

Abstract
Ergothioneine is a histidine thiol derivative. Its mycobacterial biosynthetic pathway has five steps (EgtA-E catalysis) with two novel reactions: a mononuclear nonheme iron enzyme (EgtB) catalyzed oxidative C–S bond formation and a PLP-mediated C–S lyase (EgtE) reaction. Our bioinformatic and biochemical analyses indicate that the fungus Neurospora crassa has a more concise ergothioneine biosynthetic pathway because its nonheme iron enzyme, Egt1, makes use of cysteine instead of γ-Glu-Cys as the substrate. Such a change of substrate preference eliminates the competition between ergothioneine and glutathione biosyntheses. In addition, we have identified the N. crassa C–S lyase (NCU11365) and reconstituted its activity in vitro, which makes the future ergothioneine production through metabolic engineering feasible.
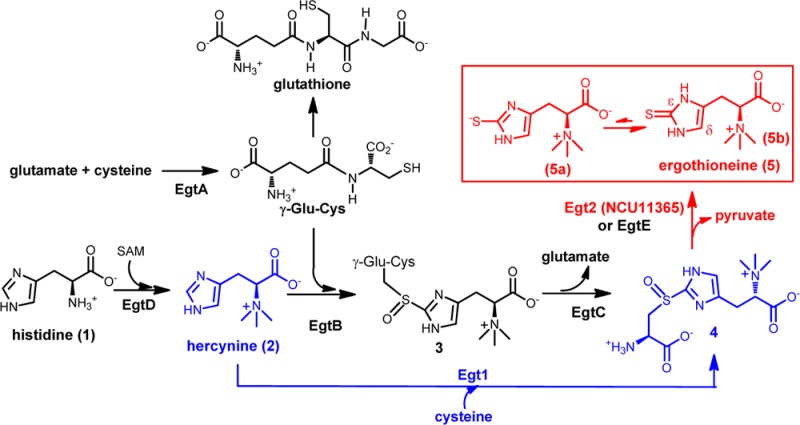
Sulfur-containing molecules are widely distributed in nature, including amino acids, enzyme cofactors, antioxidants, nucleotides, and numerous secondary metabolites.1−16 Their biosyntheses make use of many novel transformations. Ergothioneine is a thiol–imidazole-containing amino acid, and its first biosynthetic pathway in Mycobacterium smegmatis was reported in 2010 (EgtA–EgtE catalysis in Scheme 1A).17 The first step is the methylation of histidine to hercynine (2) catalyzed by EgtD. EgtA is γ-glutamylcysteine synthase catalyzing the condensation between glutamate and cysteine to γ-glutamylcysteine (γ-Glu-Cys). EgtB then catalyzes the oxidative coupling between hercynine and γ-Glu-Cys to form 3. An amidotransamidase (EgtC) then hydrolyzes 3 to form S-(β-amino-β-carboxyethyl)ergothioneine (4).17,18 The last enzyme in ergothioneine biosynthesis is a putative PLP-dependent C–S lyase (EgtE) whose activity remains to be verified in vitro.17
Scheme 1. (A) Mycobacterial and Fungal Ergothiotheine Biosynthetic Pathways. (B) Proposed Ovothiol Biosynthetic Pathway and Novel OvoA Chemistries.
Humans obtain ergothioneine from our diet and enrich it to as high as millimolar concentrations in many parts of our body using an ergothioneine-specific transporter.19 Ergothioneine’s reduction potential (E0′ = −0.06 V3) is significantly higher than glutathione (E0′ = −0.25 V) and plays many beneficial roles in human health.20 One of its roles is to scavenge reactive oxidative species. Its was also suggested to be involved in heavy metal detoxification.21,22
Due to ergothioneine’s beneficial roles to human health, there is a long-standing interest in developing efficient ergothioneine production methods.23,24 The discovery of the M. smegmatis ergothioneine biosynthetic pathway (EgtA–EgtE catalysis, Scheme 1A) in 201017 provides an opportunity for ergothioneine production through metabolic engineering. To realize this potential, two barriers need to be addressed. First, one of the EgtB substrates is γ-Glu-Cys, which is part of the glutathione biosynthesis (Scheme 1A). As one of the most important redox buffers inside the cell, glutathione has an intracellular concentration up to 10 mM, and it is essential to cellular survival. As a result, such competition is not desirable for ergothioneine production. Second, previous EgtE protein overexpression attempts were unsuccessful.25 In this work, the above two issues were resolved.
