

Abstract
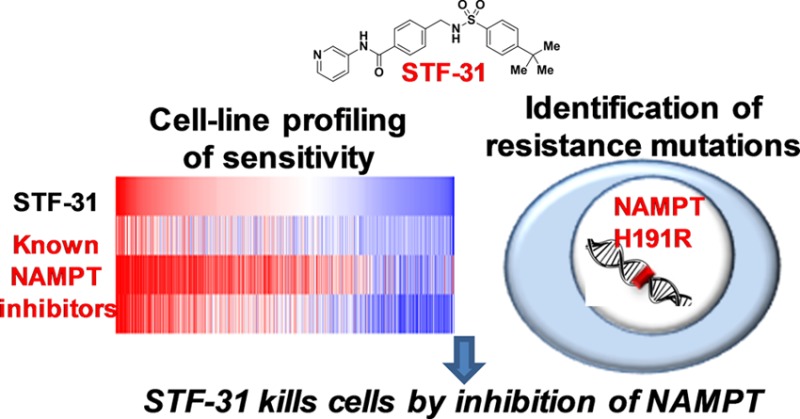
The small-molecule probes STF-31 and its analogue compound 146 were discovered while searching for compounds that kill VHL-deficient renal cell carcinoma cell lines selectively and have been reported to act via direct inhibition of the glucose transporter GLUT1. We profiled the sensitivity of 679 cancer cell lines to STF-31 and found that the pattern of response is tightly correlated with sensitivity to three different inhibitors of nicotinamide phosphoribosyltransferase (NAMPT). We also performed whole-exome next-generation sequencing of compound 146-resistant HCT116 clones and identified a recurrent NAMPT-H191R mutation. Ectopic expression of NAMPT-H191R conferred resistance to both STF-31 and compound 146 in cell lines. We further demonstrated that both STF-31 and compound 146 inhibit the enzymatic activity of NAMPT in a biochemical assay in vitro. Together, our cancer-cell profiling and genomic approaches identify NAMPT inhibition as a critical mechanism by which STF-31-like compounds inhibit cancer cells.
The small-molecule probe STF-31 was recently identified through phenotypic high-throughput screening for its ability to kill renal cell carcinoma cells deficient in the Von Hippel-Lindau tumor suppressor gene (VHL) relative to cells in which the gene had been reintroduced1 (Figure 1A). Loss of VHL has previously been associated with elevated aerobic glycolysis (the Warburg effect) and dependency on the high-affinity glucose transporter GLUT1.2,3 STF-31 and close analogues were reported to impair glucose uptake and directly associate with the glucose transporter GLUT1, suggesting that STF-31 acts as a GLUT1 antagonist.
Figure 1.

STF-31 has a cell growth inhibition profile similar to that of known NAMPT inhibitors and inhibits recombinant NAMPT. (A) Chemical structures of STF-31 and compound 146. (B) Heat-map visualization of pairwise correlations from unsupervised clustering of 496 compounds using AUC values. (C) AUC-AUC comparison between STF-31, APO-866, and CAY-10618 across 560 cell lines. Each vertical line represents a cell line, and these are aligned according to their sensitivity to STF-31. The Pearson correlation coefficient for STF-31 and each known (biochemically validated) NAMPT inhibitor is given. (D) The Spearman (rank) correlation between basal gene-expression levels and AUC values across up to 688 adherent cell lines was calculated for 18,988 transcripts, and correlation coefficients were plotted as box-and-whisker plots, with outliers (black dots) representing the 1st and 99th percentiles and NAMPT highlighted in green. (E) Recombinant NAMPT activity was measured using a coupled-enzyme system at 30 °C. Concentration–response curves were fit using non-linear regression. Each data point is mean ± SD (n = 3).
Multiple unbiased approaches have been used to identify the cellular mechanisms of action and targets of bioactive small molecules, including affinity purification coupled with quantitative proteomics, yeast genomic methods, RNAi-based modifier screening, and computational inference approaches.4 Next-generation sequencing (NGS)-based genomic or transcriptomic profiling of phenotypically resistant cell populations has also been used to elucidate drug-resistance mechanisms.5−7 Identification of unique recurrent single nucleotide variations (SNVs) or expression alterations that enable resistance can offer insights into the mechanism of action or cellular targets of compounds.
Recently, large-scale cancer cell-line (CCL) profiling of small-molecule sensitivity has enabled the correlation of cell lines’ genetic features with their sensitivity to small-molecule probes and approved drugs.8−10 Examination of patterns of sensitivity across a large collection of cell lines revealed an opportunity to use cancer cell line profiling data as another unbiased approach to identifying small-molecule mechanisms of action.
Here we use cancer cell-line profiling to provide evidence that STF-31 and its more potent analogue compound 14611 are inhibitors of NAMPT, an enzyme responsible for generation of NAD+, and confirm the hypothesis that the compounds directly inhibit NAMPT enzyme activity. Recent reports have also linked STF-31-like molecules to biochemical inhibition of NAMPT.12,13 Furthermore, we demonstrate that NAMPT is the relevant target for mediating the effects of STF-31-like small molecules on cancer cell viability through the use of unbiased NGS-based genomic biomarker identification strategies to uncover a recurrent mutation within NAMPT (H191R) that is sufficient to render cells resistant to STF-31 and compound 146.
