

Abstract
The antibody microarrays have become widespread, but their use for quantitative analyses in clinical samples has not yet been established. We investigated an immunoassay based on nanoporous silicon antibody microarrays for quantification of total prostate-specific-antigen (PSA) in 80 clinical plasma samples, and provide quantitative data from a duplex microarray assay that simultaneously quantifies free and total PSA in plasma. To further develop the assay the porous silicon chips was placed into a standard 96-well microtiter plate for higher throughput analysis. The samples analyzed by this quantitative microarray were 80 plasma samples obtained from men undergoing clinical PSA testing (dynamic range: 0.14-44ng/ml, LOD: 0.14ng/ml). The second dataset, measuring free PSA (dynamic range: 0.40-74.9ng/ml, LOD: 0.47ng/ml) and total PSA (dynamic range: 0.87-295ng/ml, LOD: 0.76ng/ml), was also obtained from the clinical routine. The reference for the quantification was a commercially available assay, the ProStatus PSA Free/Total DELFIA. In an analysis of 80 plasma samples the microarray platform performs well across the range of total PSA levels. This assay might have the potential to substitute for the large-scale microtiter plate format in diagnostic applications. The duplex assay paves the way for a future quantitative multiplex assay, which analyses several prostate cancer biomarkers simultaneously.
Keywords: quantitative, antibody microarray, total and free PSA, prostate cancer biomarker, duplex assay, porous silicon
1. Introduction
Protein or antibody microarrays are often proposed as tools for high-throughput screening for analyzing thousands of biomarkers simultaneously. In the pharmaceutical industry, high-throughput platforms are an important way to reduce assay costs. The parallel process makes it possible to drastically reduce reagent consumption compared to microtiter plate formats. Protein chip technology is becoming an increasingly established technique, not only for characterizing specific proteins or even proteomes, but also for clinical applications. Although routine clinical use of microarray technology still is in its early phase, antibody microarrays have already been developed for a number of clinical diagnostic applications [1-6].
Until now, most protein microarray applications have been used for qualitative analysis, for example to profile thousands of proteins, to quickly assess the specificity of an antibody [7, 8] or to globally analyze protein phosphorylation [9]. However, limited efforts have been put into the development of a quantitative approach. Often, protein microarrays are used for comparing the levels of large sets of proteins in two different samples [10-13]. Reverse-phase protein microarrays have been successfully used to monitor biomarkers in cancer cell lines or in laser-captured microdissections from different cancer stages [1, 4]. However, this technique must be viewed as semi-quantitative, although Pollard et al [5] described that a modified format of the technique was quantitative. For true quantitative analysis, a standard curve could be used in a similar way as in a standard microtiter plate format [14-16]. Most of the existing publications on quantitative analysis have not yet been demonstrated on larger patient cohorts. The most extensive study (Knickerbocker et al 2007) was based on cytokine measurements in 468 samples from kidney dialysis patients. It should be noted that the spot density was larger than the one we present in this paper. According to Knickerbocker et al [17] a center-to-center spacing of 250-350 μm was used as compared to 150 μm in the arrays described herein. The reason why our assay can apply such a small center-to-center spacing is the nanostructured hydrophobic surface behavior (yet hydrophilic surface chemistry) of our in-house developed porous silicon surfaces, causing an extremely small contact area for the dispensed droplets on the chip.
The clinical focus of this work is improvement of prostate cancer diagnostics. Prostate-specific antigen (PSA) concentration in plasma is widely used as an indicator of prostate disease. However, the diagnostic specificity is a concern, because an increased PSA value might be due to benign prostate hyperplasia (BPH) or prostatitis rather than prostate cancer. Before prostate cancer can be diagnosed or excluded, the patient needs to endure painful prostate biopsy. In addition, some prostate cancers progress very slowly and the patient is unlikely to die of or have any physical complications from the cancer. To improve prostate cancer diagnostics, new biomarkers are sought to distinguish BPH from prostate cancer and also indolent from rapidly developing cancer. One way to improve the diagnostics might be simultaneous analysis of multiple biomarkers, and microarray technology is compatible with multiplex analysis. However, to compete with the diagnostic immunoassays of today, the microarrays need to be quantitative.
We previously described antibody microarray methods for analyzing PSA using a sandwich immunoassay [18, 19]. The substrate used is a porous silicon surface developed in-house, produced by electrochemical dissolution of silicon wafers. These micro- and nano-structured porous silicon chips are well suited for surface-based immunoassays [18-20] and are compatible with mass spectrometry readout [21, 22]. The method has yielded sensitive and reproducible analysis of PSA-spiked sera [18, 19].
In this study we investigate whether the antibody microarray technique can quantify total PSA in routine clinical samples – 80 EDTA-plasma samples from patients undergoing clinical PSA testing. We have advanced our earlier microarray procedure by scaling down the porous silicon chip to align with a 96-well microtiter plate format (figure 1). The transition to a standard 96-well format facilitates clinical implementation of the microarray assay. The microarray data are assessed by comparing them to results from the DELFIA assay, a well characterized commercial assay widely used for clinical PSA measurements. In addition, we describe proof of principle for a duplex assay where free and total PSA are quantified on a single microarray chip – a first step towards a quantitative multiplex assay.