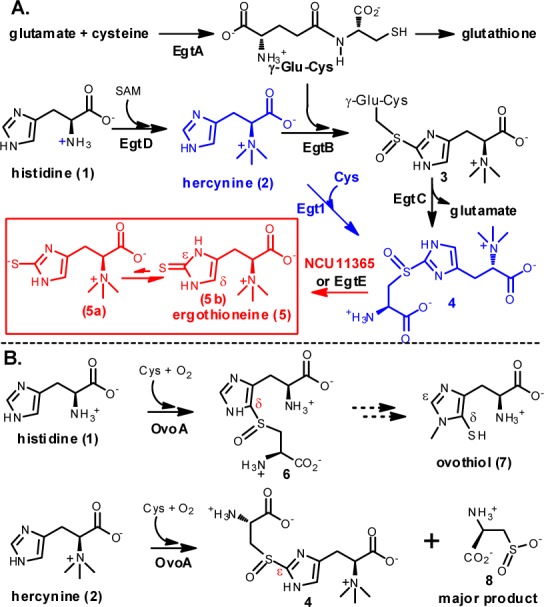
Ovothiol (7, Scheme 1B) is another thiol–imidazole-containing amino acid. The enzyme responsible for the oxidative C–S bond formation in ovothiol biosynthesis (OvoA, Scheme 1B) is also a mononuclear nonheme iron enzyme.25 Recently, we demonstrated that when OvoA’s native substrate histidine is replaced by hercynine,26 OvoA can catalyze a one-step 2 → 4 transformation (Scheme 1B). Subsequent detailed biochemical characterization of OvoA enzyme revealed that when histidine is replaced by hercynine, kobs changes by 2-fold (from 572 ± 20 min–1 to 270 ± 5 min–1), while Km for cysteine increases from 300 ± 34 μM to 3.19 ± 0.41 mM.26 In addition, OvoA also catalyzes the oxidation of cysteine to cysteine sulfinic acid (8), which is actually the major product of this reaction (Scheme 1B). The kinetic properties and the presence of side-reaction render OvoA not suitable for use in ergothioneine production through metabolic engineering. However, the presence of such a one-step 2 → 4 transformation in OvoA implies that nature might have evolved this activity as the native activity of some enzymes already. This option has also been suggested from some genetic studies,27,28 while its existence has not yet been demonstrated in vitro.
To search for enzymes capable of catalyzing a one-step 2 → 4 transformation, we compiled sequences (1886 sequences in total) related to the M. smegmatis EgtB gene from the RefSeq database. They contain either an egtB_TIGR03440 domain or both DinB_2 nonheme iron binding domain and FGE-sulfatase domains (Figure 1-inset). Among them, we randomly selected 503 for all-by-all BLAST analysis. A Cytoscape network (Figure 1) was then generated.29 The networks are arranged using the yFiles organic layout provided with Cytoscape 3.1. In these networks, each node represents an individual protein sequence, while edges indicate relatedness between two sequences.
Figure 1.
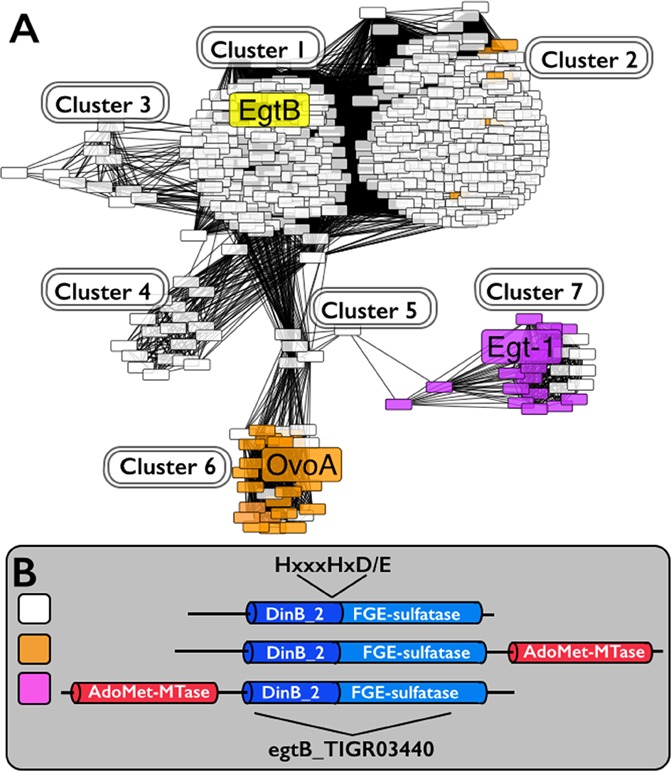
Analysis of the homologues of M. smegmatis EgtB protein. (A) Protein similarity network (PSN) for EgtB-like proteins containing the egtB_TIGR03440 domain. (B) The network is color-coded based on the presence or absence of an additional methyltransferase domain (AdoMet_MTases) either N- or C-terminal of the egtB_TIGR03440 domain. The nonheme iron binding site consensus sequence, HxxxHxD/E nonheme binding motif,34−38 is at the C-terminus of the DinB_2 domain.