Results and Discussion
The sensitivity of 679 cancer cell lines to 496 small molecules was measured in 16-point concentration–response format using ATP levels as a surrogate for growth and viability. The area under the concentration–response curve (AUC) was computed as a metric for sensitivity, and hypothesis-free unsupervised clustering of AUCs revealed groups of small molecules eliciting similar patterns of sensitivity. One cluster (Figure 1B) contained all three annotated NAMPT inhibitors included in the experiment: APO-866,14 GMX1778,15,16 and CAY-1061817 (Supporting Figure 1). This cluster also contained the previously annotated GLUT1 inhibitor STF-31 (Figure 1A). The Pearson correlation values (AUC vs AUC) between STF-31 and the three previously reported NAMPT inhibitors across 560 shared cancer cell lines ranged from 0.704 to 0.887 (Figure 1C). As with the previously reported NAMPT inhibitors, STF-31 had wide-ranging effects on cell viability across cell lines, with some lines nonresponsive at 66.7 μM and other lines sensitive at concentrations as low as 100 nM.
As these results suggested a relationship between STF-31 and NAMPT, we associated the levels of expression of individual transcripts in the cancer cell lines with the pattern of sensitivity to STF-31 and looked for associations involving NAMPT. We measured the correlation of AUC for STF-31 with transcript levels of each of 18,988 transcripts (transcript data are publicly available at http://www.broadinstitute.org/ccle).9 Low expression of the NAMPT transcript was significantly associated with sensitivity to STF-31 and the annotated NAMPT inhibitors CAY-10618, APO-866 (FK-866), and GMX1778 (CHS-828), with NAMPT the top transcript (of 18,988) correlated with response for each of the four probes (p < 6.1 × 10–5; Figure 1D). Consistent with reports that STF-31 has differential effects on viability of cell lines with varying expression of SLC2A1 (encoding GLUT1) and VHL (encoding the von Hippel-Lindau tumor suppressor),1 there was significant correlation between NAMPT expression and levels of both transcripts (ρ = 0.23 and −0.17, respectively; p < 0.001). However, the correlations between sensitivity to each of the four probes and SLC2A1 or VHL levels were substantially weaker than the correlations with NAMPT expression (Supporting Table 1). Additionally, although STF-31 was initially identified for its selective inhibition of a VHL mutant renal cell carcinoma (RCC) cell line over an isogenic line overexpressing VHL,1 in our collection of 20 RCC cell lines STF-31 shows somewhat greater inhibition of VHL wild-type lines (Supporting Figure 2).
Therefore, we hypothesized that STF-31 and its analogues could be inhibitors of NAMPT. We used a coupled-enzyme kinetic assay to measure recombinant NAMPT activity in the presence and absence of STF-31, compound 146 (Figure 1A), the STF-31 analogue that showed maximal cytotoxicity among a panel of analogues,11 GMX1778, APO-866, and CAY-10618. All five small molecules inhibited recombinant NAMPT in a dose-dependent fashion relative to a DMSO control (Figure 1E). The same compounds were inactive in a counter-assay carried out in the absence of NAMPT, precluding interference with reaction-coupling components or the detection system. These observations are consistent with a report published during the course of these studies demonstrating that STF-31 can inhibit NAMPT enzyme activity directly.12 Relative to the previously reported NAMPT inhibitors, STF-31 showed reduced potency in both the enzymatic activity assay and cell viability assays.
Collectively, these results suggested the cellular effects of STF-31 are due to inhibition of NAMPT. The cytotoxic effects of NAMPT inhibitors including GMX1778 and APO-866 can in many cases be blocked in vivo and in vitro by addition of exogenous nicotinic acid (NA), a substrate for NAMPT-independent NAD+ recycling.15,16,18,19 NA-mediated prevention of cell killing by NAMPT inhibitors requires expression of nicotinate phosphoribosyltransferase isoform 1 (NAPRT1), the enzyme that catalyzes the initial reaction in the NAMPT-independent NAD+ recycling pathway.15,16,18−20 As with GMX1778 and APO-866, exogenous NA also blocked cell growth inhibition by compound 146 (Figure 2A) in HCT116 cells that express NAPRT1 (Supporting Figure 3). Addition of NA had no effect on compound 146-mediated inhibition of HT1080 cells that express low levels of NAPRT1 (Figure 2A and Supporting Figure 3). As expected, siRNA-mediated knockdown of NAPRT1 in HCT116 restored the growth inhibition by compound 146 and APO-866 in the presence of exogenous NA (Figure 2B and Supporting Figure 3C), similar to a previous report using lung cancer cell lines.21 Additionally, culturing HCT116 cells in McCoy’s 5A media, which contains 4 μM NA, was sufficient to abrogate cell growth inhibition by compound 146, while culture in RPMI media, which contains no NA, had little effect on compound 146 sensitivity (Figure 2C). We further investigated the relationship between NAPRT1 expression and STF-31 sensitivity in the 25 cell lines from our profiling experiment grown in McCoy’s 5A media. Cancer cell lines grown in the presence of NA that remained sensitive to STF-31 invariably had low expression of NAPRT1, relative to both insensitive cells grown with NA and other cancer cell lines (both STF-31 sensitive and insensitive) grown in media not containing NA (Figure 2D), supporting the role of NAPRT1 in nicotinic acid-mediated rescue of STF-31 cytotoxicity.