Fig 1.

Schematic of the total PSA microarray procedure in the 96-well microtiter plate, starting with dispensing of anti-PSA antibodies onto the porous silicon surface. Approximately 1000 spots can be spotted onto each chip. The porous chips with physically adsorbed antibodies are then placed into microtiter plate wells, and PSA-containing plasma samples as well as standard are added; the use of a microtiter plate speeds the analysis by allowing for parallel pipetting. Subsequently, labeled antibodies are used for detection in a fluorescence microscope. The duplex assay is alike with the exception of the detector antibody is bound via biotin to Alexa Fluor® 488 labeled streptavidin.
2. Materials and Methods
2.1 Proteins and Reagents
Recombinant proPSA was produced in insect cells as described [23], and purified on Affigel 10 (Bio-Rad, Hercules, CA, USA) coupled with four monoclonal anti-PSA antibodies, 2E9, 5A10, 2C1, and 2H11. Eluted protein was further purified by gel filtration (Sephacryl S-200 HR, Pharmacia Biotech, Uppsala, Sweden). Size and purity were confirmed by SDS/PAGE and Western blot. Monoclonal antibodies 2E9, H117 and 5A10 were produced and characterized as described [24]. Detector antibody 2E9 for the PSA total sandwich antibody microarray was labeled with fluorescein isothiocyanate (FITC) isomer I-celite (Sigma, St. Louis, MO, USA), and a PD10 column (Amersham, Uppsala, Sweden) was used for separation. H117 was biotinylated by N-Hydroxysuccinimidobiotin (Sigma, St. Louis, MO, USA), dialyzed by Slide-A-Lyzer units, molecular weight cutoff 3.5 kDa (Thermo Fisher Scientific, MA, USA) and used as detector antibody in the duplex assay. Streptavidin-Alexa Flour® 488 (Invitrogen, Carlsbad, CA, USA) was chosen for detection of the duplex assay. Non-fat dry milk (Bio-Rad, Hercules, CA, USA) was used as blocking agent and ProStatus PSA Assay Buffer (DELFIA®/AutoDELFIA®, PerkinElmer, Turku, Finland) for dilution of milk, biotin coupled H117 and Streptavidin-Alexa Flour® 488. For the duplex setup to determine CVs and LOD Casein (Bio-Rad, Hercules, CA, USA) was used as an equal substitute for non-fat dry milk.
2.2 Analytical Samples
Human female plasma and serum was prepared using Greiner Bio-One K2EDTA Vacuette and BD vacutainer respectively, aliquoted, and stored at −80°C. The plasma was spiked with purified recombinant proPSA and used as a nine point standard curve for the total PSA assay, i.e. for the quantification of the 80 patient samples. A commercially available PSA Standard (DELFIA®/AutoDELFIA®, PerkinElmer, Turku, Finland), based on six concentrations, was used as standard curve in the duplex assay.
Eighty EDTA-plasma samples were collected (without retaining any patient identifiers or clinical data) from men undergoing clinical PSA testing. Samples were chosen to give equal numbers in 4 groups based on immunoassay measurements of total PSA and percentage of free PSA (%fPSA) in the human plasma. The stratification was done to make sure that our study included samples resembling those of the four most common patient groups. Group A (0.3-0.7 ng/ml totPSA, %fPSA>30) mimics the total PSA concentration and free to total PSA ratio in healthy individuals [25, 26]. Group B samples (2-8 ng/ml totPSA, %fPSA>28) were selected to resemble those of men who have elevated PSA but low risk of prostate cancer due to a high ratio of free to total PSA [25, 27]. Group C samples (3-10 ng/ml totPSA, %fPSA<15) mimics high risk of localized prostate cancer, for which PSA concentration is increased, while the free to total PSA ratio is low [25, 27, 28]. Finally, group D (totPSA>20 ng/ml, %fPSA<12) represents men with PSA levels strongly suggestive of advanced prostate carcinoma [28, 29]. We used this stratification to help us fully validate our protein microarray technique without need for clinically annotated samples. All procedures followed were in accordance with the current revision of the Helsinki Declaration.
EDTA-plasma from the clinical routine was also used for the duplex assay. Concentrations of free and total PSA were determined by the reference assay DELFIA. A titration series for the duplex assay was prepared by diluting an EDTA-plasma sample from the clinical routine in female EDTA plasma to make sure the high protein complexity of blood fluid was retained. Concentrations of free and total PSA were determined by DELFIA.
2.3 Porous Silicon Fabrication
The fabrication of micro- and nanoporous silicon by anodic dissolution of p-type, boron-doped monocrystalline silicon wafers has been described in detail [20]. Briefly, the 3-inch silicon wafer used was of 6-8 Ωcm resistivity (P-type, boron), <100>-orientation, and purchased from Addison Engineering (San Jose, CA, USA). The wafer was placed in the middle of an electrochemical etching cell. The electrolyte solution consisted of 3.6 % hydrofluoric acid and 90.7 % dimethylformamide (Merck, Darmstadt, Germany). A 45 min anodization was performed during backside illumination. The positive charge carriers in the silicon wafer migrate towards the anodic side of the wafer when the voltage is supplied. At the anodic side, silicon is solubilized and the micro- and nanopores formed. The wafer was subsequently cut into 3.5 × 3.5 mm pieces to fit the wells of the microtiter plate (Corning Costar Corporation, Cambridge, MA, USA).
