

Abstract
RNase P is a ubiquitous endonuclease that removes the 5′ leader sequence from pre-tRNAs in all organisms. In Arabidopsis thaliana, RNA-free proteinaceous RNase Ps (PRORPs) seem to be enzyme(s) for pre-tRNA 5′-end processing in organelles and the nucleus and are thought to have replaced the ribonucleoprotein RNase P variant. However, the evolution and function of plant PRORPs are not fully understood. Here, we identified and characterized three PRORP-like proteins, PpPPR_63, 67, and 104, in the basal land plant, the moss Physcomitrella patens. PpPPR_63 localizes to the nucleus, while PpPPR_67 and PpPPR_104 are found in both the mitochondria and chloroplasts. The three proteins displayed pre-tRNA 5′-end processing activity in vitro. Mutants with knockout (KO) of the PpPPR_63 gene displayed growth retardation of protonemal colonies, indicating that, unlike Arabidopsis nuclear RPORPs, the moss nuclear PpPPR_63 is not essential for viability. In the KO mutant, nuclear-encoded tRNAAsp (GUC) levels were slightly decreased, whereas most nuclear-encoded tRNA levels were not altered. This indicated that most of the cytosolic mature tRNAs were produced normally without proteinaceous RNase P-like PpPPR_63. Single PpPPR_67 or 104 gene KO mutants displayed different phenotypes of protonemal growth and chloroplast tRNAArg (ACG) accumulation. However, the levels of all other tRNAs were not altered in the KO mutants. In addition, in vitro RNase P assays showed that PpPPR_67 and PpPPR_104 efficiently cleaved chloroplast pre-tRNAArg (CCG) and pre-tRNAArg (UCU) but they cleaved pre-tRNAArg (ACG) with different efficiency. This suggests that the two proteins have overlapping function but their substrate specificity is not identical.
Introduction
RNase P is an endonuclease that removes the 5′ leader sequence from precursor tRNAs (pre-tRNAs). This endonucleolytic cleavage is an essential step in the production of mature tRNAs in all organisms, as well as in mitochondria and chloroplasts [1], [2]. Bacterial, archaeal, and eukaryotic nuclear RNase P enzymes are ribonucleoprotein complexes composed of a catalytic RNA component and one or several proteins [3]. Therefore, it was believed that 5′-maturation of tRNAs in all organisms is catalyzed by ubiquitous ribonucleoprotein (RNP) RNase P enzymes. However, human mitochondrial RNase P was identified as an RNA-free enzyme composed of three proteins called mitochondrial RNase P protein 1 (MRPP1), MRPP2, and MRPP3 [4]. MRPP3 contains two RNA-binding pentatricopeptide repeat (PPR) motifs and a conserved NYN metallonuclease domain [5], which are involved in the catalytic activity of mitochondrial RNase P. In a model plant, Arabidopsis thaliana, three MRPP3 homologs have been identified as RNase P enzymes; they are termed proteinaceous RNase P 1 (PRORP1), PRORP2, and PRORP3 [6]. In vitro cleavage assays using recombinant PRORP proteins have demonstrated that three PRORP proteins display RNase P activity [6], [7]. PRORPs are single-protein RNase P enzymes, whereas human mitochondrial RNase P is a multisubunit enzyme. PRORP1 localizes in both chloroplasts and mitochondria, whereas PRORP2 and 3 localize in the nucleus. PRORP1 is an essential gene, because prorp1 mutants are embryonic lethal [6]. Single PRORP2 or PRORP3 knockout mutant lines show wild-type phenotypes, whereas the homozygous double mutation in prorp2 prorp3 results in embryonic lethality [7]. This indicates that PRORP2 and 3 have redundant functions and are essential for embryogenesis and plant growth.
The Arabidopsis and rice genomes do not encode RNase P RNA, whereas they do code for several homologs of RNP RNase P protein subunits found in mammals and yeasts [8]. The presence of ribonucleoprotein RNase P enzyme in plants remains obscure. Recently, Gutmann et al. (2012) suggested that Arabidopsis had entirely replaced the ribonucleoprotein RNase P enzyme by PRORP enzymes for tRNA maturation during plant evolution [7]. However, functional characterization of PRORP proteins is limited in Arabidopsis.
Unlike in land plant organelles, an RNA subunit of bacterial-type RNase P is encoded in the organellar genomes of the Glaucophyta (Cyanophora paradoxa) [9], the red alga Poryphyra purpurea [10], and green algae (Nephroselmis olivacea, Ostreococcus tauri [11], [12]. These algae also possess a single nuclear gene for PRORP [6]. In C. paradoxa, RNase P activity in an extract of the photosynthesis organelle (cyanelle) is sensitive to micrococcal nuclease [13], [14], indicating the existence of bacterial-type RNase P. The nuclear-encoded PRORP in the green alga O. tauri has been shown to cleave the 5′ leader of pre-tRNA in vitro [12], [15]. Thus, algae seem to possess both a ribonuceoprotein RNase P and a PRORP. However, the latter is not known to localize in either the organelles or the nucleus.
From these differences in the status of PRORP enzymes in algae and plants, the following questions have arisen: When were plant PRORP genes duplicated during plant evolution, which PRORP protein was targeted to the organelles and which to the nucleus, and was ribonucleoprotein RNase P enzyme replaced by PRORP in plants? To answer these questions, we focused on PRORPs of the basal land plant bryophyte Physcomitrella patens. Here, we report the molecular characterization of three P. patens PRORP-like proteins (PpPPR_63, 67, and 104) that are members of the PPR protein family [16], [17]. We also report the disruption mutants of each gene and show that nuclear-localized PpPPR_63 is dispensable for moss viability and that organelle-localized PpPPR_67 and PpPPR_104 have functions that are redundant, but not completely so.
Results
The moss P. patens has three PRORP homologs
Among 105 Physcomitrella PPR proteins [17], PpPPR_63 (protein ID, Phypa_174001), PpPPR_67 (Phypa_177191) and PpPPR_104 (Phypa_152956) appeared to be PRORP homologs (Fig. 1A), because these three PPR proteins contained at least two PPR motifs and an NYN metallonuclease domain and had 42%–53% amino acid identity with Arabidopsis PRORP proteins. Although algae and Chara have a single PRORP homolog, mosses and vascular plants have two or three PRORP homologs (Fig. 1B and Table S1). This suggests that PRORP genes were duplicated after the emergence of land plants.