Figure 2.

Nicotinic acid blocks the effect of STF-31 and other NAMPT inhibitors in NAPRT1-expressing cells. (A) HCT116 and HT1080 cells were tested for viability after 72 h treatment with compound 146 and known NAMPT inhibitors in the presence or absence of 10 μM nicotinic acid (NA). Each data point is mean ± SD (n = 3). (B) Sensitivity of HCT116 cells tranfected with control or NAPRT1 siRNA to compound 146 in the presence or absence of NA. Each data point is mean ± SD (n = 3). (C) Sensitivity of HCT116 against compound 146 in different media conditions. Each data point is mean (n = 2). (D) NAPRT1-dependent effects of nicotinic acid (NA) on STF-31 sensitivity across 772 CCLs. Sensitive cell lines were defined as having an AUC < 3.5, and insensitive cell lines as an AUC > 5.5. Red bars = mean ± SD.
To elucidate the mechanisms of action for STF-31 and compound 146 further, we used next-generation sequencing to profile compound 146-resistant cells with the idea that a subset of resistance mechanisms observed may involve recurrent mutations in genes that encode the direct protein targets of the small molecule. Recently this approach has been used to illuminate potential targets that are responsible for the biological phenotypes induced by probes of interest, including inhibitors of the SF3B complex, Polo-like kinase 1, the proteasome, and the androgen receptor.5−7 This genomic approach offers an alternative path to identifying functionally relevant targets in intact cells in the context of the phenotype of interest. We generated compound 146-resistant cell lines for genomic profiling by continuous treatment of HCT116 cells with 5 times concentration of GI100 of compound 146 (200 nM) (Figure 3A). After 4 weeks of treatment, 16 resistant clones were isolated and confirmed to be completely insensitive to compound 146 at up to 10 μM following 72 h treatment by an ATPase-based viability assay (CellTiter-Glo) (Figure 3B). Importantly, these clones remained equally sensitive to paclitaxel as parental HCT116 cells, suggesting that the clones may harbor a selective mechanism conferring resistance to compound 146 (Supporting Figure 4). Whole-exome-sequencing of these 16 resistant clones and comparison to parental cells uncovered multiple unique single nucleotide variants (SNVs) in each resistant clone (Figure 3C). Focusing only on recurrent SNVs shared by all resistant clones, we identified five SNVs predicted to have “high” functional impact calculated by the MutationAssessor algorithm (Figure 3C). These SNVs included an H191R mutation in NAMPT, which has been previously reported to confer resistance to known small-molecule NAMPT inhibitors.22 Consistent with this report, we tested the sensitivity of 8 compound 146-resistant clones to NAMPT inhibitors APO-866 and CAY-10618, and all 8 were also insensitive to the treatment up to 1 μM (Figure 3D), suggesting again that NAMPT is a common target.
Figure 3.

Identification of recurrent genomic alterations in compound 146-resistant cell lines. (A) Workflow of the generation of compound 146-resistant clones in HCT116 cells. (B) Sensitivity of compound 146-resistant clones to compound 146 after 72 h incubation. Each data point is mean (n = 2). (C) Work flow to identify single nucleotide variants (SNVs) present in all resistant clones and predicted to have a high functional impact by MutationAssessor. (D) Sensitivity of compound 146-resistant cells to two known NAMPT inhibitors. Each data point is mean (n = 2).
Recurrent SNVs were also observed in PFKFB4, MCCC1, PCDHB7, and ZNF33A. However, unlike NAMPT, transcript levels of these genes were not strongly predictive of response to STF-31 or previously reported NAMPT inhibitors in our panel of 679 cancer cell lines (Figure 1D).
In contrast, the resistant clones did not have mutations in the gene encoding GLUT1 (SLC2A1). The resistant clones did not display enhanced GLUT1 protein expression relative to the parental cells and frequently showed moderately decreased levels (Supporting Figure 5). Consistent with the lack of correlation between GLUT1 protein levels and sensitivity to compound 146, we observed similar growth-inhibitory activity of compound 146 in DLD1 parental (GLUT1+/+) and isogenic GLUT1–/– cells. Taken together, these data suggest that GLUT1 does not mediate the cytotoxic effects of STF-31-like compounds (Supporting Figure 6).To assess further the relationship of the recurrent NAMPT H191R mutation and the sensitivity of cells to compound 146, STF-31, and previously reported NAMPT inhibitors, we established HCT116 cell lines stably transduced with empty vector control, wild-type-, or H191R NAMPT-expressing lentiviral vectors. We confirmed that the exogenous wild-type and H191R NAMPT proteins were expressed at a similar level by Western blotting (Figure 4A). We then assessed the sensitivity of these NAMPT-overexpressing cell lines and nonengineered parental HCT116 cells against STF-31, compound 146, and APO-866 in a viability assay. As expected, NAMPT-H191R conferred resistance to the NAMPT inhibitor APO-866 (Figure 4B) and showed no difference in response to staurosporine in HCT116 cells (Supporting Figure 7). Similarly, ectopic expression of NAMPT-H191R also abolished the growth-inhibitory activity of compound 146 as well as STF-31 (Figure 4B). Overexpression of wild-type NAMPT also caused a partial resistance to these compounds, consistent with previous reports19 and the observation that cell lines with high NAMPT expression are less sensitive to STF-31 and other NAMPT inhibitors (Figure 1D). Analogous results were obtained in a second cell line, RKO (Supporting Figure 8). These data implicate inhibition of NAMPT as the mechanism of action by which STF-31 and its analogues inhibit viability of cancer cells.
