

Abstract
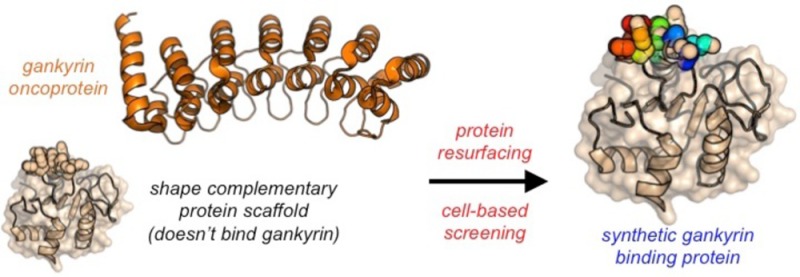
Increased cellular levels of protein–protein interactions involving the ankyrin repeat oncoprotein gankyrin are directly linked to aberrant cellular events and numerous cancers. Inhibition of these protein–protein interactions is thus an attractive therapeutic strategy. However, the relatively featureless topology of gankyrin’s putative binding face and large surface areas involved in gankyrin-dependent protein–protein interactions present a dramatic challenge to small molecule discovery. The size, high folding energies, and well-defined surfaces present in many proteins overcome some of the challenges faced by small molecule discovery. We used split-superpositive Green Fluorescent Protein (split-spGFP) reassembly to screen a 5 × 109 library of resurfaced proteins that are shape complementary to the putative binding face of gankyrin and identified mutants that potently and selectively bind this oncoprotein in vitro and in living cells. Collectively, our findings represent the first synthetic proteins that bind gankyrin and may represent a general strategy for developing protein basic research tools and drug leads that bind disease-relevant ankyrin repeats.
Historically, the vast majority of cellular probes and therapeutics have been small organic molecules (<800 Da).1,2 However, recent studies indicate that only a small percentage (approximately 15–25%) of the human proteome is susceptible to small molecule-dependent regulation.3 The fundamental limitation of small-molecule reagents is encoded in the name itself: the molecules are small and thus intrinsically unable to compete with the relatively large contact surfaces found at many biologically important ligand–receptor interfaces, such as protein–protein interactions.
One structural class that has largely evaded small molecule recognition and modulation is the ankyrin repeat.4 Gankyrin (colored, Figure 1A) is a recently identified ankyrin repeat oncoprotein, whose overexpression is directly linked to the onset, proliferation, and/or metastasis of breast,5,6 liver,7 oral,8 pancreatic,9 and colorectal cancers,10 as well as esophageal squamous cell carcinoma.11 In addition, gankyrin plays an essential role in Ras-initiated tumorigenesis, which represents ∼30% of all cancers.12
Figure 1.
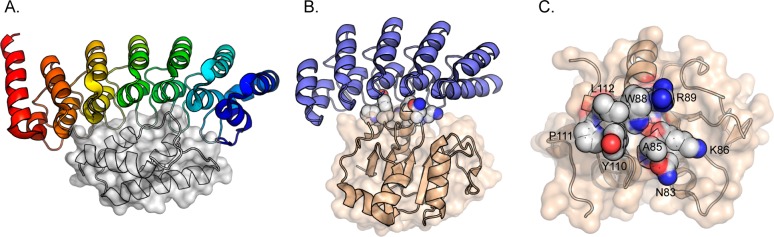
(A) Complex involving gankyrin (colored) and the C-terminal fragment of S6 ATPase (gray). (B) Complex involving Pdar (blue) and Prb (light brown). Binding face residues mutated in this work are highlighted. (C) View of the Pdar binding face of Prb. Residues randomized during library construction are highlighted and annotated.
The seven helix-turn-helix-loop ankyrin modules in gankyrin (individually colored in Figure 1A) generate a relatively featureless and extensive concave putative binding face. Gankyrin binds a number of physiological targets, including cyclin-dependent kinase 4 (CDK4),13 the E3 ubiquitin ligase MDM2,14 and the C-terminal S6 ATPase subunit of the 26S proteasome (referred to as S6 ATPase, herein).15 In forming a complex with CDK4, gankyrin regulates CDK4-dependent phosphorylation of retinoblastoma protein (pRb), ultimately leading to activation of E2F transcription factors.13,16 In forming a complex with MDM2, gankyrin regulates MDM2-dependent polyubiquitination of p53, resulting in lower cellular levels of p53 and suppression or abrogation of p53-dependent apoptosis.14 Aberrant cellular events as a result of increased levels of these protein–protein interactions, due to overexpression of gankyrin, result in decreased genome stability and the onset of oncogenic cell functions and fate. Thus, protein–protein interactions involving gankyrin, or the inhibition of these interactions, are of significant therapeutic interest.