Figure 1. PRORP proteins are widely distributed among eukaryotes.
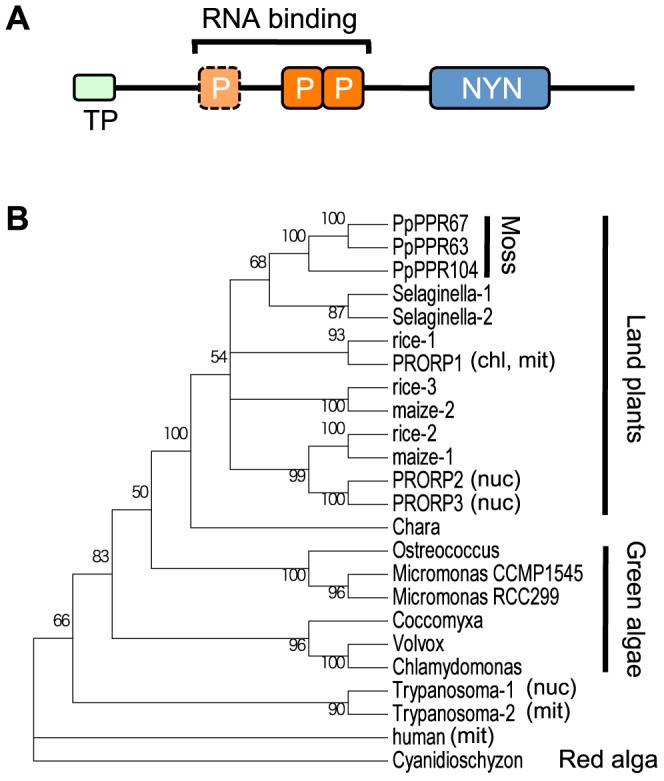
(A) Schematic diagram of plant organellar PRORPs with a transit peptide (TP), RNA binding PPR motifs (P), and NYN metallonuclease domain (NYN). (B) Neighbor-joining phylogenetic tree of PRORPs and PRORP-like proteins. Representative PRORP protein sequences from evolutionarily distant plants were used for the phylogenic analysis. Experimentally determined localization of PRORPs is presented as chloroplast (chl), mitochondria (mit), or nucleus (nuc). Bootstrap values>50 are indicated along branches. Protein ID or accession numbers of PRORPs and PRORP-like proteins are listed in Table S1.
PpPPR_63 is localized in the nucleus, whereas PpPPR_67 and 104 are found in the organelles
PpPPR_67 and 104 possess an N-terminal transit peptide-like sequence targeting the organelles, whereas PpPPR_63 does not. This suggests that PpPPR_67 and 104 are likely organelle-localized PRORP1 homologs, whereas PpPPR_63 seems to be a nuclear-localized PRORP2/PRORP3 homolog. To confirm this prediction we generated PpPPR_63-green fluorescent protein (GFP) knockin (KI) mosses. The four independent KI lines obtained showed normal growth (Fig. S1), and GFP fluorescence was observed in the nuclei (Fig. 2A, b and c). GFP fluorescence was observed in the protonemal cells, buds, and young leaves (Fig. 2A, d–i) but not in mature leaves (Fig. 2A, j and k).
Figure 2. Localization of PpPPR_63, 67, and 104.

(A) Observations of GFP fluorescence in various cells and tissues of the stable transgenic moss KI-10 line expressing PpPPR_63-GFP fusion protein. Schematic diagram of a moss plant and observed parts are in circles (a). Images are differential interference contrast (DIC) (b, d, f, h, j) and GFP (green) and chlorophyll (magenta) fluorescence (c, e, g, i, k). Single protonemal cell (b, c), protonemal cells (d, e), a bud (f, g), a young leaf (h, i), and mature leaf cells (j, k). Scale bars = 5 µm (b, c) or 20 µm (d–k). (B) Subcellular localization of N-terminal PpPPR_67-GFP fusion protein (N67-GFP, upper panels) or N-terminal PpPPR_104-GFP fusion protein (N104-GFP, bottom panels). For mitochondrial localization control, the γ subunit of mitochondrial ATPase and RFP fusion protein (Mt-RFP) was used. Fluorescence of GFP and RFP, the overlay of the two fluorescence images (Merged), and the corresponding Nomarski images (DIC) are shown.
To verify the organellar localization of PpPPR_67 and 104, their N-terminal sequences fused to GFP (N67-GFP and N104-GFP) were transiently expressed in the protonemata (Fig. 2B). GFP fluorescence of N67-GFP and N104-GFP was observed in both mitochondria and chloroplasts, coinciding with mitochondria-localized red fluorescent protein (Mt-RFP) signals and Nomarski images of chloroplasts (DIC).
PpPPR_63, 67, and 104 have RNase P activity
To investigate whether the three Physcomitrella PRORP-like PPR proteins are RNase P enzymes, we performed in vitro RNase P assays with recombinant proteins composed of the respective PRORP-like protein fused with thioredoxin at the N-terminus and six histidines and a VP5 tag at the C-terminus (Fig. 3A, B). The three recombinant proteins, r67, r104, and r63, removed the 5′ leader sequences from chloroplast pre-tRNAPhe, mitochondrial pre-tRNACys, and nuclear pre-tRNAAsp (Fig. 3C). The 5′-end of the tRNA produced was verified by primer extension analysis (Fig. 3D, E). We also confirmed the precise cleavage of pre-tRNAs by circular reverse transcription (cRT)-polymerase chain reaction (PCR) analysis (data not shown). To verify the RNase P activity of these proteins, we also tested two recombinant proteins, rM63 and rC63. rM63 possessed a mutated NYN domain, the two adjacent aspartates of which (D460 and D461) were altered to alanines. These two aspartates have been shown to be essential for the activity of Arabidopsis PRORP1 [6]. The rC63 was composed of the tag sequences and the NYN domain only. The rM63 and rC63 proteins did not cleave nuclear-encoded pre-tRNAAsp (Fig. 3C). These results indicated that the three moss PPR proteins had RNase P activity similar to that of Arabidopsis PRORPs [6], [7], [18].
Figure 3. Pre-tRNA cleavage assay of P. patens PRORP-like proteins.