Figure 4.

NAMPT H191R confers resistance to the growth inhibition effect of STF-31, compound 146, and a known NAMPT inhibitor. (A) NAMPT and GLUT1 protein expression in parental HCT116 cells, HCT116 cells engineered with either WT or H191R NAMPT, and a HCT116 cell line selected for resistance to compound 146 (α-tubulin; loading control). (B) Effects of STF-31, compound 146, and APO-866 after 72 h treatment in HCT116 cells expressing wild-type or H191R NAMPT. Each data point is mean ± SD (n = 3).
We have provided multiple independent lines of evidence indicating STF-31 and related compounds inhibit the viability of cell lines by inhibition of NAMPT. Unbiased profiling of 679 cancer cell lines initially identified strong similarities in sensitivity profiles between STF-31 and a collection of bona fide NAMPT inhibitors. Correlation of single transcript levels with cell line sensitivity revealed low NAMPT expression as the transcript best correlated with sensitivity to STF-31 and bona fide NAMPT inhibitors. STF-31 and compound 146 both inhibit NAMPT in a biochemical assay, and nicotinic acid blocks the cytotoxicity of STF-31 and other NAMPT inhibitors in cell lines that express NAPRT1. Importantly, we have identified a recurrent NAMPT H191R mutation through unbiased genomic profiling of cells selected for resistance to compound 146 and demonstrated that exogenous expression of this mutant allele in wild-type cells is sufficient to confer resistance to STF-31/compound 146 and other NAMPT inhibitors. This work highlights the power of combining multiple approaches, including large-scale cancer cell line sensitivity profiling and unbiased genomic profiling of compound-resistant clones, to reveal the mechanisms of action or the molecular targets of small-molecule probes.
Methods
Large-Scale CCL Viability Measurements
A collection of 496 small molecules was tested against 876 human CCLs spanning 23 distinct lineages. Each CCL was plated at a density of 500 cells/well in white opaque 1536-well plates in its preferred media using a highly automated, high-throughput platform. Twenty-four hours after plating, compounds were added by acoustic transfer using a Labcyte Echo 555 (Labcyte Inc., Sunnyvale, CA). Each compound was measured in 16-point, 2-fold dilution in duplicate. Sensitivity was assayed using cellular ATP levels as a surrogate for cell viability (CellTiterGlo, Promega Corporation, Madison, WI) using a ViewLux Microplate Imager (PerkinElmer, Waltham, MA) and normalized to vehicle (DMSO) treatment.
AUC-AUC Clustering
Sixteen-point concentration–response curves were fit using nonlinear fits to 3- or 4-parameter sigmoid functions. An eight-point concentration range was selected for each compound, and area-under-the-curve (AUC) measurements for each compound–CCL pair were calculated by numeric integration. Analyses were restricted to adherent CCLs because of the increased sensitivity of suspension cells. Pairwise correlations were computed by a method that accounts for missing data by restricting correlation calculations to those CCLs tested in common for both compounds. Correlations were further processed to Z-scores using a Fisher transformation to normalize pairwise correlations across different numbers of CCLs.23 Fisher-normalized values were transformed back to pairwise distances using a logarithmic scale to express two-tailed p-values around the normal distribution, then scaling the results to the interval [0,2]. All calculations were performed in MATLAB (Mathworks, Inc., Natick, MA).
Correlation of AUC with Basal Gene Expression
Publicly available gene-centric Robust Multichip Average (RMA)-normalized basal mRNA gene expression data (Affymetrix GeneChip Human Genome U133 Plus 2.0 Array) were downloaded from the Cancer Cell Line Encyclopedia (http://www.broadinstitute.org/ccle).9 Spearman (rank) correlation coefficients were calculated between gene expression and compound sensitivity (AUC) across all adherent CCLs for which both expression and sensitivity data were available (maximum n = 688 CCLs). Permutation tests (n = 16,384) were performed by randomizing CCL labels, allowing calculation of nominal p-values and ranking of z-scored transcript–AUC correlation coefficients.
