal surface of ORC–Cdc6 used for recruiting the first Mcm2–7 in OCCM remains shielded by the first hexamer in OCM and thus is not available for binding to the second Mcm2–7. It is also unlikely that ORC–Cdc6 could use the exposed C-terminal face to interact with a second Mcm2–7 because we never observed in the EM images one ORC–Cdc6 sandwiched between two Mcm2–7 hexamers. We therefore suggest that the NTD section of the first loaded Mcm2–7 may have undergone conformational changes that allow direct interaction with a second Mcm2–7 hexamer (Fig. 6, step 4).
The implication of the OCMM structure in pre-RC assembly
In the 7-min reaction sample, ∼10% of pre-RC intermediates are OCMM particles (Table 1). This is the first time the complex has been seen. Although Cdc6 is known to leave the origin at some point, we do not know whether it leaves before the double-hexamer assembly. In other words, we are uncertain whether Cdc6 is actually present as the term OCMM implies. The OCMM could be formed by interaction of two OCM complexes followed by release of ORC/Cdc6. Alternatively, the OCM may be competent to recruit the second Mcm2–7 hexamer (Fig. 2D), which would directly produce the OCMM complex as a Mcm2–7 double-hexamer precursor (Fig. 6, step 4). If so, why is the Mcm3 C terminus of the second hexamer, which is expected to be at the end of the double hexamer opposite to where ORC–Cdc6 binds, important for double-hexamer assembly (Frigola et al. 2013)? The Mcm3 C terminus influences not only the ATPase activity of ORC–Cdc6 (Frigola et al. 2013) but also the ATPase activity of the Mcm2–7 hexamer (Supplemental Fig. 3); it appears possible that the Mcm3 C terminus allosterically regulates the ATPase activity of the first loaded Mcm2–7 hexamer as well as that of the second hexamer yet to be loaded. Since Mcm2–7 ATPase is required for double-hexamer assembly (Coster et al. 2014; Kang et al. 2014), the potential allosteric function may regulate double-hexamer assembly.
The Mcm2–7 double hexamer has tilted structure
One novel finding from our double-hexamer structure is that the Mcm proteins within each hexamer are tilted toward the right by nearly 30°, making the structure twisted and appear to be spring-loaded (Fig. 5A). Discovery of the right-handed tilt was made possible due to our ability to unambiguously identify individual Mcm proteins and their respective NTDs and CTDs via the MBP insertion strategy. The twisted feature appears to be unique to the eukaryotic double hexamer. In the archaeal MCM hexamer structure modeled on a 3D EM map of the archaeal double hexamer, which is an active helicase, the individual MCM proteins are arranged nearly parallel to the long cylindrical axis (Brewster et al. 2008). Furthermore, the Mcm proteins in both the yeast OCCM structure and the Drosophila CMG helicase are not obviously twisted (Costa et al. 2011, 2014; Sun et al. 2013). Therefore, this twist in the Mcm2–7 proteins is specifically introduced during double-hexamer assembly and is relaxed in the active helicase (Fig. 6, steps 5 and 6). We suggest that the twisting promotes long-term stability of the double hexamer on the DNA, as required for a molecule that licenses DNA replication in G1 phase for initiation of DNA replication at an origin much later in S phase.