2.4 Sandwich Antibody Microarray for Total PSA Quantification
Droplets of 100 pl of monoclonal mouse capture antibody H117 (0.5 mg/ml diluted in 0.01 M PBS) were dispensed onto the porous silicon surface at a spot-to-spot distance of 150 μm. Loosely bound material was removed from the chips by washing in PBS-Tween. The microarray chips were then placed in a 96-well microtiter plate (Corning Costar Corporation, Cambridge, MA, USA). A black microtiter plate was chosen to prevent bleaching of the fluorophore. The chips, each containing around 200 spots, were blocked for 30 min on a shaker in 100 μl 5% non-fat dry milk in PBS-Tween. The microarrays were then washed 3 times in 150 μl PBS-Tween, and incubated with 30 μl plasma or standard for 75 min. Plasma samples of group A-C were added undiluted to the wells. Group D samples were diluted to 10 ng total PSA/ml in 0.1 M PBS prior to analysis. After additional washing steps, 30 μl FITC-labeled 2E9 monoclonal mouse α-total PSA-antibody (4.4 μg/ml diluted in 0.01 M PBS) was added, followed by incubation on shaker. The chips were washed 3 times in 150 μl PBS-Tween, quickly dipped in distilled water, and dried by pressurized air. Fluorescence readout was performed with a BX51WI microscope (Olympus, Japan). Fluoview 300 software was used for image analysis.
2.5 Duplex Assay for Total and Free PSA Analysis
The duplex assay was performed as the sandwich microarray described above, but with additions as follows. 50-220 droplets 0.6 mg/ml of mouse monoclonal antibody 2E9 and 50-220 droplets of 5A10 were dispensed onto the same porous silicon chip, to capture PSA total and free, respectively. The milk powder was diluted in ProStatus PSA Assay Buffer instead of BPS-Tween. The 16 EDTA-plasma samples with PSA total concentrations exceeding 2 ng/ml were diluted in 0.1 M PBS to 2 ng/ml prior to analysis. Plasma samples and standard were incubated with the microarrays for 60 minutes. Detection was performed by first incubating the chips with 30 μl 4.8 μg/ml biotinylated antibody H117 (α-total PSA-antibody), and after 60 min 5 μl 90 μg/ml streptavidin-Alexa Fluor® 488 (Invitrogen, Carlsbad, CA, USA) was added to the prefilled wells and incubation followed for another 60 min. The software used for image analysis on the 16 EDTA-plasma samples was the in-house developed Microarray iMageanalysis (MiiM) 1.8.
2.6 Mean Spot Intensity, Limit of Detection (LOD), and Lower Limit of Quantification (LLOQ)
The intensity of each spot and that of local background was quantified using Fluoview 300 or MiiM 1.8. Both Fluoview 300 and MiiM 1.8 base their measurements on signal intensity integrated over a circular area. Spot intensity was determined as the total sum of intensities within the spot minus local background. Mean spot intensities presented in the graphs and tables are calculated from nine adjacent microarray spots; the same nine spots were used to derive standard deviations and coefficients of variation (CVs).
LOD was defined as the lowest PSA concentration for which the mean spot intensity was at least two standard deviations above the mean intensity of the background, calculated from nine background locations.
LLOQ was determined from the clinical EDTA-plasma samples and defined as the lowest total PSA concentration that met all of three criteria: first, the mean spot intensity was at least 5 times that of a blank female serum sample; second, the CV was ≤ 0.2; third, the mean accuracy was ≤ 20%. Accuracy was determined as the absolute value of the difference between total PSA concentration determined by microarray analysis and DELFIA relative to the total PSA concentration by DELFIA. For each patient group a mean was calculated and denoted mean accuracy.
2.7 Recovery and Imprecision
The recovery factor (RA) of our PSA-spiked EDTA-plasma was determined according to the following formula:
| (1) |
[total PSA]spiked female plasma and [total PSA]female plasma are the analyte concentrations as measured by DELFIA after and before spiking, respectively. [total PSA]theoretical is the analyte concentration aimed at during spiking.
To address imprecision of the total PSA microarray assay, the spot intensity minus local background was analyzed for five clinical plasma samples and two samples of PSA-spiked female plasma. Within-run (sr) and total (sl) standard deviations were derived from nine replicates analyzed on two occasions. The standard deviations were divided by the overall average for each sample (X), generating relative measurements of imprecision similar to CV.
2.8 DELFIA
The ProStatus PSA Free/Total DELFIA (Perkin Elmer, Turku, Finland) was used as the reference assay for the total and free PSA concentrations. This immunoassay, which is standardized to WHO calibrator 96/670 [30, 31], is run routinely in the hospital laboratory. The assay has been demonstrated to be highly proportional and specific to the concentrations of free and total PSA in the sample [32].