(A) Schematic diagrams of various recombinant proteins. Recombinant proteins r67, r104, r63, and rM63 indicate, respectively, PpPPR_67, PpPPR_104, PpPPR_63, and the mutant PpPPR_63, the two catalytic aspartates (DD) of which were substituted with alanines (AA). rC63 consists of the tag sequences and an NYN domain only of PpPPR_63. (B) Coomassie brilliant blue (CBB)-stained recombinant proteins (1 µg each). (C) RNase P activity was assayed by using 2 µg of in vitro-transcribed chloroplast (chl) pre-tRNAPhe, mitochondrial (mit) pre-tRNACys, or nuclear (nuc) pre-tRNAAsp and 100 ng of recombinant proteins. The reaction products were separated by using 8% denaturing PAGE and stained with ethidium bromide. (D) The 5′-ends of processed pre-tRNAs in (C) were determined by primer extension analysis. Processed pre-tRNAs without proteins (lanes 1 and 5), with r67 (lanes 2 and 6), with r104 (lanes 3 and 7), or with r63 (lanes 4 and 8) were reverse transcribed from the 5′-end-labeled primers underlined in (E). (E) The nucleotide sequences of chloroplast (chl) pre-tRNAPhe and mitochondrial (mit) pre-tRNACys are shown with small letters (5′ leader and 3′ trailer sequences) and capital letters (predicted mature tRNA). Arrowheads indicate the 5′-end positions determined in (D). Underlined sequences indicate the position of primers used for primer extension.
Nuclear PpPPR_63 is dispensable for viability
To investigate the in vivo function of PpPPR_63, we generated two independent knockout (KO) mutants of PpPPR_63, Δ63-1 andΔ63-15. Both KO lines were verified to be null mutants. Gene structure of theΔ63-1 line is given as Figs. 4A and S2, and its absence of PpPPR_63 transcripts was verified by reverse transcription (RT)-PCR (Fig. 4B). Therefore, we used theΔ63-1 line for further analyses (hereafter named Δ63). In addition, to generate the complementation lines of theΔ63-1, we transformed the Δ63 moss with a transgene encoding the full length of PpPPR_63 or the mutated version and generated F63 and M63, respectively (Fig. S3). In M63, two aspartates (D460 and D461) in the NYN domain were mutated to alanines (A460 and A461). In the complementation lines F63 and M63, the introduced transgene was overexpressed (Fig. 4B). The KO mutantΔ63 and the complementation line M63 mosses displayed smaller protonemal colonies than the wild type (WT) (Fig. 4C), and they showed abnormal regeneration from leaves (especially defective growth of the sub-apical cells) (Fig. 4D). The complementation line F63 exhibited a phenotype the same as that of the WT. These results indicated that PpPPR_63 was responsible for the mutant phenotype.
Figure 4. Molecular and morphological phenotypes of the PpPPR_63 KO and complementation mosses.

(A) Schematic diagram of PpPPR_63 and construction of the KO locus. The nptII cassette was inserted into the 4th intron of the PpPPR_63 gene. (B) Detection of PpPPR_63 transcripts by RT-PCR. (C) Two-week-old protonemal colonies of the PpPPR_63 KO line (Δ63) and the complementation lines (F63 and M63). Bars = 5 mm. (D) One-week-old filamentous protonemata regenerated from cut leaves. (E) Steady-state levels of nuclear-encoded tRNAs.
Knockout of PpPPR_63 slightly affects specific nuclear-encoded tRNAs
If nuclear PpPPR_63 could function as a bona fide RNase P, knockout of the PpPPR_63 gene could result in severely reduced accumulation of nuclear-encoded tRNAs. To investigate this possibility, we analyzed the accumulation levels of several nuclear-encoded tRNAs in theΔ63 mutant. For this analysis nuclear-encoded tRNAAsp (GUC) and tRNAGln (CUG) were chosen, because expression of these tRNAs has been demonstrated to significantly decrease in the knockdown mutants of nuclear PRORP2/3 plants [7]. tRNAAsp (GUC) in theΔ63 mutant and M63 complementation line was slightly reduced to 70% of that in the WT and F63. Whereas, tRNAGln (CUG) in the Δ63 mutant, M63 and F63 complementation lines accumulated to the same level as in the WT (Fig. 4E). Similarly, nuclear-encoded tRNASer (AGA) and tRNALys (CUU) levels were not affected in the mutants. This suggests that PpPPR_63 PNase P activity may be limited to specific tRNA(s) including tRNAAsp (GUC).
Knockout of PpPPR_104 results in a significant reduction in chloroplast tRNAArg (ACG)
To investigate the in vivo function of the PpPPR_67 and 104 genes, we generated and characterized their KO mutants (Figs. 5A, S4, S5, S6). The single gene KO mutants obtained, Δ67 andΔ104, were null mutants (Fig. 5B), and the protonemal colony ofΔ104 moss was smaller than that of the WT (Fig. 5C). A peunumbra and central part of the protonemal colony mostly consists of caulonemal and chloronemal filaments, respectively. In theΔ104 moss, caulonemal filaments were poorly induced, resulting in a smaller colony. In contrast, PpPPR_67 KO moss (Δ67) showed little difference from the WT. To generate double KO mutants, we carried out four times transformation experiments, such as introducing the PpPPR_104 KO plasmid into the PpPPR_67 KO line (Δ67-1-17 line) and vice versa. However, no double KO mutants were obtained from the genotyping of 262 moss plants. This indicates that PpPPR_67 and 104 genes cannot be knocked out simultaneously. Since they may have redundant functions, single KO mutants are viable but double KO is probably lethal.
Figure 5. PpPPR_67 and PpPPR_104 KO mosses.

(A) Schematic structure of PpPPR_67 and 104 KO loci. The nptII cassette was inserted into the 5th exon of PpPPR_67 and the 4th exon of PpPPR_104. (B) Detection of PpPPR_67 and 104 transcripts by RT-PCR. (C) Two-week-old protonemata colonies of the PpPPR_67 KO line (Δ67) and the PpPPR_104 KO line (Δ104). Scale bars = 5 mm. (D) Steady-state levels of chloroplast (chl) tRNAArg isoacceptors with different anticodons, chl tRNAPhe (GAA) and mitochondrial (mit) tRNACys (GCA).