Multiple mechanisms inhibit the helicase activity in the Mcm2–7 double hexamer
Helicase loading in bacteria and eukaryotes potentially involves similar mechanisms (Arias-Palomo et al. 2013; Sun et al. 2013) but results in very different complexes. In bacteria and eukaryotic viruses such as SV40 T antigen and papillomavirus E1, the helicase is loaded in an active form; thus, initiation is controlled at the level of initial DNA unwinding (Schuck and Stenlund 2005; Skarstad and Katayama 2013). In eukaryotes, helicase loading cumulates in a Mcm2–7 double-hexamer formation, which represents an intrinsically inactive form of the replicative helicase, which only becomes activated during S phase (Tanaka and Araki 2013). If the helicase could be autoactivated and DNA polymerases would associate with the helicase, as happens in the case of the bacterial helicase, then helicase loading and helicase activation could occur simultaneously, resulting in rereplication and genome instability. In bacteria, this cannot happen, as helicase loading becomes inhibited due to highly regulated DNA unwinding and helicase loading. In eukaryotes, it is imperative that the Mcm2–7 double hexamer by itself is inactive as a helicase and only becomes activated after DNA licensing activity has been turned off. Multiple mechanisms control helicase loading during the G1–S transition and have been studied extensively (Arias and Walter 2007; Siddiqui et al. 2013). In contrast, how premature helicase activation in G1 phase is regulated is only partially understood. Here we report two mechanisms that limit helicase activity of the Mcm2–7 double hexamer. First, we observed that the Mcm2–7 subunits are arranged in an unusually tilted conformation and that the complex has, in comparison with the Mcm2–7 single hexamer, reduced ATPase activity, estimated to be two orders of magnitude lower than the ATPase of the active CMG helicase (Ilves et al. 2010). We suggest that the conserved Walker B motif in one subunit and the arginine finger motif of a neighboring subunit, which together coordinate Mcm ATP hydrolysis, are misaligned in the Mcm2–7 double hexamer (Fig. 5C). Interestingly, within the OCCM and the Drosophila CMG helicase, the Mcm subunits are not tilted (Costa et al. 2011; Sun et al. 2013). Thus, it appears possible that Orc1/Cdc6 ATP hydrolysis could facilitate this structural change in Mcm2–7 during OCM formation (Fernandez-Cid et al. 2013), which also could be a prerequisite for Mcm2–7 hexamer dimerization (Evrin et al. 2014). To our knowledge, a similar tilted conformation has not been observed in any AAA+ hexameric complex.
The second mechanism that regulates helicase activation involves the opening of the Mcm2–7 ring at a putative Mcm2/5 exit gate. Extrusion of one strand of the dsDNA is required to transform the Mcm2–7 double hexamer into an active helicase that encircles one ssDNA, while the other strand is extruded outside the center of the helicase. How the Mcm2–7 ring becomes opened during helicase activation is essentially unknown, but, clearly, Mcm2–7 ring opening at the Mcm2/5 gate would be very difficult, as the two Mcm2–7 rings are not aligned along the 2/5 axis. In summary, the Mcm2–7 helicase evolved from the bacterial helicase in multiple ways to block premature helicase activation: The Mcm2–7 helicase is loaded on dsDNA (Evrin et al. 2009; Remus et al. 2009), and the intersubunit interactions between each hexamer block the separation of the hexamers, the opening of the Mcm2–7 ring (Fig. 5D,E), and strand extrusion, while the tilted subunit arrangement restricts the ATPase activity (Fig. 5A–C).
It is known that DDK is essential for activation of the DNA helicase activity in vivo by recruitment of the GINS and Cdc45 (Siddiqui et al. 2013). The double-hexamer structure shows the juxtaposition of Mcm2 and Mcm4 NTDs from opposing Mcm2–7 hexamers, creating a docking site for DDK (Fig. 5F). This explains why DDK can only activate the Mcm2–7 hexamer that has been loaded onto DNA (Fig. 5) and why Mcm4 NSD is phosphorylated only on Mcm4 subunits associated with chromatin (Sheu and Stillman 2010). Because DDK phosphorylation per se is insufficient to separate the double hexamer (Supplemental Fig. 5; On et al. 2014), it is possible that phosphorylation promotes binding of additional factors, including Cdc45 and GINS, to untwist the double hexamer to produce two active helicases (Fig. 6, step 6).