Relatively little is known about the biological role of a complex involving gankyrin (colored) and S6 ATPase (gray space-filling depiction, Figure 1A). However, this interaction illustrates the challenge of disrupting protein–protein interactions involving this oncoprotein. Binding is stabilized by composite surfaces made from discontinuous portions of two proteins over a large surface area, which involve residues on the concave face of the ankyrin repeat.4 The binding interface between gankyrin and S6 ATPase is ∼2400 Å2, which is significantly larger than the observed average value of ∼1600 Å2 for a protein–protein interaction surface.17 Aspects of complexes involving ankyrin repeats, including featureless putative binding face surfaces and large binding interfaces, can present a substantial challenge to the development of small molecule inhibitors. For example, fragment-based drug discovery recently identified molecules that bind the Notch-1 ankyrin repeat domain with a dissociation constant (KD) of ∼10 mM.18
In comparison to their small molecule counterparts, proteins can adopt large and precisely defined three-dimensional surfaces required for binding and controlling complex biological targets that have evaded small-molecule regulation. Modern molecular biology techniques have enabled engineering, evolution, large-scale expression, and purification of diverse proteins. Additionally, multiple technologies now exist, which enable functional protein delivery to the interior of mammalian cells, or to specific subcellular environments, to the extent that multiple researchers have used exogenous natural or synthetic proteins as basic research tools or drug leads that act on intracellular targets.19−25 While a host of challenges to the broader use of proteins as basic research tools and therapeutics exist, a fundamental obstacle is one that continues to resist a general solution: protein folding. We are still largely incapable of designing functional proteins de novo. Perhaps the most sensible solution is thus one semidesign: start with a stable protein with a privileged scaffold and modify it to specifically bind a macromolecule of interest.26
A relatively modest number of protein–protein complexes involving ankyrin repeat domains has been reported. Most of these costructures contain ankyrin repeat binding partners that are large (>50 kDa) and/or unstable proteins, making them poor scaffolds for protein engineering and/or evolution. In the context of established gankyrin-binding proteins, S6 ATPase does not express in E. coli as a soluble protein in the absence of gankyrin15 and structures involving gankyrin and MDM2 or CDK4 have not been reported. Baker and co-workers recently used in silico design and in vitro evolution to generate a potent protein–protein interaction involving a PH1109-derived protein called Prb (Figure 1B, light brown) and a synthetic thermostable ankyrin repeat called Pdar (Figure 1B, blue).27 PH1109 is a bacterial CoA-binding protein from the hyperthermophile Pyrococcus horikoshii. In contrast to many structurally characterized ankyrin repeat binding proteins, PH1109 and mutants thereof are relatively small proteins (∼16 kDa) that are thermostable and express very well in E. coli. Additionally, PH1109 can reliably be mutated at the putative ankyrin repeat binding interface without fear of distorting the overall fold.27 All of these are valuable features when considering a scaffold for generating novel protein–protein interactions.