2.9 Regression Analysis
Graph Pad Prism software was used for regression analysis and for calculating 95 % prediction interval [33] and 95 % confidence interval.
3. RESULTS
3.1 96-well Compatible Microarrays
The porous silicon wafer was cut into 3.5 × 3.5 mm pieces to fit into microtiter plate wells (figure 2). Up to around 1000 antibody droplets could be arrayed on each piece.
Fig 2.

Porous silicon chips compatible with microtiter plates. A) After electrochemical porosification, the wafer was cut into 3.5 × 3.5 mm pieces. B) Close-up of the “disco-ball” structure of the backside of the cut wafer. C) For the microarray assay, the chips were placed into a 96-well plate, and then loaded with PSA standards and the plasma samples from patients undergoing clinical PSA testing. D) Scanning electron microscope side view image of the micro- and nano-structured porous silicon surface. Each pore is 10-15 μm deep and 1-2 μm wide. In addition, the pores have nanostructured side branching [18], offering a vastly increased surface area.
3.2 Standard Curve of the 96-well Compatible Microarray for Total PSA Quantification
A titration series was created by spiking known amounts of PSA into human female plasma. These samples were also analyzed for total PSA levels by DELFIA. To create a standard curve, the DELFIA results were used for calculating the absolute total PSA concentrations, and a sigmoid curve fit (eq 2) was generated by MatLab.
| (2) |
The constants for the sigmoid fit were: a=13.0 × 107; b=3.38 and c=3.43. The coefficient of correlation was 0.997. The experimental data as well as the curve fit can be seen in figure 3. The inset in the figure shows that the spot intensity remained responsive to changes in quantity for the four lowest total PSA concentrations on the standard curve. This establishes the analytical sensitivity of the assay, defined by the International Union of Pure and Applied Chemistry as the ability of an analytical procedure to produce a change in signal for a defined change of the quantity.
Fig 3.

Total PSA standard curve of PSA spiked into female EDTA-plasma. The magnitudes of the error bars represent one standard deviation calculated from nine adjacent spots for each total PSA concentration. PSA concentrations were determined by DELFIA. The inset graph in the lower right corner displays the four lowest concentrations, plotted on a linear scale.
The LOD of the total PSA microarray assay was found to be 0.14 ng/ml, which corresponds well with our earlier findings [18]. The dynamic range was 2-3 orders of magnitude (0.14-44 ng/ml). Female serum without any added PSA did not give rise to any detectable signals in the assay (data not shown). In addition to this, the recovery (RA) was around 30 % for PSA spiked into female EDTA-plasma over a concentration range from 1 to 420 ng/ml. The recovery seemed to be equally distributed over all PSA levels (data not shown).
3.3 Quantification of Total PSA in Clinical Plasma Samples
The mean spot intensities, calculated from nine spots for each sample, were subsequently used to derive the corresponding total PSA concentration from the standard curve (figure 3). We note that before the final PSA concentration could be derived from the mean spot intensities, the generated x-value (log [total PSA]) should be recalculated to [total PSA].
Mean spot intensities, CVs, and total PSA concentrations derived from the microarray data, as well as corresponding total PSA concentrations by DELFIA, are listed in figure 4A. One chip from group B and one from group C were lost during analysis.
Fig 4.

4A: Mean spot intensities, CVs, and total PSA concentrations derived from the total PSA microarray and the sigmoid curve fit, with the corresponding total PSA concentration by DELFIA. 4B: The total PSA concentrations derived from the antibody microarray assay plotted against the DELFIA reference values. The dashed lines outline: the 95 % confidence interval (inner lines) and the 95 % prediction interval (outer lines). 4C: Total PSA concentration by antibody microarray plotted against total PSA concentration by DELFIA using a logarithmic scale on both axes. 4D: Coefficients of variation in microarray readout for the 80 clinical plasma samples. The mean CV (12 %) is represented by the dashed line.
Next, to compare the performance of our microarray platform to that of DELFIA, we plotted the microarray-derived total PSA concentrations for the 80 clinical plasma samples against the concentrations as determined by DELFIA (figure 4B). Using linear regression, we found the correlation coefficient between the microarray results and DELFIA results to be above 0.97 and the slope to be 1.005 ± 0.027 (standard deviations of residuals, Sx/y=28.24). The y-intercept (when x=0) was −2.05 ± 3.47, indicating the microarray assay generates a slightly lower total PSA concentration than the DELFIA. However, this could easily be corrected for by calibration. Figure 4B also shows two zones of uncertainty. The inner zone represents the 95 % confidence interval for the mean total PSA concentration by the microarray. The outer zone shows the 95 % prediction or tolerance interval, i.e. the uncertainty in predicting values of the total PSA concentration from the microarray assay for an individual value of the total PSA concentration from the DELFIA assay [33]. In figure 4C, the same dataset as in figure 4B was plotted on a log-log scale, to better separate the data. The two methods show a clear linearity.
LLOQ for the total PSA microarray was set to above 0.7 ng/ml, according to the definition stated in Materials and Methods. The LLOQ requirements were not quite fulfilled by the group A samples ([total PSA] ≤ 0.7 ng/ml), since the mean accuracy was above 20 %, but they were fulfilled by the group B samples ([total PSA] 2.1-8 ng/ml, mean accuracy = 20 %).