The twisted Mcm2–7 double-hexamer structure suggests a possible dsDNA-melting mechanism
Because the dsDNA is known to be untwisted by the Mcm2–7 double hexamer (Fu et al. 2011), we suggest that the right-hand twisted double-hexamer architecture may play a role in initial DNA melting. It is possible that when the double hexamer untwists itself as a result of DDK phosphorylation and binding of helicase-activating factors (e.g., Cdc45 and GINS), it may unwind the dsDNA, causing the dsDNA to melt, and the separated ssDNA to extrude from the central channel of the double hexamer (Fig. 6, step 6).
To summarize, we captured and visualized by EM two new Mcm2–7 loading intermediates (OCM and OCMM) in pre-RC assembly and established the detailed architecture of the Mcm2–7 double hexamer, which identified key mechanisms that inhibit the Mcm2–7 helicase activity in G1 phase. The structure of the double hexamer also explains how recruitment of DDK is restricted to Mcm2–7 on DNA in vivo as well as in vitro. Combined with the previously reported OCCM architecture, the structures of the four helicase loading steps have improved our understanding of the multistep pre-RC assembly and will guide future biochemical and structural studies on DNA licensing and helicase activation.
Materials and methods
Expression and purification of proteins
ORC was expressed by using baculovirus-infected cells and was purified as described (Klemm et al. 1997). Cdc6 and Cdt1 were expressed in bacteria and purified as described (Speck et al. 2005; Evrin et al. 2009). Mcm2–7 and MBP–Mcm2–7 were expressed in S. cerevisiae and purified as described (Evrin et al. 2009; Fernandez-Cid et al. 2013). DDK was expressed and purified as described (Evrin et al. 2014). Most MBP–Mcm2–7 constructs were described in Evrin et al. (2013) and Fernandez-Cid et al. (2013) with the exception of Mcm2–MBP. During its construction using site-directed mutagenesis, a restriction site was inserted after amino acid 712 in Mcm2. MBP was amplified from pMAL-c2X (New England Biolabs) with primers incorporating flexible linkers (sequences available on request) and inserted in the restriction sites, generating pCS624 (pESC-LEU-Mcm2–MBP/Mcm7).
In vitro assembly of pre-RC intermediates
The pre-RC intermediates were assembled in a one-step reaction: 40 nM ORC, 80 nM Cdc6, 40 nM Cdt1, and 40 nM MCM2-7 in buffer A (50 mM HEPES-KOH at pH 7.5, 100 mM KGlu, 10 mM MgAc, 50 mM ZnAc, 3 mM ATP or ATP-γS, 5 mM DTT, 0.1% Triton X-100, 5% glycerol) were added to 6 nM pUC19-ARS1 plasmid beads at 24°C (Evrin et al. 2009). To capture the pre-RC assembly intermediates in the presence of ATP, the reaction was stopped at various time points ranging from 2 to 30 min. For polyacrylamide gel electrophoresis (PAGE) and silver-staining analysis, beads were washed three times with buffer A plus 1 mM EDTA or buffer B (50 mM HEPES-KOH at pH 7.5, 1 mM EDTA, 500 mM NaCl, 5% glycerol, 0.1% Triton X-100, 5 mM DTT) before digestion with 12 μL of DNase I (1 U) in buffer A plus 5 mM CaCl2 for 6 min at 24°C. For EM analysis, the beads were washed once with buffer C (50 mM HEPES-KOH at pH 7.5, 100 mM potassium acetate, 5 mM magnesium acetate, 5 mM CaCl2) and eluted with 0.003 U of DNase I in 10 μL of buffer C, which was followed by the addition of glutaraldehyde to a final concentration of 0.2% for 15 min at 4°C.