To investigate whether organellar tRNAs were affected in the PpPPR_67 and 104 KO mutants, we performed northern blot analyses by using all organelle-encoded tRNA probes. The chloroplast tRNAArg (ACG) level significantly decreased inΔ104 but not inΔ67 orΔ63 (Fig. 5D). All the other tRNAs, including chloroplast tRNAPhe (GAA) and mitochondrial tRNACys (GCA) that were used for the in vitro RNase P cleavage assay, were not altered in the WT and the three KO mutants (Figs. 5D, S7, S8). This result suggests that PpPPR_67 and PpPPR_104 have redundant functions as RNase P in moss organelles.
PpPPR_67 and 104 cleave chloroplast pre-tRNAArg (ACG) with different efficiency
Because chloroplast tRNAArg (ACG) levels decreased in the Δ104 but not in theΔ67, we assumed that PpPPR_104 and PpPPR_67 would have distinct RNase P activity against chloroplast pre-tRNAArg (ACG). To test this possibility, three chloroplast pre-tRNAArg isoacceptors were used as substrates for in vitro RNase P cleavage assay. This assay revealed that PpPPR_67 and 104 efficiently cleaved pre-tRNAArg (CCG) and pre-tRNAArg (UCU) but they cleaved pre-tRNAArg (ACG) with different efficiency (Fig. 6). These results suggest that the function of their RNase P is not completely redundant.
Figure 6. Pre-tRNA cleavage assay of the recombinant proteins r67 and r104.
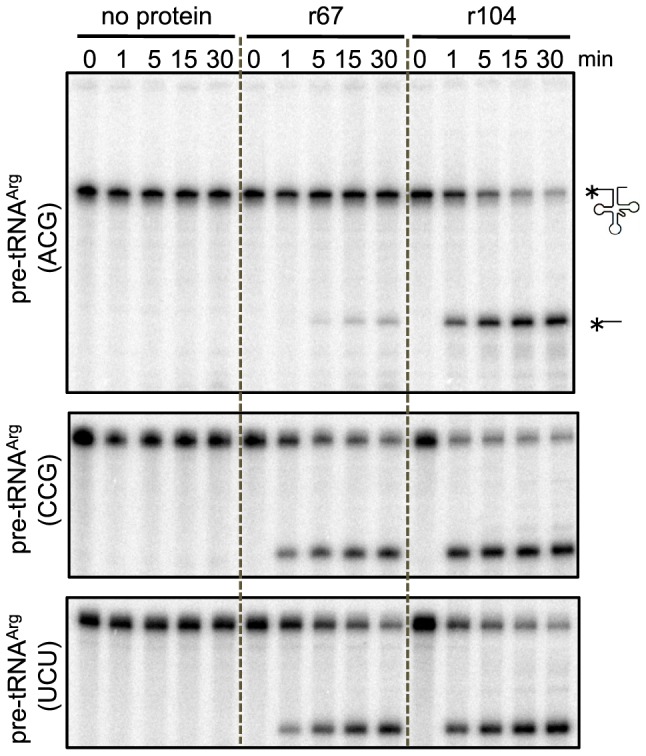
Reactions containing 500 nM of chloroplast 5′-32P-pre-tRNAArg (ACG) (upper), tRNAArg (CCG) (middle), or tRNAArg (UCU) (lower) and 500 nM of recombinant protein were quenched at 1, 5, 15, and 30 min points, resolved on 8% denaturing PAGE, and analyzed with a phosphoimager.
Discussion
The first two questions to be addressed here were: When were plant PRORP genes duplicated during plant evolution and where were the respective PRORP proteins localized or targeted to? Our present study provides evidence that PRORP genes were duplicated after the emergence of the early land plants, i.e. mosses. The moss P. patens has three PRORP-like proteins displaying RNase P activity in vitro. Arabidopsis PRORP1 is localized in both the mitochondria and chloroplasts, and PRORP2 and 3 are localized in the nucleus [6]. On the other hand, the moss P. patens has two PRORP1-like proteins, PpPPR_67 and 104, both of which are dual-targeted to the mitochondria and chloroplasts, and one nuclear-localized PRORP2/3-like PpPPR_63. Thus, duplicated PRORP gene products are likely targeted to either the organelles or the nucleus.
Nuclear PpPPR_63 is involved in normal plant growth and branch formation of protonemata
In Arabidopsis, the double KO mutation of PRORP2 and 3 resulted in embryonic lethality [6]. In contrast, a null mutant of PpPPR_63 displayed smaller protonemal colonies than the WT and also showed abnormal branch formation of filamentous protonemata (Fig. 4). This indicates that unlike Arabidopsis nuclear RPORPs, the moss nuclear PpPPR_63 is dispensable for plant viability. The nuclear-encoded tRNAAsp (GUC) level in the PpPPR_63 KO mutant was slightly decreased (to 70% of that in WT mosses), but most nuclear-encoded tRNA levels were not altered. This result indicates that most of the cytosolic mature tRNAs were produced normally, without proteinaceous RNase P-like PpPPR_63. This does not concur with the results observed from functional analysis of Arabidopsis PRORP2 and 3 [6], [7]. In Arabidopsis mutant plants where PRORP3 was absent and the PRORP2 level was decreased, the steady-state levels of five nuclear-encoded tRNAs investigated were significantly reduced, to 30% of the tRNA levels in control plants [7]. These observations indicate that Arabidopsis nuclear PRORP2 and 3 function as master RNase P enzymes, while the moss PpPPR_63 is not the sole RNase P enzyme in the nucleus.
It is unlikely that the slight reduction of the tRNAAsp (GUC) level resulted in growth retardation and abnormal branch formation in the PpPPR_63 KO mutant. PpPPR_63 may have a versatile function rather than pre-tRNA 5′-end processing. Arabidopsis PRORP proteins are involved in the maturation of not only tRNAs but also mRNA or snoRNA [7]. Similarly, PpPPR_63 is probably involved in the maturation of unknown mRNA or small RNA-containing tRNA-like structures.