Figure 4D shows the reproducibility, reported as CVs, for each of the 80 clinical samples analyzed by the total PSA microarray. The mean CV was 12 % and the reproducibility was found consistent across the four theoretical patient groups.
3.4 Imprecision of the Total PSA Microarray Assay
Within-run (sr) and total (sl) standard deviations divided by the overall mean for the sample are reported in table 1.
TABLE 1.
Imprecision of the total PSA microarray assay determined for seven PSA-containing samples at two different occasions.
| Sample | [total PSA] by DELFIA (ng/ml) |
sr/X (%) |
sl/X (%) |
|---|---|---|---|
| PSA-spiked female EDTA-plasma | |||
| Sample 1 | 7.0 | 9.4 | 11 |
| Sample 2 | 24.0 | 12 | 23 |
| Clinical EDTA-plasma | |||
| Sample 1 | 5.0 | 15 | 16 |
| Sample 2 | 7.4 | 15 | 16 |
| Sample 3 | 7.4 | 16 | 17 |
| Sample 4 | 9.0 | 20 | 19 |
| Sample 5 | 9.1 | 14 | 28 |
3.5 Duplex Assay
Two standard curves were created for total and free PSA, by plotting the mean spot intensity against the corresponding PSA concentrations as measured by DELFIA. Linear regression was performed in MATLAB and the following equations were generated for total and free PSA, respectively: y=0.914x+8.54 and y=1.06x+7.33 (data not shown). To compare the performance of our duplex microarray platform to that of DELFIA, we plotted the standard curve microarray-derived total and free PSA concentrations for the 16 clinical plasma samples against the concentrations as determined by DELFIA (figure 5). According to DELFIA analysis, the total and free PSA concentrations ranged from 0.87 to 295 ng/ml and from 0.40 to 74.9 ng/ml, respectively.
Fig 5.
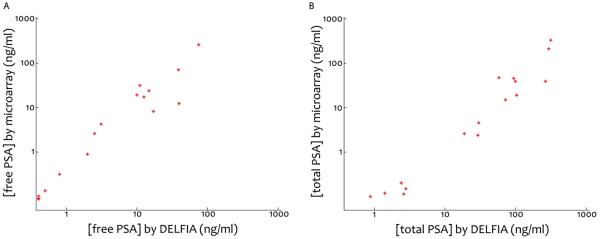
Graphs presenting the concentrations of free PSA (5A) and total PSA (5B) detected by the duplex microarray and plotted against the concentrations as measured by DELFIA.
Table 2 and figure 6 report the duplex microarray results from a titration series of a plasma sample from the clinic diluted in female plasma. Mean spot intensities, CVs and total and free PSA concentrations derived from the duplex microarray data, as well as corresponding total and free PSA concentrations by DELFIA, are listed in Table 2. According to DELFIA analysis, the total and free PSA concentrations ranged from 0.67 to 630 ng/ml and from 0.05 to 58 ng/ml, respectively. The lowest free PSA concentration (0.05 ng/ml) was not detectable by the antibody microarray, resulting in a LOD for the PSA total of 0.76 ng/ml and 0.47 for PSA free. Mean CV of PSAtotal and PSAfree, were determined to 0.16 and 0.19 respectively. The results of the blanks, i.e. pure female EDTA plasma, are included in the graph and table. Neither of the blanks gave any detectable spots.
TABLE 2.
Mean spot intensity and CV for total and free PSA measured in undiluted EDTA plasma samples by the duplex microarray assay. Corresponding DELFIA concentrations are also listed. A.u. denotes arbitrary units.
| PSA total DELFIA (ng/ml) |
MSI (A.u.) |
CV (%) |
PSA free DELFIA (ng/ml) |
MSI (A.u.) |
CV (%) |
|---|---|---|---|---|---|
| 630 | 12·106 | 0.19 | 58 | 11·106 | 0.15 |
| 66 | 11·106 | 0.14 | 4.7 | 4.7·106 | 0.17 |
| 6.3 | 5.3·106 | 0.19 | 0.47 | 0.55·106 | 0.24 |
| 0.76 | 0.52·106 | 0.11 | 0.05 | - | - |
| 0 (Blank) | - | - | 0 (Blank) | - | - |
Fig 6.

Graph presenting the mean spot intensities of total and free PSA detected by the duplex microarray and plotted against the concentrations as measured by DELFIA. The error bars represent standard deviations calculated from nine adjacent spots for each PSA concentration. A1-A5 and B1-B5 are fluorescence microscope images from the titration series of total and free PSA, respectively. Images 1 represent the blank (female EDTA plasma) and 2-5 represent the dilution series 0.67-630 ng total PSA/ml and 0.05-58 ng free PSA/ml. Each spot is approximately 50 μm in diameter, and the spot-to-spot distance is 150 μm.