ATPase assay
The ATPase assay was performed as described (Speck and Stillman 2007; Fernandez-Cid et al. 2013). We used 2.5 pmol of Cdt1 and wild-type or mutant Mcm2–7, which was incubated for 30 min on ice in 12 μL of ATPase buffer (25 mM HEPES at pH 7.6, 100 mM KGlu, 5 mM MgAc, 1 mM DTT, 1 mM EDTA, 1 mM EGTA, 0.1% [v/v] Triton X-100, 10% glycerol) containing 1 mM ATP. After the incubation, 5 μCi of [α-32P] ATP (3000 Ci/mmol) was added, and the reaction was started by moving the tubes from ice into a 24°C water bath. Two-microliter aliquots were collected at 15, 30, 45, and 60 min and stopped with 0.5 μL of stop solution (2% SDS, 50 mM EDTA). One microliter of the samples was consequently spotted on TLC plates and developed.
For the double-hexamer analysis, 12 pre-RC assay reactions were prepared as described with minor modifications (Evrin et al. 2013). A one-step reaction was used. For a single reaction, 40 nM ORC, 80 nM Cdc6, 40 nM Cdt1, and 40 nM Mcm2–7 in buffer A (50 mM HEPES-KOH at pH 7.5, 100 mM KGlu, 10 mM MgAc, 50 μM ZnAc, 3 mM ATP, 5 mM DTT, 0.1% NP40, 5% glycerol) were added to 6 nM linear pUC19-ARS1 DNA coupled to magnetic beads for 15 min at 24°C. Beads were washed twice with buffer A containing 300 mM NaCl and rinsed once with ATPase buffer (25 mM HEPES at pH 7.6, 100 mM KGlu, 5 mM MgAc, 1mM DTT, 0.1% [v/v] NP40, 5% glycerol) before digestion with 1 U of DNase I in ATPase buffer plus 5 mM CaCl2 and 1 mM ATP for 5 min at 24°C. The amount of released Mcm2–7 double hexamer was quantified by SDS-PAGE and densitometry. About 0.8 pmol of Mcm2–7 single hexamer or double hexamer was used in the assay. Error bars represent the standard deviation from at least three independent experiments.
In vitro DDK kinase assays
The pre-RCs were formed and washed with low salt or high salt as described above. Next, pre-RCs or purified Mcm2–7 were incubated in buffer P (50 mM Hepes-KOH at pH 7.5, 100 mM KGlu, 10 mM MgAc, 50 μM ZnAc, 3 mM ATP, 5 mM DTT, 0.1% Triton X-100, 5% glycerol) for nonradioactive assays or in buffer P+(50 mM Hepes-KOH at pH 7.5, 100 mM KGlu, 10 mM MgAc, 50 μM ZnAc, 0.1 mM ATP, 5 mM DTT, 0.1% Triton X-100, 5% glycerol, 100 μCi/mL γ-32P ATP) containing 5–80 nM DDK (40 nM in the case of Fig. 5G) for 32P-containing assays and incubated for 15 min at 27°C. For EM analysis, we used 80 nM DDK-treated samples. After an additional wash with buffer A plus 5 mM CaCl2 (for PAGE analysis) or buffer C (for EM analysis), the DNA was digested by 1 U of DNase I for 6 min at 24°C. Proteins were analyzed by EM or separated on a 7.5% SDS–polyacrylamide gel and silver-stained. The incorporation of 32P-phosphate was detected by autoradiography of the silver-stained gel.
In vitro assembly and purification of the wild-type and MBP-inserted Mcm2–7 double hexamers for single-particle EM
The pre-RCs were assembled in a one-step reaction: 40 nM ORC, 80 nM Cdc6, 40 nM Cdt1, and 40 nM MCM2–7 or MBP–Mcm2–7 in buffer A (50 mM HEPES-KOH at pH 7.5, 100 mM KGlu, 10mM MgAc, 50 mM ZnAc, 3 mM ATP, 5 mM DTT, 0.1% Triton X-100, 5% glycerol) were added to 6 nM pUC19-ARS1 plasmid beads at 24°C (Evrin et al. 2009). After 15 min, the beads were washed three times with buffer B and three times with buffer C and eluted with 1 U of DNase I in 5 μL of buffer C. For the cryo-EM analysis, 20 elutions were pooled and concentrated 10× with an Amicon concentrator (Ultracel 100K).