Organelle-localized PpPPR_67 and 104 have functions not completely redundant for tRNA accumulation
In this study, we generated single KO mutants of PpPPR_67 and PpPPR_104 but failed to obtain double KO mutants. Similarly, KO mutation of Arabidopsis PRORP1 was shown to result in embryonic lethality [6]. This suggests that PpPPR_67 and 104 have redundant functions. However, it is interesting to observe that a single mutant of PpPPR_67 and 104 displayed different phenotypes of protonemal growth and chloroplast tRNAArg (ACG) accumulation. Chloroplast tRNAArg (ACG) significantly decreased in the PpPPR_104 KO mutant but not in PpPPR_67 KO mutants. The P. patens chloroplast genome codes for three tRNAArg isoacceptors [19]. An in vitro RNase P cleavage assay revealed that pre-tRNAArg (ACG) was more efficiently cleaved by PpPPR_104 than by PpPPR_67, whereas pre-tRNAArg (UCU) and pre-tRNAArg (CCG) were cleaved with similar efficiency by both proteins. This is the first evidence that PRORP enzymes have substrate specificity of tRNA processing. The anticodon of P. patens tRNAArg (ACG) is modified that including inosine in chloroplasts, and tRNAArg (ICG) can recognize all four codons. Chloroplast tRNAArg (CCG) recognizing only the GGC codon is dispensable [20]. Processing of chloroplast pre-tRNAArg (ACG) by PpPPR_104 is essential for the accumulation of mature tRNAArg (ACG). Therefore, the PpPPR_104 KO mutant may have displayed small protonemal colonies because of a great reduction in mature tRNAArg (ACG) levels.
Unlike Arabidopsis [6], [7], pre-tRNAs were hardly detected in the moss organelles (Figs. S7 and S8). It is conceivable that precursors might be turned over faster than mature tRNAs in mosses.
Was RNP-type RNase P enzyme entirely replaced by PRORP protein in mosses?
The last question is whether ribonucleoprotein RNase P enzyme was replaced by PRORP in both the organelles and the nucleus. In this study, we showed that disruption of PpPPR_63 did not result in a significant reduction in nuclear-encoded tRNAs. This raises the possibility that unidentified RNase P activity may contribute to pre-tRNA maturation in the moss nucleus (Fig. 7). However, it remains unknown whether 5′-end processing of nuclear pre-tRNAs is supported by RNP-type RNase P. In P. patens, an RNA component of RNP-type RNase P was not identified, yet but protein components (Pop4, Rpp1, Pop1, and Pop5) are encoded in the genome [21]. Identification of RNase P activity rather than PRORP1-type PpPPR_63 will be a challenge for the future.
Figure 7. Schematic summarizing the localization of three PRORP homologs,
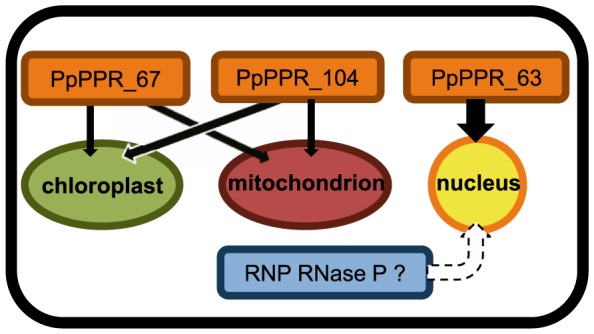
PpPPR_63, 67, and 104 in P. patens. This study suggested that unidentified RNase P enzyme(s) including RNP-type RNase P may be present at least in the nucleus.
Materials and Methods
Accession numbers
The sequences reported in this paper have been deposited in the DDBJ/GenBank/EMBL databases with accession numbers: PpPPR_63 mRNA, AB983707; PpPPR_67 mRNA, AB983708; PpPPR_104 mRNA, AB808584.
Plant material and preparation of DNA and RNA
The moss P. patens subsp. patens was grown at 25°C under continuous light (30 µmol photon m−2s−1), and genomic DNA and total cellular RNA were prepared from the moss protonemata as described previously [22].
Preparation of recombinant proteins
DNAs corresponding to the proteins (1–595 residues for PpPPR_63, 119–791 for PpPPR_67, and 125–993 for PpPPR104) were amplified by using the primers listed in Table S2 and cloned into pBAD/Thio-TOPO vector (Invitrogen). Proteins were expressed for 6 h at 18°C in E. coli BL21 and purified as described previously [23].
Preparation of pre-tRNA
DNAs representing precursors of chloroplast tRNAPhe, mitochondrial tRNACys, and nuclear tRNAAsp were amplified by PCR from P. patens genomic DNA with oligonucleotide pairs T7-cpF5 and cpF3, T7-mtC5 and mtC3, and T7-nuDGTC5 and nuDGTC, respectively (Table S2). The 5′ oligonucleotides contained the T7 promoter sequence. The pre-tRNAPhe, pre-tRNACys, and nuclear pre-tRNAAsp were respectively 142, 153, and 140 nt long and were designed with respective 5′ leaders of 42, 51, and 40 nt and 3′ trailers of 27, 32, and 25 nt. Amplified DNAs (200 ng) were transcribed with T7 RNA polymerase (TaKaRa) for 1 h at 37°C, then digested with 5 U of RNase-free DNase I (TaKaRa) and 20 U of RNase Inhibitor (TaKaRa) for 15 min at 37°C. Synthesized pre-tRNAs were purified by phenol chloroform extraction.
For preparation of chloroplast pre-tRNAArg isoacceptors, the corresponding genes were cloned and in vitro transcribed. The obtained pre-tRNAs were dephosphorylated with Thermo Sensitive Alkaline Phosphatase (Promega) and were 5′-end radiolabeled with [γ32P] ATP and polynucleotidekinase (TaKaRa). Labeled RNAs were purified from the gel after 8% polyacrylamide gel electrophoresis (PAGE) containing 7M urea.
Pre-tRNA cleavage assay
Cleavage assays were performed as described previously [7]. Recombinant protein (100 ng) and in vitro-synthesized pre-tRNA (2 µg) were incubated for 1 h at 25°C in a 20 µl mixture of 20 mM Tris-HCl (pH 8.0), 30 mM KCl, 4.5 mM MgCl2, 20 µg/ml bovine serum albumin, and 2 mM DTT. A half of the reaction mixture was separated on 8% polyacrylamide-7 M urea gels and stained with ethidium bromide.
For cleavage assays of three chloroplast pre-tRNAArg isoacceptors, the 5′ end-radiolabeled pre-tRNAs (500 nM each) and the recombinant proteins (500 nM each) were incubated and aliquots were withdrawn at the indicated times. Dried PAGE gels were analyzed by Storm 820 (GE Healthcare).
Determination of the 5′-end of tRNA
The 32P 5′-end-labeled oligonucleotides cp-F3 or mt-C3 (Table S2) and 200 ng of in vitro-cleaved RNAs were mixed and reverse transcribed with ReverTraAce (ToYoBo) for 1 h at 42°C, then digested with RNase A and precipitated with ethanol. The sequence ladders were obtained by using template DNA, the radiolabled primer cp-F3 or mt-C3, as described previously [22].