Enzymatic NAMPT Assays
Recombinant NAMPT activity was measured using a coupled-enzyme reaction system (CycLex NAMPT Colorimetric Assay Kit; MBL International). The assay was carried out in 384-well plate format using the two-step method with the following modification: compounds dissolved in DMSO were pin-transferred (100 nL/well, CyBio Vario) onto a recombinant NAMPT solution prior to addition of cofactors. The formation of the colored product WST-1-formazan was measured at 450 nm on a SpectraMax M5 microplate reader using the SoftMax Pro software (Molecular Devices) for 30 min at 30 °C for all compounds, with DMSO as a vehicle control. The reaction rate for each enzymatic reaction during the linear range was determined by linear regression, and then the slope of regression lines plotted against compound concentration and curves were fit using four-parameter nonlinear regression (GraphPad Prism, GraphPad Software, Inc.).
To exclude the possibility that compounds were interfering with subsequent enzymatic coupling steps, the assay was carried out as described above in the absence of NAMPT and NAMPT substrates but in the presence of 5 μM nicotinamide mononucleotide (NMN), the product of the NAMPT reaction.
Cell Culture
HCT116 and RKO were purchased from ATCC and cultured in RPMI1640 (Life Technologies) plus 10% FBS (Life Technologies) or EMEM (ATCC) plus 10% FBS (Life Technologies) at 37 °C with 5% CO2. Lenti-X 293T was purchased from Clontech and cultured in DMEM (Life Technologies) plus 10% FBS (Life Technologies), 2 mM l-glutamine (Life Technologies), and 1 mM sodium pyruvate (Life Technologies).
Small Molecules
STF-31 was purchased from TOCRIS Biosciences. Compound 146 was synthesized as previously described.11
HCT116/HT1080/RKO Cell Viability Assay
Cells were plated at 2000 cells/well in 96-well plates and incubated overnight. Spent media was removed, and fresh media added containing compound at 100 μL/well (9 doses, 3-fold dilution, 0.1% DMSO) in duplicate or triplicate. After incubation for 72 h, cell viability was measured by using CellTiter-Glo. A time-zero (TZ) plate was generated and utilized for GI50/LD50 calculations.24
Generation of Small-Molecule-Resistant Clones
HCT116 cells (2 × 106) seeded in 10 cm dishes were treated with compound 146 (0.1% DMSO) at 200 nM (5-fold of GI100 obtained from 72 h cell viability assay). The media containing compound 146 was refreshed twice a week. After 4 weeks, resistant colonies were developed, and those sized at 1–2 mm in diameter were picked up and transferred to 96-well plates to expand with continuous compound 146 treatment. Genomic DNA was isolated from each resistant clone using the DNeasy blood and tissue kit (Qiagen) and then subjected to whole-exome-sequencing using the Illumina HiSeq 2000 at Beijing Genome Institute (Shenzhen, China).
Plasmids
NAMPT cDNA was purchased from GeneCopoeia (GC-A1275). The H191R mutation was introduced into wild-type NAMPT in the pDONR Vector using the QuikChange II XL site-directed mutagenesis kit (Agilent) with primer pair 5′-GGTCTGGAATACAAGTTACGTGATTTTGGCTACAGAGGA (forward) and 5′-TCCTCTGTAGCCAAAATCACGTAACTTGTATTCCAGACC (reverse). Both wild-type and H191R mutant NAMPT were then cloned into the pLenti6.3/V5 vector (Life Technologies) using the Gateway LR Clonase II enzyme mix kit (Life Technologies). All constructs were confirmed by sequencing. All plasmids DNAs were prepared using QIAprep Spin Miniprep Kit (Qiagen).
Viral Packaging and Infection
The cDNA-expressing lentiviral vectors were packaged in HEK293 cells. Briefly, 2.4 μg of pLenti-NAMPT, 2.4 μg of Δ8.91, and 0.6 μg of VSVG plasmids (Sigma) were mixed with 16.2 μL of TransIT-293 (Mirus, MIR2700) in 583.2 μL of Opti-MEM (Life Technologies) for 15 min. Then, the mixture was added to Lenti-X 293T (Clontech) cells in a 10 cm dish (2.5 × 106 cells) with 9.5 mL of culture media. After 24 h incubation, the media was refreshed (6 mL/dish), and after an additional 24 h incubation, the media containing lentivirus was collected and filtered through a 0.2 μm filter to remove cell debris. HCT116 or RKO cells were plated in 6-well plates (1 × 105 cells/well) and incubated overnight. The spent media was removed, fresh media was added containing 8 μg/mL Polybrene (Millipore), and 100 or 900 μL of NAMPT lentivirus containing 8 μg/mL Polybrene was added into cell plates (2 mL/well media in total). After 24 h of incubation, the media was changed to media containing 15 μg/mL blastcidin, and cells were incubated for 5 days. Selected cells were transferred to new plates and expanded.
siRNA Transfection
NAPRT1 siRNA (GE Dharmacon, L-016912-01) or control siRNA (GE Dharmacon, D-001810-10) was transfected into HCT116 cells by DharmaFECT (GE Dharmacon, T2002) using the vendor’s protocol (http://dharmacon.gelifesciences.com/uploadedFiles/Resources/basic-dharmafect-protocol.pdf). For Western blot analysis, cell lysates were collected 72 h after transfection. For cell viability assays compounds were added 48 h after transfection.