EM
For negative stain EM, ∼3-μL samples were deposited on the EM grids; after 60 sec, the grids were blotted with a piece of filter paper, and a 4-μL drop of 1% uranyl acetate aqueous solution was deposited. After waiting for 30 sec, the stain solution was blotted, and a second 4-μL drop of stain solution was deposited; after 60 sec, it was blotted and left to air-dry. To achieve ideal staining, we found it was important to leave a thin layer of stain solution on the grid during the last blotting such that there was enough stain salt to fully embed the particles but not too much to make the grid too dark in EM. For cryo-EM of a plasmid-bound double hexamer, we glow-discharged the holey carbon grids coated with a thin continuous carbon film just before use, pipetted 3 μL of sample on a EM grid, waited for 1 min before blotting for 5 sec, and plunge-froze the grid into liquid ethane. The sample was vitrified in an FEI Vitrobot with sample chamber temperature set to 11°C, relative humidity set to 70%, and blotting pad position offset −1 mm. We used the JEM-2010F to record images at a low electron dose of 15 e−/Å2 at a magnification of 50,000× in a 4kx4k Gatan ultrascan CCD camera. The EM image sampling size corresponds to 2.12 Å per pixel at the specimen level. Negative stain EM was performed at room temperature, and cryo-EM was done using a Gatan 626 cryo-specimen holder with the specimen temperature maintained below −170°C.
Single-particle image analysis and 3D reconstruction
Individual particle images were manually selected on a computer display with the e2boxer in the EMAN2 package (Tang et al. 2007). The contrast transfer function (CTF) parameters were determined separately by ctfit in EMAN for all particles from each CCD frame. The particle images were phase-flipped, high-pass-filtered (hp = 1), normalized with the edgenorm option, and pooled into one image stack. We then carried out reference-free 2D image classification and averaging with refine2d.py. We found that binning the particle images by a factor of two and then low-pass filtering to 20 Å produced the best results. Particle selection of the Mcm2–7 loading intermediates in ATP relied on the fact that they are large protein complexes with a distinct parallel line feature in the side views. The manually selected particle images were subjected to multiple rounds of reference-free classification. The particle images belonging to classes that were not in the near-perfect side views were removed.
For 3D reconstruction of the purified wild-type Mcm2–7 double hexamer, a data set of ∼22,630 raw particle images was used. A 3D volume was calculated using the startcsym program with the C6 symmetry in EMAN, taking advantage of the pseudo sixfold symmetry of the structure. The volume was first refined in the D6 symmetry, and the resulting 3D volume was then low-pass-filtered to 100 Å to minimize initial model bias; the volume was rotated by 90° so the true twofold axis of the double hexamer was aligned to the Z-axis. This starting model is essentially a 3D blob with the correct size but without any structural features. The starting model was then refined by projection matching with the raw particle images in EMAN with the C2 symmetry and without amplitude correction. The final 3D map has a resolution of ∼15 Å as estimated by the traditional Fourier shell correlation at a 0.5 threshold and has a resolution of ∼20 Å as estimated by the gold standard Fourier shell correlation at a 0.143 threshold. The negative stain 3D EM density map of the S. cerevisiae Mcm2–7 double hexamer was deposited in the Electron Microscopy Data Bank under accession code EMD-5857.