4. Discussion
In this paper, we have advanced our microarray for total PSA analysis by making it microtiter plate compatible, further characterized the assay for clinical samples, and shown proof of principle for quantitative duplex detection of both free and total PSA on the same chip. The main goal was to be able to quantify the total PSA concentration in a set of 80 patient samples obtained from the clinical routine, and to compare the microarray assay to DELFIA, a clinically well-known and commercially available 96-well immunoassay. The correlation between the assays, for the eighty patients, was 0.97 and the slope was 1.005 ± 0.027. Both dynamic range and LOD of our total PSA microarray assay were very similar to those of DELFIA; dynamic range is 0.14-44 ng/ml for the current microarray assay and 0.5-50 ng/ml for DELFIA [32], and LOD is 0.14 ng/ml for the microarray and 0.1 ng/ml for DELFIA [32]. Both assays are clearly sensitive enough to cover the diagnostic cutoff levels of 0.6 to 4 ng total PSA/ml that are frequently used for identifying men with elevated risk of malignant prostate disease. In addition, we note that no signal could be observed when undiluted female serum or plasma was analyzed on the microarrays, indicating no interference from other molecules. These results indicate the potential of the microarray analysis to be used as a quantitative tool within the clinic.
Our total PSA standard curve, derived from female EDTA-plasma spiked with PSA, gave excellent fit to a sigmoid curve (R=0.997). We note the importance of running the standard along with the plasma samples, to avoid introducing assay-to-assay differences. In our experiment, the 80 clinical plasma samples were run along with the standard, using the same solutions of capture and detector antibody as well as the same blocking and washing procedure for all microarrays. It should also be noted that the mean spot intensities of the 80 plasma samples as a function of total PSA concentration showed the same sigmoid trend as the standard curve of figure 3 (data not shown). In addition, the linearity of our protein microarrays has been addressed in earlier publications [18, 19].
When spiking purified PSA into female EDTA-plasma or serum, we have earlier observed that the total PSA level measured in immunoassays is lower than the amount theoretically added [18]. Differences between the theoretical and measured total PSA concentrations could of course be explained partly by imprecise pipetting. However, since we find a clear pattern of lower-than-expected total PSA levels, the effect might be explained by a plasma phenomenon. It is known that α2-macroglobulin binds PSA in blood, and any macroglobulin-bound PSA is unrecognizable to conventional immunoassays [34]. PSA recovery trends similar to ours have also been reported before [24, 35].
The 80 plasma samples were chosen to represent the full range of analyte values likely to be encountered in clinical PSA testing. The total PSA microarray platform performed well for all four of the theoretical patient groups (figure 4C). The microarray analysis was made on crude plasma samples without any dilution, except for group D. The group D samples had to be diluted to fit the dynamic range of the assay. However, those samples also had to be diluted when analyzed by the DELFIA.
The benefit of sandwich immunoassays for clinical applications should be emphasized. A direct labeled assay, i.e. where the whole protein solution to be analyzed is fluorescently labeled, is not preferred since labeling every patient sample would be too time-consuming. In addition, using the sandwich approach gives improved specificity, since two different monoclonal antibodies are used.
On protein microarrays, the slides or surfaces are often divided into cells by a hydrophobic pen to allow separate samples to be added to the various cells. Our solution was instead to use porous silicon surfaces placed into a 96-well microtiter plate, into which the standard and plasma samples were loaded (figure 1 and 2). The concept of printing antibodies on the bottom of 96-well microtiter plates was demonstrated in a study of receptor tyrosine kinase activation [36], and also for PSA and a cytokine [16]. The 96-well format then allows for parallel pipetting using multi-pipettes, and is compatible with existing automated microtiter plate handling. In addition, the plate can be put on shaker to improve the binding kinetics.
Assay development was focused on creating a technology platform on which a large number of plasma samples as well as a large number of analytes could be measured. We have shown quantitative duplex detection of free and total PSA on the same porous silicon chip (figure 5 and 6). As can be seen in the plots (figure 5) the microarray results corresponds well the DELFIA values. Mean CVs for the duplex assay (mean CVfree PSA= 0.19 and mean CVtotal PSA=0.16) were below 20 %, which is in accordance with that of the total PSA assay (12 %). LOD for the duplex assay (LODfree PSA=0.47 And LODtotal PSA=0.76) was slightly larger than for the PSA total assay but they are not stretched to the limit. In the longer run we aim to develop our microchips for quantitative analysis of not only free and total PSA but also other prostate cancer biomarkers in a multiplex format. The duplex assay is a first step towards a multiplex chip. Using the micro scale format both sample and reagent volume are reduced. Since approximately 1000 spots could be arrayed onto each chip, up to 1000 times more information could be derived using the microarrays as compared to an ordinary 96-well assay. However, in reality probably no more than 50 biomarkers could realistically be measured on a diagnostic chip. For sandwich immunoassays, Schweitzer and McBeath [15, 37] have recommended analysis of 40-50 biomarkers per microarray to avoid the risk of cross reactivity. Even so, the reduction in assay costs would be a clear benefit.