An EgtB homologue sequence similarity network was generated using a BLAST e value cutoff of 1 × 10–50 (Figure 1). This e value was used because at this e value, OvoA and EgtB are clearly segregated into different clusters (cluster 1 and cluster 6), which is consistent with current biochemical data (Scheme 1).17,25,26,30 OvoA and EgtB are related in sequence, while they are biochemically distinct. Ergothioneine’s thiol group is at its imidazole ε-carbon and ovothiol’s thiol is at its imidazole δ-carbon. In addition, EgtB and OvoA have different substrate specificities. EgtB catalyzes the oxidative coupling between hercynine (2) and γ-Glu-Cys, while OvoA preferentially uses histidine and cysteine. OvoA can also catalyze a one-step 2 → 4 transformation when its native substrate histidine is replaced by hercynine (Scheme 1B). One of the interesting discoveries from the EgtB homologue cytoscape analysis is that the fungal genes (cluster 7) are clearly separated from both EgtB and OvoA clusters. Among the fungal genes, one of the genes (Egt1 from N. crassa(31)) was proposed to be involved in fungal ergothioneine biosynthesis because its knockout in N. crassa makes the mutant much more sensitive to oxidative stress.28 Thus far, none of the proposed fungal ergothioneine biosynthetic enzymes has been biochemically characterized in vitro.
To examine whether fungal EgtB homologues are indeed biochemically distinct from both EgtB and OvoA as suggested by the sequence similarity network analysis (Figure 1), we subcloned the fungus N. crassa Egt1 gene to pASK-IBA3+ vector and overexpressed it in E. coli. The purified protein does not have clear UV–vis spectroscopic features, which is consistent with the properties of nonheme iron enzymes (Figure 1S, Supporting Information). The amount of iron was quantified using iron titration method and Fe2+ reconstituted Egt1 has ∼0.90 ± 0.05 Fe per Egt1 monomer.32 Using hercynine and cysteine as substrates, Egt1 was analyzed by three different assays (Figure 2). NeoFox oxygen electrode was used to directly measure the oxygen consumption rate. In air-saturated HEPES buffer (∼250 μM of oxygen), the Egt1 kinetic parameters are kobs of 136 ± 4 min–1 and Km of 436 ± 30 μM for hercynine, Km of 603 ± 40 μM for Cys (Figure 2S, Supporting Information). More importantly, the 1H NMR assay suggested that the oxidative coupling between cysteine and hercynine occurs at the imidazole ε-position (Egt1 reaction in Scheme 1A and Figure 2A). The signals with chemical shifts of 6.84 and 7.62 ppm are from the hercynine imidazole H atoms. When hercynine and cysteine were used as the Egt1 substrates, the coupling product has a chemical shift at 7.12 ppm (Figure 2A), which is consistent with a one-step 2 → 4 transformation (Egt1 catalysis in Scheme 1A, Figure 2A, Figures 3S–5S, Supporting Information).
Figure 2.

Egt1 analysis using three different assays. (A) 1H NMR assay for EgtB-, OvoA- and Egt1-reactions. The 1H NMR chemical shifts of the imidazole hydrogen atoms are indicative of the C–S bond regio-selectivity. (B) Correlation between the oxygen consumption assay and 1H NMR assay, which indicated that the formation of the oxidative coupling product 4 is the dominant reaction in Egt1-catalysis (>90% of the oxygen consumption). (C) 13C NMR assay to monitor the presence of other cysteine related oxygen consumption reactions, which suggested the formation of cysteine sulfinic acid 8 as a minor reaction.