The development of the Pdar–Prb complex highlights both the power and current limitations of in silico methods, as well as the utility of high-throughput screening and/or macromolecular evolution. For example, while rigid-body docking in silico provided a valuable protein scaffold and in silico design of the complex provided a valuable starting point for its optimization, the reported crystal structure of this protein–protein complex is significantly different from the in silico design. Additionally, highest affinity complexes were identified through the application of macromolecular evolution experiments.27
Using shape complementarity and privileged scaffold resurfacing as design principles, we hypothesized that Prb-derived proteins could be generated to selectively recognize gankyrin. The amino acid backbone of gankyrin and Pdar align with a backbone root-mean-square deviation (rmsd) value of 0.69 Å over all Pdar residues (Supporting Information, Figure S1). If a Prb-derived protein binds gankyrin in a manner that is similar to the Pdar–Prb complex, the binding face residues on the Prb-derived protein would likely engage large regions of gankyrin and therefore might be able to compete with, or inhibit, disease-relevant complexes involving gankyrin. However, residues on the concave binding face and loop regions of Pdar and gankyrin are only ∼12% sequence homologous (Supporting Information, Figure S2). Furthermore, analysis of the binding face on Pdar reveal a large number of hydrophobic residues and an extensive hydrophobic patch. In contrast, solvent exposed residues on the putative binding face of gankyrin are primarily polar or charged. This suggests that extensive resurfacing of Prb is required to achieve selective and potent recognition of gankyrin. Additionally, these observations indicate that the generation of a novel gankyrin-binding protein is a substantial molecular recognition challenge since binding is unlikely to driven, primarily, by hydrophobic effects. Evaluation of the Prb–Pdar complex revealed eight Prb residues that directly engage, or are nearby, the surface of Pdar (N83, A85, K86, W88, R89, Y110, P111, and L112; Figure 1C). We reasoned that if a binding mode similar to that observed in the Pdar–Prb interaction is utilized, mutation of these residues might result in new proteins that selectively recognize gankyrin. The remaining question is how to best identify new gankyrin-binding proteins from this library.
We recently described split-superpositive green fluorescent protein (split-spGFP) reassembly as a robust and efficient method for identifying protein–protein interactions in living cells (Figure 2A).28 In previously reported split-GFP systems,29−31 the split-GFP fragments are susceptible to aggregation, resulting in relatively low efficiency of protein–protein interaction-dependent reassembly. In contrast, supercharged proteins, including supercharged GFP, are resistant to aggregation by virtue of their high theoretical net charge.32 Thus, split-spGFP fragments likely resist aggregation, which allows for efficient interaction-dependent reassembly.
Figure 2.

(A) Interaction-dependent reassembly of split-superpositive GFP fragments to generate active (fluorescent) GFP. (B) Flow cytometry data showing GFP fluorescence in E. coli coexpressing Gankyrin-CspGFP and GBP 1–7-NspGFP or a Pdar/Prb positive control. (C) ELISA data. Targets of GBPs are colored as follows: gankyrin (blue); Pdar (green); Notch-1 (red). (D) Coomassie-stained PAGE following copurification of Gankyrin-His6x and untagged Gankyrin binding protein 3, 5, or 7 (GBP 3, 5, or 7). (E) Flow cytometry data showing GFP fluorescence in E. coli that express Gankyrin-CspGFP/S6 ATPase and GBP 5-NspGFP or GBP 7-NspGFP. Error bars in panel C represent the standard deviation from three independent experiments.
Using standard molecular biology techniques, we prepared a DNA library that encodes Prb with eight randomized binding face residues (shown in Figure 1C). This DNA library was cloned into a pET plasmid as a fusion to N-spGFP. We also cloned gankyrin into a pBad plasmid as a fusion to C-spGFP. These two plasmids were sequentially transformed into E. coli, generating a library of ∼5 × 109 transformants. Sequencing ∼50 library plasmids from our library suggested very efficient randomization of the Prb binding face, as we did not observe any duplicate sequences in this region. Doubly transformed E. coli were made to concomitantly express the Prb library-NspGFP fusion and gankyrin-CspGFP fusion proteins, and incubated at 30 °C for 6 h. After such time, E. coli with the highest levels of GFP (indicating interaction-dependent GFP reassembly) were isolated by fluorescence-activated cell sorting (FACS). Following two rounds of screening, we individually rescreened seven resurfaced shape complementary proteins, which bind gankyrin living cells (in E. coli, Figure 2B). These proteins are herein referred to as gankyrin binding protein 1–7 (GBP 1–7). While all seven of these resurfaced proteins bind gankyrin (as determined by split-spGFP reassembly), we focused on the five best performing proteins (GBPs 1, 3, 4, 5, and 7).