Western Blotting
Cell lysates were prepared in RIPA buffer (Boston BioProducts) with protease inhibitor cocktail (Roche). Harvested lysate was centrifuged (12000 rpm, 5 min, 4 °C) to remove debris, and resulting supernatant was collected. Cell lysates were then mixed with LDS sample buffer (Life Technologies) and sample reducing agent (Life Technologies), according to the manufacture’s protocol. After incubation for 10 min at 70 °C, the sample was separated by SDS-PAGE (Novex 4–12% Bis-Tris gel, Life Technologies), transferred to a nitrocellulose membrane by iBlot (Life Technologies), and blocked with Odyssey Blocking Buffer (Li-COR) with 50% TBS-T buffer, according to the manufacturer’s instructions. Primary antibodies were purchased and treated with the indicated dilution in blocking buffer at 4 °C overnight: anti-NAMPT antibody (abcam, ab45890, 1:250), anti-NAPRT1 (Thermo/Pierce, PA5-31880, 1:1000), anti-GLUT1 (Epitomics, 2944-1, 1:5000), anti-GAPDH (Sigma, G9545, 1:5000), anti-α-tublin (Sigma, T6199, 1:2000), and eIF4E (Cell signaling technology, 9742, 1:1000). Secondary antibodies were purchased from Li-COR (IRDye 800CW Infrared Dye and 680RD, 1:10000) and treated for 1 h at RT. Images were developed using the ODYSSEY system (Li-COR).
Genomic DNA Extraction for Sequencing
Genomic DNA was extracted from cells (2 × 106 cells) by using the DNeasy Blood & Tissue Kit (Promega).
Sequence Data Processing
Unmapped sequencing reads were aligned to the NCBI human reference genome build GRCh37 using BWA.25 Duplicate molecules in aligned read data were then flagged using the MarkDuplicates algorithm in the Picard suite of utilities.26 To improve the accuracy of variant calling, base quality scores were empirically recalibrated using a set of covariates that take into account read group, initial quality score, read cycle, and dinucleotide context. To reduce the amount of false-positive single nucleotide variants (SNVs), reads were locally realigned at regions that potentially harbor small insertions or deletions. Base quality score recalibration and local realignment were performed using the Genome Analysis Toolkit.27 SNVs in each resistant clone were identified using MuTect, using the parental cell line HCT116 as the matched “normal” sample.28 Variants were annotated using Oncotator.29 Variants detected in each resistant clone were then merged into a single MAF file and filtered for recurrent mutations found only in all resistant clones. To identify recurrent SNVs likely to have a functional impact, mutations were filtered down to only variants with “high” (functional impact score >3.5) prediction scores from the MutationAssessor algorithm.30
Acknowledgments
Drs. M.-H. Hao, L. Yu, P. Smith, Y. Mizui, Y. Wang, and V. Viswanathan are thanked for valuable discussion. Drs. D. Reynolds and K. Arai are thanked for assistance with experiments using chemical probes. We thank Horizon Discovery Group plc (Cambridge, U.K.) for kindly providing the DLD1 GLUT1+/+ and GLUT1–/– isogenic cell lines. This work was supported by the National Institutes of Health (U01CA176152, to S.L.S.) and H3 Biomedicine. S.L.S. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
Eight supporting figures, one supporting table, and AUCs for STF-31 and three additional NAMPT inhibitors in 563-634 cancer cell lines. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors contributed equally to this work. J.H.C., A.F.S., and S.L.S. designed and led acquisition of cell-line sensitivity profiling experiments. D.J.A., M.G.R., B.S.-L., P.A.C., A.F.S., and S.L.S. analyzed cell-line sensitivity profiling data. D.J.A. performed NAMPT biochemical activity assays. M.W., P.Z., and D.I. designed and executed experiments to identify and characterize effects of NAMPT mutations in cells. X.P. contributed to these experiments. A.H.R. analyzed the whole-exome sequencing data. D.J.A., D.I., M.G.R., B.S.-L., A.H.R., P.A.C., P.Z., A.F.S., and S.L.S. wrote the manuscript.