The importance of a quantitative microarray assay should be noted. In our opinion, microarrays need to be quantitative to compete with existing diagnostic assays. Although only two prostate cancer markers are addressed in this work, the quantitative approach is a step forward towards an automated method with similar quantitative status as a commercial 96-well assay. Knickerbocker et al. [14] have earlier demonstrated a true quantitative multiplex microarray approach to determine cytokine concentrations in blood samples from patients initiating kidney dialysis. Our aim is a similar quantitative multiplex approach for prostate cancer biomarkers, performed on our in-house developed porous silicon surfaces.
In order to extend the microarray platform to also enable monitoring of patients that have undergone radical prostatectomy a considerably reduced PSA LOD is requested. We note that the LOD of the standard DELFIA assay (0.1 ng/ml) does not reach the desired sensitivity in the range of 10 pg/ml. Common ways of improving LOD is done by signal amplifications such as rolling circle amplification [15, 38], enzymatic signal amplification [4] or alternative capture molecules [39]. As an alternative approach we are driving novel developments that targets a total PSA LOD of 1-10 pg/ml where the key to the improved sensitivity is that the binding capacity of each microarray spot has been increased several orders of magnitude by entrapping the capture antibody in a vastly surface enlarging 3-dimensional matrix [40].
5. CONCLUSIONS
In conclusion, we have addressed a more extensive quantitative antibody microarray study of total PSA concentration in plasma samples obtained from patients undergoing clinical PSA testing. The microarray format was chosen to allow multiplex detection of biomarkers related to prostate cancer. An initial proof of concept was shown by the quantitative duplex analysis of both free and total PSA on the same 96 well compatible porous silicon chip. Several other proteins in the kallikrein family have shown strong correlation with malignant prostate disease and are thus prime candidates for our on-going efforts to develop a multiplex microarray.
Acknowledgements
This study was supported by a joint grant from the Swedish Research Council, Vinnova, SSF, under the program Biomedical Engineering for Better Health [2006-7600]. The Swedish Research Council [Medicine; no. 20095 and 2006-6020], Swedish Cancer Society [11-0624], National Cancer Institute [R33 CA 127768-03], Crafoord Foundation, Foundation Federico SA, Carl Trygger Foundation, Knut and Alice Wallenberg Foundation, the Royal Physiological Society of Lund, Sten Lexner Foundation, Hecht Foundation, National Cancer Institute Specialized Programs of Research Excellence [P50-CA92629], Sidney Kimmel Center for Prostate and Urologic Cancers, and David H. Koch through the Prostate Cancer Foundation are also acknowledged for their support. We thank Janet Novak, PhD, for substantive editing of the manuscript; this work was paid for by MSKCC. The authors also thank Gun-Britt Eriksson and Mona Hassan Al-Battat for providing critical reagents (antibodies and PSA) and expert technical assistance.
Abbreviations
- PSA
prostate-specific antigen
- BPH
benign prostate hyperplasia
- totPSA
total PSA
- %fPSA
percentage of free PSA
- FITC
fluorescein isothiocyanate
- LOD
limit of detection
- LLOQ
lower limit of quantification
Footnotes
Conflict of interest
Dr. Hans Lilja holds patents for free PSA, intact PSA, and hK2 assays.
References
- [1].Grubb RL, Calvert VS, Wulkuhle JD, et al. Signal pathway profiling of prostate cancer using reverse phase protein arrays. Proteomics. 2003;3:2142–6. doi: 10.1002/pmic.200300598. [DOI] [PubMed] [Google Scholar]
- [2].Horn S, Lueking A, Murphy D, et al. Profiling humoral autoimmune repertoire of dilated cardiomyopathy (DCM) patients and development of a disease-associated protein chip. Proteomics. 2006;6:605–13. doi: 10.1002/pmic.200401293. [DOI] [PubMed] [Google Scholar]
- [3].Ingvarsson J, Wingren C, Carlsson A, et al. Detection of pancreatic cancer using antibody microarray-based serum protein profiling. Proteomics. 2008;8:2211–9. doi: 10.1002/pmic.200701167. [DOI] [PubMed] [Google Scholar]
- [4].Nishizuka S, Charboneau L, Young L, et al. Proteomic profiling of the NCI-60 cancer cell lines using new high-density reverse-phase lysate microarrays. Proc Natl Acad Sci U S A. 2003;100:14229–34. doi: 10.1073/pnas.2331323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Pollard HB, Srivastava M, Eidelman O, et al. Protein microarray platforms for clinical proteomics. Proteomics Clinical Applications. 2007;1:934–52. doi: 10.1002/prca.200700154. [DOI] [PubMed] [Google Scholar]