Subcellular localization
The DNA region from the 6th intron to the last codon of the PpPPR_63 gene was inserted in-frame into the GFP coding region of pGFPmutNTPII (http://moss.nibb.ac.jp). The 3′-flanking region of PpPPR_63 was inserted downstream of the nptII cassette of pGFPmutNTPII. The resultant plasmid p63-GFPKI (Fig. S1) was linearized with NotI and introduced into P. patens by particle bombardment as described previously [23]. For N67-GFP and N104-GFP, cDNAs corresponding to an N-terminal part of the proteins (residues 1–112 and 1–105, respectively) were amplified by using the appropriate primers (Table S2) and cloned into pKSPGFP9 [23]. The resultant plasmid was cotransfected with pMt-RFP into P. patens protonemata and GFP and RFP fluorescence were observed as described previously [24].
Generation of KO lines
The 5′ region (2960 bp) of PpPPR_63, a 2958-bp region of PpPPR_67, and a 3032-bp region of PpPPR_104 were amplified from moss genomic DNA by PCR using the appropriate primers (Table S2) and cloned into pGEM-T Easy (Promega). The KO plasmid p63KO4-13A (Fig. S2) was generated by insertion of the nptII cassette into the 4th intron of the PpPPR_63 gene by using the GPS-M mutagenesis System (NEB). The KO plasmids p67KO-1 and p67KO-2 (Figs. S4 and S5) were generated by insertion of the nptII cassette into HindIII in the 4th exon, or into StuI in the 5th exon, respectively, of the PpPPR_67 gene. The KO plasmid p104KO (Fig. S6) was generated by insertion of the nptII cassette into the BglII site in the 4th exon of the PpPPR_104 gene. These KO plasmids were digested with NotI and introduced into P. patens by particle bombardment.
Generation of complementation lines
For F63, the PpPPR_63 cDNA coding region (1785 bp) amplified by PCR using specific primers (Table S2) was cloned into the SwaI site of the overexpression vector pOX9WZ1 [25]. The mutated PpPPR_63 (M63: D460A/D461A) was amplified from the F63 construct by using the primers 63DA-F and 63DA-R. Both constructs were linearized with NotI and introduced into the KO line Δ63.
Northern blot analysis
Total cellular RNA (5 µg) was separated on 10% polyacrylamide-7 M urea gels and transferred to nylon membranes. Hybridization and washing were performed at 42°C. The gene-specific oligonucleotide probes (Table S2) were labeled with DIG-ddUTP and terminal deoxynucleotidyl transferase (Roche). Signals were acquired with EZ-capture (ATTO).
Supporting Information
Generation of PpPPR_63 - GFP knockin (KI) mosses. (A) Schematic of insertion of p63-GFPKI into the 3′ terminal end of the PpPPR_63 coding region. Translated and untranslated regions are represented by black and white boxes, respectively. Primers P1 to P3 used for PCR, and the expected amplified DNA sizes, are shown. (B) PCR analysis using the indicated primer pairs. The predicted 4.5- and 1.8-kb fragments were amplified from the KI lines. (C) Protonemal colonies of wild-type (WT) and KI mosses grown for 2 weeks on BCDATG medium plates without antibiotics. KI-10 was used for further analyses (Fig. 2). Scale bars = 5 mm.
(ZIP)
Generation of PpPPR_63 knockout (KO) moss. (A) Schematic diagrams of the PpPPR_63 gene, the KO construct (p63KO4-13), and the generated knockout locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the 4th intron of PpPPR_63. Translated and untranslated regions are represented by black and white boxes, respectively. Primers P4 to P7 used for PCR and the expected amplified DNA sizes are shown. The DNA probe used in (C) is represented by grey bars. (B) PCR analysis using the indicated primer pairs. The predicted 1.9-, 2.2- and 5.5-kb fragments were amplified from the KO lines; 3.0 kb was amplified from the WT. (C) Total cellular DNAs (20 µg) from wild type (WT) and KO mosses were digested with SphI and hybridized with the PpPPR_63 probe in (A). The probe was labeled with DIG-dUTP. Predicted hybridized fragments were detected in WT and KO mosses.
(EPS)
Generation of PpPPR_63 complementation lines intoΔ 63 -1. (A) Schematic diagrams of PpPPR_63 loci of wild type andΔ63-1. Primers P8 and P9 and the expected fragment sizes for PCR analysis are shown. (B) The PpPPR_63 coding region (1.8 kb) amplified from the wild type cDNA was cloned into the SwaI site of the overexpression vector, pOX9WZ1, which harbors a rice actin promoter, a c-myc tag and the Zeocin resistance cassette. The mutated PpPPR_63 (M63: D460A/D461A) gene was modified from the F63 construct. (C) The predicted 1.8- and 3.0-kb fragments derived from the transgenic lines and wild type (WT), respectively, were amplified. The predicted 5.0-kb fragments were not amplified fromΔ63-1 under the PCR conditions. F63-3 and M63-8 plants were used for further analysis as F63 and M63, respectively, in Fig. 4. Transgenic lines V-7 and V-8 were generated by transferring the vector only into theΔ63-1 moss.
(EPS)
Generation of PpPPR_67 knockout (KO) lines. (A) Schematic diagrams of the PpPPR_67 gene (WT), the KO construct (p67KO-1), and the generated KO locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the HindIII site of the 4th exon of PpPPR_67. Primers used for PCR and the expected amplified DNA sizes are shown. (B) The predicted 5.0-, 2.1- and 1.6-kb fragments were amplified from the transgenic lines, and 3.0 kb was amplified from the WT. The KO line 67-1-17 was used in Figs. S7 and S8.
(EPS)
Generation of PpPPR_67 knockout (KO) lines. (A) Schematic diagrams of the PpPPR_67 locus, the KO construct (p67KO-2), and the targeted KO locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the StuI site of the 5th exon of PpPPR_67. Primers and the amplified DNA sizes for PCR analysis are shown. (B) The predicted 5.0-, 2.3- and 1.5-kb fragments were amplified from the transgenic lines, and 3.0 kb was amplified from the WT. The KO line 67-2-2 was used asΔ67 in Figs. 5, S7, and S8.