In oxygenase- and oxidase-catalyzed reactions, it is common to have uncoupling between the oxygen consumption and the production of the desired product. When hercynine and cysteine were used as the substrates, the formation of cysteine sulfinic acid (8) is the major OvoA reaction and accounts for ∼60% of oxygen consumption (Scheme 1B).30 When compound 4 formation rate and oxygen consumption rate in Egt1-catalysis were analyzed quantitatively, under the assay conditions used, the oxygen consumption rate (84 ± 2 min–1) is slightly faster than the oxidative product 4 formation rate (77 ± 2 min–1, Figure 2B), which suggests that in Egt1-catalysis, the 2 → 4 transformation is the dominant reaction (∼92% of the oxygen consumed). To examine whether the remaining ∼8% of the oxygen consumption is due to cysteine oxidation reactions, Egt1-catalysis was further characterized using 13C NMR and [β-13C]-Cys were used to replace Cys. Indeed, besides the oxidative coupling product 4 (54.5 ppm, Figure 2C), cysteine sulfinic acid (8, 57.9 ppm, Figure 2C) was the other product (Figures 6S–8S, Supporting Information). The ratio between 4 and 8 is ∼12:1 (Figure 9S, Supporting Information).
To provide further evidence supporting the 2 → 4 transformation as the native Egt1-chemistry, Egt1 substrate specificities were examined using hercynine, histidine, cysteine, or γ-Glu-Cys as the substrates and in various combinations. In air-saturated HEPES buffer (∼250 μM of oxygen), the reaction kinetics was measured by NeoFox oxygen electrode. The kinetic parameters for the various combinations are as follows: (A) when cysteine and hercynine are the substrates, kobs of 136 ± 4 min–1 and a Km of 436 ± 30 μM for hercynine, a Km of 603 ± 40 μM for Cys; (B) when histidine and cysteine are the substrates, kobs is at least 100-fold less than the case using the Cys and hercynine combination (for these reasons, the detailed kinetic parameters were not measured for this case); (C) when γ-Glu-Cys and hercynine are the substrates, kobs of 27.7 ± 1.6 min–1 and Km of 7.68 ± 1.11 mM for γ-Glu-Cys, Km of 390 ± 58 μM for hercynine; (D) when γ-Glu-Cys and histidine are the substrates, similar to the case of histidine and cysteine combination, the rate is close to background level and the kinetic parameters were not measured (Figure 10S, Supporting Information). These comparative kinetic studies highly suggest that the native substrates for Egt1 enzyme are hercynine and cysteine (Egt1 catalysis in Scheme 1A). When hercynine is replaced by histidine, the reaction rate reduces by at least 2 orders of magnitude. In addition, Egt1 has ∼62-fold greater specificity (kobs/Km) for l-cysteine relative to γ-Glu-Cys.
In ergothioneine biosynthesis, there are two novel reactions: the oxidative C–S bond formation and the subsequent C–S bond cleavage reaction, which result in a net transfer of the sulfur atom form cysteine to histidine imidazole side-chain (Scheme 1 and Supporting Information). In the M. smegmatis ergothioneine biosynthetic pathway, the in vitro activities of EgtA, EgtB, EgtC, and EgtD enzymes have been established. The proposed C–S lyase activity (EgtE) remains to be verified because EgtE has not yet been successfully expressed in E. coli.17 To identify the N. crassa EgtE homologue, we searched for genes that are predicted to interact with Egt1 using the String database based on gene co-occurrence, fusion, and gene neighbor hood information (http://string-db.org/);33 and the NCU01256 gene was identified, and it was predicted to be a PLP-dependent enzyme. We subcloned it into pASK-IBA3+ vector and overexpressed it in E. coli BL21(DE3) strain; the purified NCU01256 protein does not have the desired C–S lyase activity. This result implies that a homologue of NCU01256 in N. crassa might be the desired C–S lyase. We then searched the N. crassa genome (http://mips.helmholtz-muenchen.de/genre/proj/ncrassa/)31 using NCU01256 as the query sequence, and two potential PLP-containing proteins (NCU04636 and NCU11365, Supporting Information) were identified. Similarly, we have cloned them into a pASK-IBA3+ vector, and the proteins were overexpressed in E. coli BL21(DE3) strain. Subsequent biochemical characterization revealed that NCU11365 is the C–S lyase (Figures 13S −15S, Supporting Information). The UV–vis spectrum of the isolated NCU11365 is consistent with the presence of a PLP cofactor (Figure 13S, Supporting Information). NCU11365 protein was characterized by two different assays: 1H NMR assay and a colorimetric assay. In the 1H NMR spectrum, the imidazole hydrogen in compound 4 has a chemical shift of 7.12 ppm. Once the C–S bond is cleaved by NCU11365, the resulting ergothioneine thiol-imidazole has a chemical shift of 6.67 ppm (Figure 14S, Supporting Information). To further confirm the product identity, it was isolated from the NCU11365 reaction mixture and characterized by NMR and high-resolution mass spectrometry (Figure 14S, Supporting Information). NCU11365 has also been characterized using a colorimetric assay by coupling the C–S lyase reaction with lactate dehydrogenase. In this coupled assay, the pyruvate produced from NCU11365-catalysis is reduced into lactate by NADH (Figure 15S, Supporting Information). The reaction rate can then be monitored by measuring the NADH consumption rate at 340 nm. When compound 4 was used as the substrate, the kinetic parameters are kobs of 684.4 ± 12.2 min–1 and Km of 194.7 ± 9.8 μM.