We further characterized binding by an enzyme-linked immunosorbant assay (ELISA), which, in our hands, is more stringent than split-spGFP reassembly. As seen in Figure 2C, GBP 3, GBP 5, and GBP 7 appear to strongly bind gankyrin, while other GBPs are much poorer binders. Importantly, GBP 5 and GBP 7 do not appreciably bind off-target ankyrin repeats Pdar (green bars) and Notch-1 (red bars), which exhibit very high structural homology with gankyrin (backbone atom rmsd = 0.69 and 1.27 Å, respectively; Figure S3, Supporting Information), but differ dramatically with respect to the makeup of amino acids on their concave binding face. Pdar and Notch-1 exhibit ∼12% and ∼9% sequence homology, respectively, with the concave binding face of Gankyrin (Figures S2 and S4, Supporting Information).
Binding was further confirmed by measuring the amount of GBP that is copurified with His6x-tagged gankyrin from E. coli cell lysate.33E. coli was induced to coexpress His6x-tagged gankyrin and untagged GBP 3, 5, or 7. Cleared cell lysate was incubated with Ni-NTA agarose, followed by washing steps and release of His6x-tagged gankyrin by the addition of imidazole. Gankyrin or gankyrin-GBP copurified complexes were identified by gel electrophoresis and coomassie staining. As seen in Figure 2D, appreciable levels of copurified GBP 5 and GBP 7 were observed, while much lower levels of GBP 3 copurified with gankyrin, suggesting that GBP 5 and 7 are the highest affinity GBPs and warrant further study. The relative absence of other copurified cellular proteins further demonstrates the high level of selectivity that is achieved in these newly identified protein–protein interactions.
As stated previously, S6 ATPase does not express independently as a soluble protein. The gankyrin-S6 ATPase complex is only generated by coexpressing these two proteins from a single pET-DUET plasmid.15 In order to determine if GBP 5 or GBP 7 bind gankyrin in the presence of S6 APTase or inhibit this physiological interaction, we performed a modified split-spGFP experiment. We coexpressed gankyrin-CspGFP and S6 ATPase from pET-DUET and GBP 5- or GBP7-NspGFP from pBad, in E. coli. Since gankyrin and S6 ATPase assemble when coexpressed, we reasoned that gankyrin-GBP interaction-dependent reassembly of the fused spGFP fragments would only occur if GBP 5 or GBP 7 bind gankyrin over S6 ATPase or recognize a region of gankryrin that differs from S6 ATPase. We observe virtually identical amounts of gankyrin interaction-dependent GFP signal in E. coli that coexpress gankyrin-CspGFP/S6 ATPase and GBP5-NspGFP or GBP7-NspGFP (Figure 2E).
To assess the contribution of each residue on the resurfaced region of GBP 5 and GBP 7, we performed pull-down experiments from E. coli cell lysate containing His6x-tagged gankyrin and untagged alanine mutants of each gankyrin-binding protein. In each pull-down experiment, a single residue that was randomized in construction of the protein library was mutated to alanine (with the exception of glycine 83 in GBP 5, which we viewed as a minor change unlikely to dramatically alter complex stability). Consistent with our ELISA data, gankyrin does not bind appreciable levels of Prb (Figure 3A, lane 1), but does copurify with GBP 5 (Figure 3A, lane 2). Three mutations to the resurfaced region, R85A, N110A and W111A, significantly decreased the amount of copurified mutant GBP (Figure 3A, lanes 3, 7, and 8, respectively), suggesting these residues are particularly critical for gankyrin recognition. For GBP 7, Y83A, I85A, and W86A, mutations resulted in significantly decreased levels of copurified mutant GBP (Figure 3B, lanes 3, 4, and 5). For GBP 5 and GBP 7, mutations that result in significantly lower levels of copurification are tightly grouped and different, suggesting unique recognition “hot spots” (Figures 3C,D).
Figure 3.

(A) Coomassie-stained PAGE following copurification of gankyrin-His6x and untagged Prb, gankyrin binding protein 5 (GBP 5), and alanine mutants thereof (stated below the gel). (B) Coomassie-stained PAGE following copurification of gankyrin-His6x and untagged Prb, gankyrin binding protein 7 (GBP 7), or alanine mutants thereof (stated below the gel). (C) Binding face of GBP 5, with key gankyrin-binding residues highlighted in green. (D) Binding face of GBP 7, with key gankyrin-binding residues highlighted in green. Structures shown in panels C and D are of the putative binding face of Prb, which is the starting point for our protein resurfacing. These representations are not intended to provide any information on structural features of GBP 5 or GBP 7, or alanine mutants thereof, but rather to graphically represent where mutations deleterious to gankyrin binding reside on GBP 5 and GBP 7. Taken together, these depictions indicate where binding “hot spots” are on the resurfaced proteins GBP 5 and GBP 7, as determined by our pull-down data in panels A and B.