(EPS)
Generation of PpPPR_104 knockout (KO) line. (A) Schematic diagrams of the PpPPR_104 locus, the KO construct (p104KO), and the targeted KO locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the BglII site of the 4th exon of PpPPR_104. Primers used for PCR and the expected fragment sizes are shown. (B) The predicted 5.0-, 1.8- and 1.3-kb fragments were amplified from the transgenic lineΔ104-7, and 3.0 kb was amplified from the wild type (WT). The KO line 104-7 was used asΔ104 in Figs. 5, S7, and S8.
(EPS)
Northern blot analysis of chloroplast tRNAs. Total cellular RNAs (5 µg) from wild type (WT), Δ67-1-17, Δ67-2-2, andΔ104-7 protonemata were separated on 8% polyacrylamide containing 7 M urea and transferred to nylon membranes. Chloroplast tRNA gene-specific oligonucleotide probes (Table S2) were labeled with DIG-ddUTP and terminal deoxynucleotidyl transferase (Roche).
(TIF)
Northern blot analysis of mitochondrial tRNAs. Total cellular RNAs (5 µg) were subjected to northern blot analysis as described in Fig. S7. Mitochondrial tRNA gene-specific oligonucleotide probes (Table S2) were labeled as described in Fig. S7.
(TIF)
List of PRORP and PRORP-like proteins from various organisms.
(XLSX)
Oligonucleotides used in this study.
(XLSX)
Acknowledgments
We thank Mitsuyasu Hasebe (National Institute of Basic Biology, Okazaki) for the gift of a plasmid pGFPmutNTPII. We are grateful to Tomoaki Nishiyama (Kanazawa University) and Hidetoshi Sakayama (Kobe University) for a TBLASTN search of a PRORP homolog to unpublished genome database of Chara braunii. We also thank Haruka Nagashima for technical assistance.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by JSPS KAKENHI Grant Numbers 2057003, 23657003, 25291059, 25660292 (MS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Frank DN, Pace NR (1998) Ribonuclease P: unity and diversity in a tRNA processing ribozyme. Ann Rev Biochem 67: 153–180. [DOI] [PubMed] [Google Scholar]
- 2. Schön A (1999) Ribonuclease P: the diversity of a ubiquitous RNA processing enzyme. FEMS Microbiol Rev 23: 391–406. [DOI] [PubMed] [Google Scholar]
- 3. Hartmann E, Hartmann RK (2003) The enigma of ribonuclease P evolution. Trends Genet 19: 561–569. [DOI] [PubMed] [Google Scholar]
- 4. Holzmann J, Frank P, Löffler E, Bennett KL, Gerner C, et al. (2008) RNase P without RNA: identification and functional reconstitution of the human mitochondrial tRNA processing enzyme. Cell 135: 462–474. [DOI] [PubMed] [Google Scholar]
- 5. Anantharaman V, Aravind L (2006) The NYN domains: novel predicted RNAses with a PIN domain-like fold. RNA Biol 3: 18–27. [DOI] [PubMed] [Google Scholar]
- 6. Gobert A, Gutmann B, Taschner A, Gössringer M, Holzmann J, et al. (2010) A single Arabidopsis organellar protein has RNase P activity. Nat Struct Mol Biol 17: 740–744. [DOI] [PubMed] [Google Scholar]
- 7. Gutmann B, Gobert A, Giegé P (2012) PRORP proteins support RNase P activity in both organelles and the nucleus in Arabidopsis. Genes Dev 26: 1022–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rosenblad MA, López MD, Piccinelli P, Samuelsson T (2006) Inventory and analysis of the protein subunits of the ribonucleases P and MRP provides further evidence of homology between the yeast and human enzymes. Nucleic Acids Res 18: 5145–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stirewalt VL, Michalowski CB, Löffelhardt W, Bohnert HJ, Bryoant DA (1995) Nucleotide sequence of the cyanelle genome from Cyanophora paradoxa . Plant Mol Biol Rep 13: 327–332. [Google Scholar]
- 10. Reith ME, Munholland J (1995) Complete nucleotide sequence of the Porphyra purpurea chloroplast genome. Plant Mol. Biol Rep 13: 333–335. [Google Scholar]
- 11. Turmel M, Lemieux C, Burger G, Lang BF, Otis C, et al. (1999) The complete mitochondrial DNA sequences of Nephroselmis olivacea and Pedinomonas minor. Two radically different evolutionary patterns within green algae. Plant Cell 11: 1717–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lai LB, Bernal-Bayard P, Mohannath G, Lai SM, Gopalan V, et al. (2011) A functional RNase P protein subunit of bacterial origin in some eukaryotes. Mol Genet Genomics 286: 359–369. [DOI] [PubMed] [Google Scholar]
- 13. Baum M, Cordier A, Schön A (1996) RNase P from a photosynthetic organelle contains an RNA homologous to the cyanobacterial counterpart. J Mol Biol 257: 43–52. [DOI] [PubMed] [Google Scholar]
- 14. Cordier A, Schön A (1999) Cyanelle RNase P: RNA structure analysis and holoenzyme properties of an organellar ribonucleoprotein enzyme. J Mol Biol 289: 9–20. [DOI] [PubMed] [Google Scholar]
- 15. Lai LB, Vioque A, Kirsebom LA, Gopalan V (2010) Unexpected diversity of RNase P, an ancient tRNA processing enzyme: challenges and prospects. FEBS Lett 584: 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O'Toole N, Hattori M, Andres C, Iida K, Lurin C, et al. (2008) On the expansion of the pentatricopeptide repeat gene family in plants. Mol Biol Evol 25: 1120–1128. [DOI] [PubMed] [Google Scholar]
- 17. Sugita M, Ichinose M, Ide M, Sugita C (2013) Architecture of the PPR gene family in the moss Physcomitrella patens . RNA Biol 10: 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Howard MJ, Lim WH, Fierke CA, Koutmos M (2012) Mitochondrial ribonuclease P structure provides insight into the evolution of catalytic strategies for precursor-tRNA 5′ processing. Proc Natl Acad Sci USA 109: 16149–16154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sugiura C, Kobayashi Y, Aoki S, Sugita C, Sugita M (2003) Complete chloroplast DNA sequence of the moss Physcomitrella patens: evidence for the loss and relocation of rpoA from the chloroplast to the nucleus. Nucl Acids Res 31: 5324–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sugiura C, Sugita M (2004) Plastid transformation reveals that moss tRNAArg-CCG is not essential for plastid function. Plant J 40: 314–321. [DOI] [PubMed] [Google Scholar]