The authors declare the following competing financial interest(s): S.L.S. is a founder of and advisor to H3 Biomedicine. He is also an H3B shareholder.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Chan D. A.; Sutphin P. D.; Nguyen P.; Turcotte S.; Lai E. W.; Banh A.; Reynolds G. E.; Chi J. T.; Wu J.; Solow-Cordero D. E.; Bonnet M.; Flanagan J. U.; Bouley D. M.; Graves E. E.; Denny W. A.; Hay M. P.; Giaccia A. J. (2011) Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 3, 94ra70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell P. H.; Wiesener M. S.; Chang G. W.; Clifford S. C.; Vaux E. C.; Cockman M. E.; Wykoff C. C.; Pugh C. W.; Maher E. R.; Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275. [DOI] [PubMed] [Google Scholar]
- Iliopoulos O.; Levy A. P.; Jiang C.; Kaelin W. G. Jr.; Goldberg M. A. (1996) Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. U.S.A. 93, 10595–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenone M.; Dancik V.; Wagner B. K.; Clemons P. A. (2013) Target identification and mechanism of action in chemical biology and drug discovery. Nat. Chem. Biol. 9, 232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoi A.; Kotake Y.; Takahashi K.; Kadowaki T.; Matsumoto Y.; Minoshima Y.; Sugi N. H.; Sagane K.; Hamaguchi M.; Iwata M.; Mizui Y. (2011) Biological validation that SF3b is a target of the antitumor macrolide pladienolide. FEBS J. 278, 4870–4880. [DOI] [PubMed] [Google Scholar]
- Wacker S. A.; Houghtaling B. R.; Elemento O.; Kapoor T. M. (2012) Using transcriptome sequencing to identify mechanisms of drug action and resistance. Nat. Chem. Biol. 8, 235–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpal M.; Korn J. M.; Gao X.; Rakiec D. P.; Ruddy D. A.; Doshi S.; Yuan J.; Kovats S. G.; Kim S.; Cooke V. G.; Monahan J. E.; Stegmeier F.; Roberts T. M.; Sellers W. R.; Zhou W.; Zhu P. (2013) An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide). Cancer Discovery 3, 1030–1043. [DOI] [PubMed] [Google Scholar]
- Basu A.; Bodycombe N. E.; Cheah J. H.; Price E. V.; Liu K.; Schaefer G. I.; Ebright R. Y.; Stewart M. L.; Ito D.; Wang S.; Bracha A. L.; Liefeld T.; Wawer M.; Gilbert J. C.; Wilson A. J.; Stransky N.; Kryukov G. V.; Dancik V.; Barretina J.; Garraway L. A.; Hon C. S.; Munoz B.; Bittker J. A.; Stockwell B. R.; Khabele D.; Stern A. M.; Clemons P. A.; Shamji A. F.; Schreiber S. L. (2013) An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J.; Caponigro G.; Stransky N.; Venkatesan K.; Margolin A. A.; Kim S.; Wilson C. J.; Lehar J.; Kryukov G. V.; Sonkin D.; Reddy A.; Liu M.; Murray L.; Berger M. F.; Monahan J. E.; Morais P.; Meltzer J.; Korejwa A.; Jane-Valbuena J.; Mapa F. A.; Thibault J.; Bric-Furlong E.; Raman P.; Shipway A.; Engels I. H.; Cheng J.; Yu G. K.; Yu J.; Aspesi P. Jr.; de Silva M.; Jagtap K.; Jones M. D.; Wang L.; Hatton C.; Palescandolo E.; Gupta S.; Mahan S.; Sougnez C.; Onofrio R. C.; Liefeld T.; MacConaill L.; Winckler W.; Reich M.; Li N.; Mesirov J. P.; Gabriel S. B.; Getz G.; Ardlie K.; Chan V.; Myer V. E.; Weber B. L.; Porter J.; Warmuth M.; Finan P.; Harris J. L.; Meyerson M.; Golub T. R.; Morrissey M. P.; Sellers W. R.; Schlegel R.; Garraway L. A. (2012) The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett M. J.; Edelman E. J.; Heidorn S. J.; Greenman C. D.; Dastur A.; Lau K. W.; Greninger P.; Thompson I. R.; Luo X.; Soares J.; Liu Q.; Iorio F.; Surdez D.; Chen L.; Milano R. J.; Bignell G. R.; Tam A. T.; Davies H.; Stevenson J. A.; Barthorpe S.; Lutz S. R.; Kogera F.; Lawrence K.; McLaren-Douglas A.; Mitropoulos X.; Mironenko T.; Thi H.; Richardson L.; Zhou W.; Jewitt F.; Zhang T.; O’Brien P.; Boisvert J. L.; Price S.; Hur W.; Yang W.; Deng X.; Butler A.; Choi H. G.; Chang J. W.; Baselga J.; Stamenkovic I.; Engelman J. A.; Sharma S. V.; Delattre O.; Saez-Rodriguez J.; Gray N. S.; Settleman J.; Futreal P. A.; Haber D. A.; Stratton M. R.; Ramaswamy S.; McDermott U.; Benes C. H. (2012) Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutphin P.; Chan D.; Turcotte S.; Denny W. A.; Hay M. P.; Giddens A. C.; Bonnet M.; Giaccia A.. Heteroaryl benzamides, compositions and methods of use. Patent WO2011011514 A1, 2011
- Dragovich P. S.; Zhao G.; Baumeister T.; Bravo B.; Giannetti A. M.; Ho Y. C.; Hua R.; Li G.; Liang X.; Ma X.; O’Brien T.; Oh A.; Skelton N. J.; Wang C.; Wang W.; Wang Y.; Xiao Y.; Yuen P. W.; Zak M.; Zhao Q.; Zheng X. (2014) Fragment-based design of 3-aminopyridine-derived amides as potent inhibitors of human nicotinamide phosphoribosyltransferase (NAMPT). Bioorg. Med. Chem. Lett. 24, 954–962. [DOI] [PubMed] [Google Scholar]