In the overwhelming majority of our data (split-spGFP reassembly, ELISA, and His6x copurification) GBP 7 appears to have the highest affinity for gankyrin. We measured the solution phase dissociation constant (KD) between gankyrin and GBP 7 by isothermal titration calorimetry (ITC). This resurfaced shape complementary protein binds gankyrin with good affinity (KD ≈ 6.1 μM; Figure 4A). The observed change in enthalpy (ΔH) and entropy (ΔS) for this binding interaction were −2.78 kcal/mol and 14.6 cal/mol·K, respectively. Consistent with our previous data, GBP 5 binds gankryin, but with lower affinity. We observed an unsaturated binding isotherm under identical conditions that were used to measure the GBP 7–gankyrin interaction (data not shown). Since GBP 5 and GBP 7 are derived from a protein natively expressed in the hyperthermophile Pyrococcus horikoshii, these proteins are likely to be very thermostable, a desired characteristic of protein reagents. We measured the thermostability of Prb (our original PH1109-derived scaffold) and resurfaced gankyrin-binding mutants GBP 5 and GBP 7 by differential scanning calorimetry (DSC). Impressively, Prb exhibits a very high melting temperature (Tm) of 91.1 °C. Despite extensive mutagenesis, both resurfaced mutants GBP 5 and GBP 7 retain excellent thermostability (Tm ≈ 86.8 and 87.1 °C, respectively; Figure 4B).
Figure 4.

(A) Isothermal titration calorimetry binding isotherm for gankyrin and gankyrin binding protein 7 (GBP 7). (B) Differential scanning calorimetry data for ankyrin repeat-binding proteins Prb (black), gankyrin binding protein 7 (GBP 7, red), and gankyrin binding protein 5 (GBP 5, blue).
In conclusion, limitations to small molecule reagents and drug leads require fundamentally new approaches to the recognition of disease-relevant receptors. The size, electrostatic complexity, and relatively featureless surfaces associated with many protein–protein interactions involving disease-relevant ankyrin repeat domains present a particularly difficult challenge for small molecule reagents. Synthetic proteins offer a unique opportunity to recognize–and potentially modulate the activity–of challenging macromolecular targets such as ankyrin repeats. Here, we described novel synthetic proteins that selectively and potently recognize the oncoprotein ankyrin repeat gankyrin. Split-superpositive GFP reassembly, ELISA, and cell lysate pull-down experiments suggest that these interactions occur in living cells and are highly selective. These new gankyrin-binding proteins are thermostable, express well in E. coli as soluble proteins, and represent the first synthetic proteins that recognize gankyrin in vitro and in complex cellular environments. These proteins likely represent valuable starting points for further optimizing affinity to gankyrin and modulating gankyrin-dependent oncogenic cell function and fate. Efforts toward this end are currently underway and will be reported in due course.
Methods
See the Supporting Information for complete methods.
Acknowledgments
This work was supported by the Colorado State University Cancer Supercluster and the National Institutes of Health (R01GM107520). A.C. was supported in part by a fellowship named in honor of Professor L. Hegedus. We thank M. Beach (GE Healthcare) for assistance with ITC and DSC experiments.