For the purified MBP-inserted Mcm2–7 double-hexamer samples, the final negative stain EM data set contained 5358, 9917, 8867, 8224, 1368, and 28,474 particle images for double hexamers with MBP insertion in the N-terminal region of Mcm2, Mcm3, Mcm6, and Mcm7 and the C-terminal region of Mcm2 and Mcm5, respectively. The raw images were processed similarly to the wild-type double-hexamer images. 3D refinement of the MBP-insertion double hexamers used the wild-type double-hexamer 3D map as the starting model. We relaxed and optimized the mask size during refinement to avoid cutting off the protruding MBP densities while minimizing noise. The 3D maps of the MBP insertion double hexamers had a resolution ranging from 25 Å to 35 Å. The DDK-modified double-hexamer data set had 3063 raw particles, and the 3D map had a resolution of 34 Å. EM map surface rendering, density segmentation, and docking with crystal structures were carried out in the University of California at San Francisco Chimera (Pettersen et al. 2004).
Supplementary Material
Acknowledgments
We acknowledge the help of Cecile Evrin and Stefan Samel in the early stages of this project. This work was supported by National Institutes of Health grant numbers GM45436 (to B.S.) and GM74985 (to H.L.) and the United Kingdom Medical Research Council (to C.S.). J.S., A.R. A.F.-C., and S.T. performed the specimen preparation and biochemistry. J.S. and Z.Y. collected the EM data and performed the EM reconstructions. J.S., A.R., A.F.-C., B.S., C.S., and H.L. designed experiments and wrote the manuscript.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.242313.114.
References
- Arias EE, Walter JC. 2007. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes Dev 21: 497–518 [DOI] [PubMed] [Google Scholar]
- Arias-Palomo E, O’Shea VL, Hood IV, Berger JM. 2013. The bacterial DnaC helicase loader is a DnaB ring breaker. Cell 153: 438–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Dutta A. 2002. DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Bell SP, Stillman B. 1992. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature 357: 128–134 [DOI] [PubMed] [Google Scholar]
- Bochman ML, Schwacha A. 2008. The Mcm2–7 complex has in vitro helicase activity. Mol Cell 31: 287–293 [DOI] [PubMed] [Google Scholar]
- Bochman ML, Schwacha A. 2009. The Mcm complex: unwinding the mechanism of a replicative helicase. Microbiol Mol Biol Rev 73: 652–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman ML, Schwacha A. 2010. The Saccharomyces cerevisiae Mcm6/2 and Mcm5/3 ATPase active sites contribute to the function of the putative Mcm2–7 ‘gate.’ Nucleic Acids Res 38: 6078–6088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochman ML, Bell SP, Schwacha A. 2008. Subunit organization of Mcm2–7 and the unequal role of active sites in ATP hydrolysis and viability. Mol Cell Biol 28: 5865–5873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster AS, Wang G, Yu X, Greenleaf WB, Carazo JM, Tjajadi M, Klein MG, Chen XS. 2008. Crystal structure of a near-full-length archaeal MCM: functional insights for an AAA+ hexameric helicase. Proc Natl Acad Sci 105: 20191–20196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, de Vries MA, Bell SP. 2007. Orc6 is required for dynamic recruitment of Cdt1 during repeated Mcm2–7 loading. Genes Dev 21: 2897–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JP, Hayashi MK, Simon MN, Xu RM, Stillman B. 2000. A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicase. Proc Natl Acad Sci 97: 1530–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, Berger JM. 2011. The structural basis for MCM2–7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol 18: 471–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa A, Renault L, Swuec P, Petojevic T, Pesavento J, Ilves I, MacLellan-Gibson K, Fleck RA, Botchan MR, Berger JM. 2014. DNA binding polarity, dimerization, and ATPase ring remodeling in the CMG helicase of the eukaryotic replisome. eLife 3: 03273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coster G, Frigola J, Beuron F, Morris EP, Diffley JF. 2014. Origin licensing requires ATP binding and hydrolysis by the MCM replicative helicase. Mol Cell 55: 666–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey MJ, Indiani C, O’Donnell M. 2003. Reconstitution of the Mcm2–7p heterohexamer, subunit arrangement, and ATP site architecture. J Biol Chem 278: 4491–4499 [DOI] [PubMed] [Google Scholar]