In summary, our bioinformatic analysis and biochemical characterizations indicate that bacteria and fungi make use of different ergothioneine biosynthetic pathways (EgtA–EgtE catalysis vs Egt1–Egt2 catalysis in Scheme 1A). The three mononuclear nonheme iron enzymes in ergothioneine and ovothiol biosyntheses (EgtB, OvoA, and Egt1) are related in sequence. However, more detailed investigation using sequence-similarity network analysis indicated that they are at different subgroups in sequence space at an E value cutoff of 1 × 10–50. Biochemical analysis results are fully consistent with the bioinformatic predictions. The differences among them are their substrate preferences and product C–S bond regioselectivity. EgtB of the M. smegmatis pathway (Scheme 1A) uses hercynine and γ-Glu-Cys as its substrates, and it is extremely specific in terms of substrate specificity.26 OvoA in ovothiol biosynthesis has a relaxed substrate specificity (Scheme 1B).25,26 Interestingly, Egt1 from fungus N. crassa prefers hercynine and cysteine and catalyzes a one-step 2 → 4 transformation (Scheme 1A). Such discovery is of practical importance because the ergothioneine and glutathione biosynthetic pathways are uncoupled and they do not compete with each other anymore in N. crassa. Thus, the N. crassa ergothioneine biosynthetic pathway is a more suitable platform than the mycobacterial one for ergothioneine production through metabolic engineering. In addition, the C–S lyase (NCU11365) was successfully identified through genome mining, and the whole ergothioneine biosynthetic pathway was now reconstituted in vitro. Besides its potential application in ergothioneine production through metabolic engineering, the mononuclear nonheme iron enzymes catalyzed oxidative C–S bond formations (EgtB, OvoA, and Egt1) are distinct from currently known biological C–S bond formation reactions.17,25,34−38 Future studies will focus on detailed mechanistic studies of these novel transformations.
Acknowledgments
This work is partially supported by an NSF award (CHE-1309148) and an NIH award (GM093903) to P.L.
Supporting Information Available
Experimental procedure, enzyme characterization data, 1H and 13C NMR of compounds 4 and 5, and kinetic data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ W.H. and H.S. contributed equally to this work.
The authors declare the following competing financial interest(s): A patent related to this work has been submitted
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fontecave M.; Ollagnier-de-Choudens S.; Mulliez E. Chem. Rev. 2003, 103, 2149. [DOI] [PubMed] [Google Scholar]
- Kessler D. FEMS Microbiol. Rev. 2006, 30, 825. [DOI] [PubMed] [Google Scholar]
- Hand C. E.; Honek J. F. J. Nat. Prod. 2005, 68, 293. [DOI] [PubMed] [Google Scholar]
- Fahey R. C. Annu. Rev. Microbiol. 2001, 55, 333. [DOI] [PubMed] [Google Scholar]
- Lin C. I.; McCarty R. M.; Liu H. W. Chem. Soc. Rev. 2013, 42, 4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry R. J. In Comprehensive Natural Products Chemistry (I); Barton D., Nakanishi K., Meth-Cohn O., Eds.; Pergamon: Oxford,, 1999; Vol. 1, p 825. [Google Scholar]
- Wang L. R.; Chen S.; Xu T. G.; Taghizadeh K.; Wishnok J. S.; Zhou X. F.; You D. L.; Deng Z. X.; Dedon P. C. Nat. Chem. Biol. 2007, 3, 709. [DOI] [PubMed] [Google Scholar]
- Mueller E. G. Nat. Chem. Biol. 2006, 2, 185. [DOI] [PubMed] [Google Scholar]
- Jacob C. Nat. Prod. Rep. 2006, 23, 851. [DOI] [PubMed] [Google Scholar]