Supporting Information Available
Experimental Methods and supplementary data (Figures S1–S4). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Stockwell B. R. (2000) Chemical genetics: ligand-based discovery of gene function. Nat. Rev. Genet. 1, 116–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell B. R. (2000) Frontiers in chemical genetics. Trends Biotechnol. 18, 449–455. [DOI] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. (2006) How many drug targets are there?. Nat. Rev. Drug Discovery 5, 993–996. [DOI] [PubMed] [Google Scholar]
- Li J.; Mahajan A.; Tsai M. D. (2006) Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45, 15168–15178. [DOI] [PubMed] [Google Scholar]
- Kim Y. H.; Kim J. H.; Choi Y. W.; Lim S. K.; Yim H.; Kang S. Y.; Chung Y. S.; Lee G. Y.; Park T. J. (2013) Gankyrin is frequently overexpressed in breast cancer and is associated with ErbB2 expression. Exp. Mol. Pathol. 94, 360–365. [DOI] [PubMed] [Google Scholar]
- Zhen C.; Chen L.; Zhao Q.; Liang B.; Gu Y. X.; Bai Z. F.; Wang K.; Xu X.; Han Q. Y.; Fang D. F.; Wang S. X.; Zhou T.; Xia Q.; Gong W. L.; Wang N.; Li H. Y.; Jin B. F.; Man J. H. (2013) Gankyrin promotes breast cancer cell metastasis by regulating Rac1 activity. Oncogene 32, 3452–3460. [DOI] [PubMed] [Google Scholar]
- Fu X. Y.; Wang H. Y.; Tan L.; Liu S. Q.; Cao H. F.; Wu M. C. (2002) Overexpression of p28/gankyrin in human hepatocellular carcinoma and its clinical significance. World J. Gastroenterol. 8, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Knobloch T. J.; Kresty L. A.; Zhang Z.; Lang J. C.; Schuller D. E.; Weghorst C. M. (2011) Gankyrin, a biomarker for epithelial carcinogenesis, is overexpressed in human oral cancer. Anticancer Res. 31, 2683–2692. [PubMed] [Google Scholar]
- Meng Y.; He L.; Guo X.; Tang S.; Zhao X.; Du R.; Jin J.; Bi Q.; Li H.; Nie Y.; Liu J.; Fan D. (2010) Gankyrin promotes the proliferation of human pancreatic cancer. Cancer Lett. 297, 9–17. [DOI] [PubMed] [Google Scholar]
- Tang S.; Yang G.; Meng Y.; Du R.; Li X.; Fan R.; Zhao L.; Bi Q.; Jin J.; Gao L.; Zhang L.; Li H.; Fan M.; Wang Y.; Wu K.; Liu J.; Fan D. (2010) Overexpression of a novel gene gankyrin correlates with the malignant phenotype of colorectal cancer. Cancer Biol. Ther. 9, 88–95. [DOI] [PubMed] [Google Scholar]
- Ortiz C. M.; Ito T.; Tanaka E.; Tsunoda S.; Nagayama S.; Sakai Y.; Higashitsuji H.; Fujita J.; Shimada Y. (2008) Gankyrin oncoprotein overexpression as a critical factor for tumor growth in human esophageal squamous cell carcinoma and its clinical significance. Int. J. Cancer 122, 325–332. [DOI] [PubMed] [Google Scholar]
- Man J. H.; Liang B.; Gu Y. X.; Zhou T.; Li A. L.; Li T.; Jin B. F.; Bai B.; Zhang H. Y.; Zhang W. N.; Li W. H.; Gong W. L.; Li H. Y.; Zhang X. M. (2010) Gankyrin plays an essential role in Ras-induced tumorigenesis through regulation of the RhoA/ROCK pathway in mammalian cells. J. Clin. Invest. 120, 2829–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J. N.; Tsai M. D. (2002) Novel insights into the INK4-CDK4/6-Rb pathway: Counter action of gankyrin against INK4 proteins regulates the CDK4-mediated phosphorylation of Rb. Biochemistry 41, 3977–3983. [DOI] [PubMed] [Google Scholar]
- Higashitsuji H.; Higashitsuji H.; Itoh K.; Sakurai T.; Nagao T.; Sumitomo Y.; Masuda T.; Dawson S.; Shimada Y.; Mayer R. J.; Fujita J. (2005) The oncoprotein gankyrin binds to MDM2/HDM2, enhancing ubiquitylation and degradation of p53 (vol 8, pg 75, 2005). Cancer Cell (8), 169–169. [DOI] [PubMed] [Google Scholar]
- Nakamura Y.; Nakano K.; Umehara T.; Kimura M.; Hayashizaki Y.; Tanaka A.; Horikoshi M.; Padmanabhan B.; Yokoyama S. (2007) Structure of the oncoprotein gankyrin in complex with S6 ATPase of the 26S proteasome. Structure 15, 179–189. [DOI] [PubMed] [Google Scholar]