- Diffley JF. 2001. DNA replication: building the perfect switch. Curr Biol 11: R367–R370 [DOI] [PubMed] [Google Scholar]
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. 2009. A double-hexameric MCM2–7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proc Natl Acad Sci 106: 20240–20245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Fernandez-Cid A, Zech J, Herrera MC, Riera A, Clarke P, Brill S, Lurz R, Speck C. 2013. In the absence of ATPase activity, pre-RC formation is blocked prior to MCM2–7 hexamer dimerization. Nucleic Acids Res 41: 3162–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evrin C, Fernandez-Cid A, Riera A, Zech J, Clarke P, Herrera MC, Tognetti S, Lurz R, Speck C. 2014. The ORC/Cdc6/MCM2–7 complex facilitates MCM2–7 dimerization during prereplicative complex formation. Nucleic Acids Res 42: 2257–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Cid A, Riera A, Tognetti S, Herrera MC, Samel S, Evrin C, Winkler C, Gardenal E, Uhle S, Speck C. 2013. An ORC/Cdc6/MCM2–7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol Cell 50: 577–588 [DOI] [PubMed] [Google Scholar]
- Fletcher RJ, Bishop BE, Leon RP, Sclafani RA, Ogata CM, Chen XS. 2003. The structure and function of MCM from archaeal M. thermoautotrophicum. Nat Struct Biol 10: 160–167 [DOI] [PubMed] [Google Scholar]
- Francis LI, Randell JC, Takara TJ, Uchima L, Bell SP. 2009. Incorporation into the prereplicative complex activates the Mcm2–7 helicase for Cdc7–Dbf4 phosphorylation. Genes Dev 23: 643–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frigola J, Remus D, Mehanna A, Diffley JF. 2013. ATPase-dependent quality control of DNA replication origin licensing. Nature 495: 339–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, Walter JC. 2011. Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146: 931–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy CF, Dryga O, Seematter S, Pahl PM, Sclafani RA. 1997. mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7p. Proc Natl Acad Sci 94: 3151–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. 2011. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell 146: 80–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilves I, Petojevic T, Pesavento JJ, Botchan MR. 2010. Activation of the MCM2–7 helicase by association with Cdc45 and GINS proteins. Mol Cell 37: 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Warner MD, Bell SP. 2014. Multiple functions for Mcm2–7 ATPase motifs during replication initiation. Mol Cell 55: 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RD, Austin RJ, Bell SP. 1997. Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell 88: 493–502 [DOI] [PubMed] [Google Scholar]
- Labib K. 2010. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev 24: 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labib K. 2011. Building a double hexamer of DNA helicase at eukaryotic replication origins. EMBO J 30: 4853–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei M, Kawasaki Y, Young MR, Kihara M, Sugino A, Tye BK. 1997. Mcm2 is a target of regulation by Cdc7–Dbf4 during the initiation of DNA synthesis. Genes Dev 11: 3365–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubimov AY, Costa A, Bleichert F, Botchan MR, Berger JM. 2012. ATP-dependent conformational dynamics underlie the functional asymmetry of the replicative helicase from a minimalist eukaryote. Proc Natl Acad Sci 109: 11999–12004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehanna A, Diffley JF. 2012. Pre-replicative complex assembly with purified proteins. Methods 57: 222–226 [DOI] [PubMed] [Google Scholar]
- On KF, Beuron F, Frith D, Snijders AP, Morris EP, Diffley JF. 2014. Prereplicative complexes assembled in vitro support origin-dependent and independent DNA replication. EMBO J 33: 605–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Pospiech H, Grosse F, Pisani FM. 2010. The initiation step of eukaryotic DNA replication. Subcell Biochem 50: 79–104 [DOI] [PubMed] [Google Scholar]