- Marine Sulfur-Containing Natural Products; Atta-ur-Rahman, Ed.; Elsevier Science Ltd.: Kidlington, 2001; Vol. 25, pp 811–917. [Google Scholar]
- Sulfur-Containing Natural Products from Marine Invertebrates; Atta-ur-Rahman, Ed.; Elsevier Science Ltd.: Kidlington, 2003; Vol. 28, pp 617–751. [Google Scholar]
- Knerr P. J.; van der Donk W. A. Annu. Rev. Biochem. 2012, 81, 479. [DOI] [PubMed] [Google Scholar]
- Xie Y. C.; Li Q. L.; Song Y. X.; Ma J. Y.; Ju J. H. ChemBioChem 2014, 15, 1183. [DOI] [PubMed] [Google Scholar]
- Okeley N. M.; van der Donk W. A. Chem. Biol. 2000, 7, R159. [DOI] [PubMed] [Google Scholar]
- Li B.; Wever W. J.; Walsh C. T.; Bowers A. A. Nat. Prod. Rep. 2014, 31, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q.; Song F.; Xiao X.; Huang P.; Li L.; Monte A.; Abdel-Mageed W. M.; Wang J.; Guo H.; He W.; Xie F.; Dai H.; Liu M.; Chen C.; Xu H.; Piggott A. M.; Liu X.; Capon R. J.; Zhang L. Angew. Chem. Int. .Ed. 2013, 52, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebeck F. P. J. Am. Chem. Soc. 2010, 132, 6632. [DOI] [PubMed] [Google Scholar]
- Ishikawa Y.; Israel S. E.; Melville D. B. J. Biol. Chem. 1974, 249, 4420. [PubMed] [Google Scholar]
- Grundemann D.; Harlfinger S.; Golz S.; Geerts A.; Lazar A.; Berkels R.; Jung N.; Rubbert A.; Schomig E. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah I. K.; Halliwell B. BBA-Mol. Basis Dis. 2012, 1822, 784. [DOI] [PubMed] [Google Scholar]
- Speisky H.; Gomez M.; Carrasco-Pozo C.; Pastene E.; Lopez-Alarcon C.; Olea-Azar C. Bioorg. Med. Chem. 2008, 16, 6568. [DOI] [PubMed] [Google Scholar]
- Zhu B. Z.; Mao L.; Fan R. M.; Zhu J. G.; Zhang Y. N.; Wang J.; Kalyanaraman B.; Frei B. Chem. Res. Toxicol. 2011, 24, 30. [DOI] [PubMed] [Google Scholar]
- Erdelmeier I.; Daunay S.; Lebel R.; Farescour L.; Yadan J.-C. Green Chem. 2012, 14, 2256. [DOI] [PubMed] [Google Scholar]
- Xu J. Z.; Yadan J. C. J. Org. Chem. 1995, 60, 6296. [Google Scholar]
- Braunshausen A.; Seebeck F. P. J. Am. Chem. Soc. 2011, 133, 1757. [DOI] [PubMed] [Google Scholar]
- Song H.; Leninger M.; Lee N.; Liu P. Org. Lett. 2013, 15, 4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluskal T.; Ueno M.; Yanagida M. PLoS One 2014, 9, e97774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello M. H.; Barrera-Perez V.; Morin D.; Epstein L. Fungal Genet. Biol. 2012, 49, 160. [DOI] [PubMed] [Google Scholar]
- Atkinson H. J.; Morris J. H.; Ferrin T. E.; Babbitt P. C. PLoS One 2009, 4, e4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H.; Sae Her A.; Raso F.; Zhen Z.; Huo Y.; Liu P. Org. Lett. 2014, 16, 2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galagan; et al. Nature 2003, 422, 859. [DOI] [PubMed] [Google Scholar]
- Fish W. W. Methods Enzymol. 1988, 158, 357. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D.; Franceschini A.; Kuhn M.; Simonovic M.; Roth A.; Minguez P.; Doerks T.; Stark M.; Muller J.; Bork P.; Jensen L. J.; von Mering C. Nucleic Acids Res. 2011, 39, D561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs C.; Fujimori D. G.; Walsh C. T.; Bollinger J. M. Acc. Chem. Res. 2007, 40, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Omar M. M.; Loaiza A.; Hontzeas N. Chem. Rev. 2005, 105, 2227. [DOI] [PubMed] [Google Scholar]
- Kovaleva E. G.; Lipscomb J. D. Nat. Chem. Biol. 2008, 4, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S. K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y. S.; Zhou J. Chem. Rev. 2000, 100, 235. [DOI] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Chem. Rev. 2004, 104, 939. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.