- Matheny C. J.; Wei M. C.; Bassik M. C.; Donnelly A. J.; Kampmann M.; Iwasaki M.; Piloto O.; Solow-Cordero D. E.; Bouley D. M.; Rau R.; Brown P.; McManus M. T.; Weissman J. S.; Cleary M. L. (2013) Next-generation NAMPT inhibitors identified by sequential high-throughput phenotypic chemical and functional genomic screens. Chem. Biol. 20, 1352–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmann M.; Schemainda I. (2003) FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 63, 7436–7442. [PubMed] [Google Scholar]
- Olesen U. H.; Christensen M. K.; Bjorkling F.; Jaattela M.; Jensen P. B.; Sehested M.; Nielsen S. J. (2008) Anticancer agent CHS-828 inhibits cellular synthesis of NAD. Biochem. Biophys. Res. Commun. 367, 799–804. [DOI] [PubMed] [Google Scholar]
- Watson M.; Roulston A.; Belec L.; Billot X.; Marcellus R.; Bedard D.; Bernier C.; Branchaud S.; Chan H.; Dairi K.; Gilbert K.; Goulet D.; Gratton M. O.; Isakau H.; Jang A.; Khadir A.; Koch E.; Lavoie M.; Lawless M.; Nguyen M.; Paquette D.; Turcotte E.; Berger A.; Mitchell M.; Shore G. C.; Beauparlant P. (2009) The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 29, 5872–5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombano G.; Travelli C.; Galli U.; Caldarelli A.; Chini M. G.; Canonico P. L.; Sorba G.; Bifulco G.; Tron G. C.; Genazzani A. A. (2010) A novel potent nicotinamide phosphoribosyltransferase inhibitor synthesized via click chemistry. J. Med. Chem. 53, 616–623. [DOI] [PubMed] [Google Scholar]
- Tan B.; Young D. A.; Lu Z. H.; Wang T.; Meier T. I.; Shepard R. L.; Roth K.; Zhai Y.; Huss K.; Kuo M. S.; Gillig J.; Parthasarathy S.; Burkholder T. P.; Smith M. C.; Geeganage S.; Zhao G. (2013) Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD+ biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. J. Biol. Chem. 288, 3500–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerna D.; Li H.; Flaherty S.; Takebe N.; Coleman C. N.; Yoo S. S. (2012) Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) activity by small molecule GMX1778 regulates reactive oxygen species (ROS)-mediated cytotoxicity in a p53- and nicotinic acid phosphoribosyltransferase1 (NAPRT1)-dependent manner. J. Biol. Chem. 287, 22408–22417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien T.; Oeh J.; Xiao Y.; Liang X.; Vanderbilt A.; Qin A.; Yang L.; Lee L. B.; Ly J.; Cosino E.; LaCap J. A.; Ogasawara A.; Williams S.; Nannini M.; Liederer B. M.; Jackson P.; Dragovich P. S.; Sampath D. (2013) Supplementation of nicotinic acid with NAMPT inhibitors results in loss of in vivo efficacy in NAPRT1-deficient tumor models. Neoplasia 15, 1314–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shames D. S.; Elkins K.; Walter K.; Holcomb T.; Du P.; Mohl D.; Xiao Y.; Pham T.; Haverty P. M.; Liederer B.; Liang X.; Yauch R. L.; O’Brien T.; Bourgon R.; Koeppen H.; Belmont L. D. (2013) Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin. Cancer Res. 19, 6912–6923. [DOI] [PubMed] [Google Scholar]
- Olesen U. H.; Petersen J. G.; Garten A.; Kiess W.; Yoshino J.; Imai S.; Christensen M. K.; Fristrup P.; Thougaard A. V.; Bjorkling F.; Jensen P. B.; Nielsen S. J.; Sehested M. (2010) Target enzyme mutations are the molecular basis for resistance towards pharmacological inhibition of nicotinamide phosphoribosyltransferase. BMC Cancer 10, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancik V.; Carrel H.; Bodycombe N. E.; Seiler K. P.; Fomina-Yadlin D.; Kubicek S. T.; Hartwell K.; Shamji A. F.; Wagner B. K.; Clemons P. A. (2014) Connecting small molecules with similar assay performance profiles leads to new biological hypotheses. J. Biomol. Screening 19, 771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCI60 In Vitro Cell Line Screening Project. http://dtp.nci.nih.gov/branches/btb/ivclsp.html.
- Li H.; Durbin R. (2010) Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard Home Page. http://picard.sourceforge.net.
- McKenna A.; Hanna M.; Banks E.; Sivachenko A.; Cibulskis K.; Kernytsky A.; Garimella K.; Altshuler D.; Gabriel S.; Daly M.; DePristo M. A. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K.; Lawrence M. S.; Carter S. L.; Sivachenko A.; Jaffe D.; Sougnez C.; Gabriel S.; Meyerson M.; Lander E. S.; Getz G. (2013) Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oncotator Home Page. http://www.broadinstitute.org/oncotator/.
- Reva B.; Antipin Y.; Sander C. (2011) Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 39, e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.