- 21. Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, et al. (2008) The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319: 64–69. [DOI] [PubMed] [Google Scholar]
- 22. Hattori M, Miyake H, Sugita M (2007) A pentatricopeptide repeat protein is required for RNA processing of clpP pre-mRNA in moss chloroplasts. J Biol Chem 282: 10773–10782. [DOI] [PubMed] [Google Scholar]
- 23. Tasaki E, Hattori M, Sugita M (2010) The moss pentatricopeptide repeat protein with a DYW domain is responsible for RNA editing of mitochondrial ccmFc transcript. Plant J 62: 560–570. [DOI] [PubMed] [Google Scholar]
- 24. Uchida M, Ohtani S, Ichinose M, Sugita C, Sugita M (2011) The PPR-DYW proteins are required for RNA editing of rps14, cox1 and nad5 transcripts in Physcomitrella patens mitochondria. FEBS Lett 585: 2367–2371. [DOI] [PubMed] [Google Scholar]
- 25. Sugita C, Kato Y, Yoshioka Y, Tsurumi N, Iida Y, et al. (2012) CRUMPLED LEAF (CRL) homologs of Physcomitrella patens are involved in the complete separation of dividing plastids. Plant Cell Physiol 53: 1124–1133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Generation of PpPPR_63 - GFP knockin (KI) mosses. (A) Schematic of insertion of p63-GFPKI into the 3′ terminal end of the PpPPR_63 coding region. Translated and untranslated regions are represented by black and white boxes, respectively. Primers P1 to P3 used for PCR, and the expected amplified DNA sizes, are shown. (B) PCR analysis using the indicated primer pairs. The predicted 4.5- and 1.8-kb fragments were amplified from the KI lines. (C) Protonemal colonies of wild-type (WT) and KI mosses grown for 2 weeks on BCDATG medium plates without antibiotics. KI-10 was used for further analyses (Fig. 2). Scale bars = 5 mm.
(ZIP)
Generation of PpPPR_63 knockout (KO) moss. (A) Schematic diagrams of the PpPPR_63 gene, the KO construct (p63KO4-13), and the generated knockout locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the 4th intron of PpPPR_63. Translated and untranslated regions are represented by black and white boxes, respectively. Primers P4 to P7 used for PCR and the expected amplified DNA sizes are shown. The DNA probe used in (C) is represented by grey bars. (B) PCR analysis using the indicated primer pairs. The predicted 1.9-, 2.2- and 5.5-kb fragments were amplified from the KO lines; 3.0 kb was amplified from the WT. (C) Total cellular DNAs (20 µg) from wild type (WT) and KO mosses were digested with SphI and hybridized with the PpPPR_63 probe in (A). The probe was labeled with DIG-dUTP. Predicted hybridized fragments were detected in WT and KO mosses.
(EPS)
Generation of PpPPR_63 complementation lines intoΔ 63 -1. (A) Schematic diagrams of PpPPR_63 loci of wild type andΔ63-1. Primers P8 and P9 and the expected fragment sizes for PCR analysis are shown. (B) The PpPPR_63 coding region (1.8 kb) amplified from the wild type cDNA was cloned into the SwaI site of the overexpression vector, pOX9WZ1, which harbors a rice actin promoter, a c-myc tag and the Zeocin resistance cassette. The mutated PpPPR_63 (M63: D460A/D461A) gene was modified from the F63 construct. (C) The predicted 1.8- and 3.0-kb fragments derived from the transgenic lines and wild type (WT), respectively, were amplified. The predicted 5.0-kb fragments were not amplified fromΔ63-1 under the PCR conditions. F63-3 and M63-8 plants were used for further analysis as F63 and M63, respectively, in Fig. 4. Transgenic lines V-7 and V-8 were generated by transferring the vector only into theΔ63-1 moss.
(EPS)
Generation of PpPPR_67 knockout (KO) lines. (A) Schematic diagrams of the PpPPR_67 gene (WT), the KO construct (p67KO-1), and the generated KO locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the HindIII site of the 4th exon of PpPPR_67. Primers used for PCR and the expected amplified DNA sizes are shown. (B) The predicted 5.0-, 2.1- and 1.6-kb fragments were amplified from the transgenic lines, and 3.0 kb was amplified from the WT. The KO line 67-1-17 was used in Figs. S7 and S8.
(EPS)
Generation of PpPPR_67 knockout (KO) lines. (A) Schematic diagrams of the PpPPR_67 locus, the KO construct (p67KO-2), and the targeted KO locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the StuI site of the 5th exon of PpPPR_67. Primers and the amplified DNA sizes for PCR analysis are shown. (B) The predicted 5.0-, 2.3- and 1.5-kb fragments were amplified from the transgenic lines, and 3.0 kb was amplified from the WT. The KO line 67-2-2 was used asΔ67 in Figs. 5, S7, and S8.
(EPS)
Generation of PpPPR_104 knockout (KO) line. (A) Schematic diagrams of the PpPPR_104 locus, the KO construct (p104KO), and the targeted KO locus. A Geneticin G418-resistance gene (nptII) cassette was inserted into the BglII site of the 4th exon of PpPPR_104. Primers used for PCR and the expected fragment sizes are shown. (B) The predicted 5.0-, 1.8- and 1.3-kb fragments were amplified from the transgenic lineΔ104-7, and 3.0 kb was amplified from the wild type (WT). The KO line 104-7 was used asΔ104 in Figs. 5, S7, and S8.
(EPS)
Northern blot analysis of chloroplast tRNAs. Total cellular RNAs (5 µg) from wild type (WT), Δ67-1-17, Δ67-2-2, andΔ104-7 protonemata were separated on 8% polyacrylamide containing 7 M urea and transferred to nylon membranes. Chloroplast tRNA gene-specific oligonucleotide probes (Table S2) were labeled with DIG-ddUTP and terminal deoxynucleotidyl transferase (Roche).
(TIF)
Northern blot analysis of mitochondrial tRNAs. Total cellular RNAs (5 µg) were subjected to northern blot analysis as described in Fig. S7. Mitochondrial tRNA gene-specific oligonucleotide probes (Table S2) were labeled as described in Fig. S7.
(TIF)
List of PRORP and PRORP-like proteins from various organisms.
(XLSX)
Oligonucleotides used in this study.
(XLSX)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.