- Ramer MD, Suman ES, Richter H, Stanger K, Spranger M, Bieberstein N, Duncker BP. 2013. Dbf4 and Cdc7 proteins promote DNA replication through interactions with distinct Mcm2–7 protein subunits. J Biol Chem 288: 14926–14935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randell JC, Bowers JL, Rodriguez HK, Bell SP. 2006. Sequential ATP hydrolysis by Cdc6 and ORC directs loading of the Mcm2–7 helicase. Mol Cell 21: 29–39 [DOI] [PubMed] [Google Scholar]
- Randell JC, Fan A, Chan C, Francis LI, Heller RC, Galani K, Bell SP. 2010. Mec1 is one of multiple kinases that prime the Mcm2–7 helicase for phosphorylation by Cdc7. Mol Cell 40: 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus D, Diffley JF. 2009. Eukaryotic DNA replication control: lock and load, then fire. Curr Opin Cell Biol 21: 771–777 [DOI] [PubMed] [Google Scholar]
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. 2009. Concerted loading of Mcm2–7 double hexamers around DNA during DNA replication origin licensing. Cell 139: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samel SA, Fernandez-Cid A, Sun J, Riera A, Tognetti S, Herrera MC, Li H, Speck C. 2014. A unique DNA entry gate serves for regulated loading of the eukaryotic replicative helicase MCM2–7 onto DNA. Genes Dev 28: 1653–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck S, Stenlund A. 2005. Assembly of a double hexameric helicase. Mol Cell 20: 377–389 [DOI] [PubMed] [Google Scholar]
- Schwacha A, Bell SP. 2001. Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol Cell 8: 1093–1104 [DOI] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. 2006. Cdc7–Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol Cell 24: 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu YJ, Stillman B. 2010. The Dbf4–Cdc7 kinase promotes S phase by alleviating an inhibitory activity in Mcm4. Nature 463: 113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui K, On KF, Diffley JF. 2013. Regulating DNA replication in eukarya. Cold Spring Harb Perspect Biol 5: a012930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstad K, Katayama T. 2013. Regulating DNA replication in bacteria. Cold Spring Harb Perspect Biol 5: a012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, Stillman B. 2007. Cdc6 ATPase activity regulates ORC × Cdc6 stability and the selection of specific DNA sequences as origins of DNA replication. J Biol Chem 282: 11705–11714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck C, Chen Z, Li H, Stillman B. 2005. ATPase-dependent cooperative binding of ORC and Cdc6 to origin DNA. Nat Struct Mol Biol 12: 965–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Kawakami H, Zech J, Speck C, Stillman B, Li H. 2012. Cdc6-induced conformational changes in ORC bound to origin DNA revealed by cryo-electron microscopy. Structure 20: 534–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Evrin C, Samel SA, Fernandez-Cid A, Riera A, Kawakami H, Stillman B, Speck C, Li H. 2013. Cryo-EM structure of a helicase loading intermediate containing ORC–Cdc6–Cdt1–MCM2–7 bound to DNA. Nat Struct Mol Biol 20: 944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takara TJ, Bell SP. 2011. Multiple Cdt1 molecules act at each origin to load replication-competent Mcm2–7 helicases. EMBO J 30: 4885–4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Araki H. 2013. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb Perspect Biol 5: a010371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157: 38–46 [DOI] [PubMed] [Google Scholar]
- Tsakraklides V, Bell SP. 2010. Dynamics of pre-replicative complex assembly. J Biol Chem 285: 9437–9443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayraghavan S, Schwacha A. 2012. The eukaryotic Mcm2–7 replicative helicase. Subcell Biochem 62: 113–134 [DOI] [PubMed] [Google Scholar]
- Wendler P, Ciniawsky S, Kock M, Kube S. 2012. Structure and function of the AAA+ nucleotide binding pocket. Biochim Biophys Acta 1823: 2–14 [DOI] [PubMed] [Google Scholar]
- Yardimci H, Walter JC. 2014. Prereplication-complex formation: a molecular double take? Nat Struct Mol Biol 21: 20–25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.