- Higashitsuji H.; Itoh K.; Nagao T.; Dawson S.; Nonoguchi K.; Kido T.; Mayer R. J.; Arii S.; Fujita J. (2000) Reduced stability of retinoblastoma protein by gankyrin, an oncogenic ankyrin-repeat protein overexpressed in hepatomas. Nat. Med. 6, 96–99. [DOI] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. (1996) Principles of protein-protein interactions. Proc. Natl. Acad. Sci., U.S.A. 93, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Rahman N.; Martinez-Arias A.; Blundell T. L. (2011) Probing the druggability of protein-protein interactions: targeting the Notch1 receptor ankyrin domain using a fragment-based approach. Biochem. Soc. Trans. 39, 1327–1333. [DOI] [PubMed] [Google Scholar]
- Cronican J. J.; Beier K. T.; Davis T. N.; Tseng J. C.; Li W.; Thompson D. B.; Shih A. F.; May E. M.; Cepko C. L.; Kung A. L.; Zhou Q.; Liu D. R. (2011) A class of human proteins that deliver functional proteins into mammalian cells in vitro and in vivo. Chem. Biol. 18, 833–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronican J. J.; Thompson D. B.; Beier K. T.; McNaughton B. R.; Cepko C. L.; Liu D. R. (2010) Potent delivery of functional proteins into mammalian cells in vitro and in vivo using a supercharged protein. ACS Chem. Biol. 5, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePorter S. M.; Lui I.; Bruce V. J.; Gray M. A.; Lopez-Islas M.; McNaughton B. R. (2014) Mutagenesis modulates the uptake efficiency, cell-selectivity, and functional enzyme delivery of a protein transduction domain. Mol. BioSyst. 10, 18–23. [DOI] [PubMed] [Google Scholar]
- DePorter S. M.; Lui I.; Mohan U.; McNaughton B. R. (2013) A protein transduction domain with cell uptake and selectivity profiles that are controlled by multivalency effects. Chem. Biol. 20, 434–444. [DOI] [PubMed] [Google Scholar]
- Deshayes S.; Morris M. C.; Divita G.; Heitz F. (2005) Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell. Mol. Life. Sci. 62, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S. M.; Raines R. T. (2005) Polyarginine as a multifunctional fusion tag. Protein Sci. 14, 1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S. M.; Raines R. T. (2007) Arginine grafting to endow cell permeability. ACS Chem. Biol. 2, 167–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton D. R.; Weiss R. A. (2010) A boost for HIV vaccine design. Science 329, 770–773. [DOI] [PubMed] [Google Scholar]
- Karanicolas J.; Corn J. E.; Chen I.; Joachimiak L. A.; Dym O.; Peck S. H.; Albeck S.; Unger T.; Hu W.; Liu G.; Delbecq S.; Montelione G. T.; Spiegel C. P.; Liu D. R.; Baker D. (2011) A de novo protein binding pair by computational design and directed evolution. Mol. Cell 42, 250–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeley B. D.; Chapman A. M.; McNaughton B. R. (2012) Split-superpositive GFP reassembly is a fast, efficient, and robust method for detecting protein-protein interactions in vivo. Mol. BioSyst. 8, 2036–2040. [DOI] [PubMed] [Google Scholar]
- Magliery T. J.; Regan L. (2006) Reassembled GFP: detecting protein-protein interactions and protein expression patterns. Methods Biochem. Anal. 47, 391–405. [DOI] [PubMed] [Google Scholar]
- Magliery T. J.; Wilson C. G.; Pan W.; Mishler D.; Ghosh I.; Hamilton A. D.; Regan L. (2005) Detecting protein–protein interactions with a green fluorescent protein fragment reassembly trap: scope and mechanism. J. Am. Chem. Soc. 127, 146–157. [DOI] [PubMed] [Google Scholar]
- Wilson C. G.; Magliery T. J.; Regan L. (2004) Detecting protein–protein interactions with GFP-fragment reassembly. Nat. Methods 1, 255–262. [DOI] [PubMed] [Google Scholar]
- Lawrence M. S.; Phillips K. J.; Liu D. R. (2007) Supercharging proteins can impart unusual resilience. J. Am. Chem. Soc. 129, 10110–10112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a similar pull-down experiment, seeRoot M. J.; Kay M. S.; Kim P. S. (2001) Protein design of an HIV-1 entry inhibitor. Science 291, 884–888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.