- [6].Srivastava M, Bubendorf L, Srikantan V, et al. ANX7, a candidate tumor suppressor gene for prostate cancer. Proc Natl Acad Sci U S A. 2001;98:4575–80. doi: 10.1073/pnas.071055798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Michaud GA, Salcius M, Zhou F, et al. Analyzing antibody specificity with whole proteome microarrays. Nature Biotechnology. 2003;21:1509–12. doi: 10.1038/nbt910. [DOI] [PubMed] [Google Scholar]
- [8].Michaud GA, Snyder M. Proteomic approaches for the global analysis of proteins. Biotechniques. 2002;33:1308–16. doi: 10.2144/02336pt04. [DOI] [PubMed] [Google Scholar]
- [9].Ptacek J, Devgan G, Michaud G, et al. Global analysis of protein phosphorylation in yeast. Nature. 2005;438:679–84. doi: 10.1038/nature04187. [DOI] [PubMed] [Google Scholar]
- [10].Hudelist G, Pacher-Zavisin M, Singer CF, et al. Use of high-throughput protein array for profiling of differentially expressed proteins in normal and malignant breast tissue. Breast Cancer Res Treat. 2004;86:281–91. doi: 10.1023/b:brea.0000036901.16346.83. [DOI] [PubMed] [Google Scholar]
- [11].Knezevic V, Leethanakul C, Bichsel VE, et al. Proteomic profiling of the cancer microenvironment by antibody arrays. Proteomics. 2001;1:1271–8. doi: 10.1002/1615-9861(200110)1:10<1271::AID-PROT1271>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- [12].Miller JC, Zhou H, Kwekel J, et al. Antibody microarray profiling of human prostate cancer sera: antibody screening and identification of potential biomarkers. Proteomics. 2003;3:56–63. doi: 10.1002/pmic.200390009. [DOI] [PubMed] [Google Scholar]
- [13].Tannapfel A, Anhalt K, Hausermann P, et al. Identification of novel proteins associated with hepatocellular carcinomas using protein microarrays. J Pathol. 2003;201:238–49. doi: 10.1002/path.1420. [DOI] [PubMed] [Google Scholar]
- [14].Knickerbocker T, Chen JR, Thadhani R, MacBeath G. An integrated approach to prognosis using protein microarrays and nonparametric methods. Molecular Systems Biology. 2007;3 doi: 10.1038/msb4100167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Schweitzer B, Roberts S, Grimwade B, et al. Multiplexed protein profiling on microarrays by rolling-circle amplification. Nat Biotechnol. 2002;20:359–65. doi: 10.1038/nbt0402-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wiese R, Belosludtsev Y, Powdrill T, Thompson P, Hogan M. Simultaneous multianalyte ELISA performed on a microarray platform. Clin Chem. 2001;47:1451–7. [PubMed] [Google Scholar]
- [17].Knickerbocker T, MacBeath G. Detecting and quantifying multiple proteins in clinical samples in high-throughput using antibody microarrays. Methods in molecular biology. 2011;723:3–13. doi: 10.1007/978-1-61779-043-0_1. [DOI] [PubMed] [Google Scholar]
- [18].Jaras K, Ressine A, Nilsson E, et al. Reverse-phase versus sandwich antibody microarray, technical comparison from a clinical perspective. Anal Chem. 2007;79:5817–25. doi: 10.1021/ac0709955. [DOI] [PubMed] [Google Scholar]
- [19].Jaras K, Tajudin AA, Ressine A, et al. ENSAM: Europium Nanoparticles for Signal enhancement of Antibody Microarrays on nanoporous silicon. J Proteome Res. 2008;7:1308–14. doi: 10.1021/pr700591j. [DOI] [PubMed] [Google Scholar]
- [20].Ressine A, Ekstrom S, Marko-Varga G, Laurell T. Macro-/nanoporous silicon as a support for high-performance protein microarrays. Anal Chem. 2003;75:6968–74. doi: 10.1021/ac034425q. [DOI] [PubMed] [Google Scholar]
- [21].Finnskog D, Jaras K, Ressine A, et al. High-speed biomarker identification utilizing porous silicon nanovial arrays and MALDI-TOF mass spectrometry. Electrophoresis. 2006;27:1093–103. doi: 10.1002/elps.200500751. [DOI] [PubMed] [Google Scholar]
- [22].Finnskog D, Ressine A, Laurell T, Marko-Varga G. Integrated protein microchip assay with dual fluorescent- and MALDI read-out. J Proteome Res. 2004;3:988–94. doi: 10.1021/pr0499287. [DOI] [PubMed] [Google Scholar]
- [23].Lovgren J, Rajakoski K, Karp M, Lundwall a, Lilja H. Activation of the zymogen form of prostate-specific antigen by human glandular kallikrein 2. Biochem Biophys Res Commun. 1997;238:549–55. doi: 10.1006/bbrc.1997.7333. [DOI] [PubMed] [Google Scholar]
- [24].Lilja H, Christensson A, Dahlen U, et al. Prostate-specific antigen in serum occurs predominantly in complex with alpha 1-antichymotrypsin. Clin Chem. 1991;37:1618–25. [PubMed] [Google Scholar]
- [25].Christensson A, Bjork T, Nilsson O, et al. Serum prostate specific antigen complexed to alpha 1-antichymotrypsin as an indicator of prostate cancer. J Urol. 1993;150:100–5. doi: 10.1016/s0022-5347(17)35408-3. [DOI] [PubMed] [Google Scholar]
- [26].Finne P, Auvinen A, Maattanen L, et al. Diagnostic value of free prostate-specific antigen among men with a prostate-specific antigen level of <3.0 microg per liter. Eur Urol. 2008;54:362–70. doi: 10.1016/j.eururo.2007.10.056. [DOI] [PubMed] [Google Scholar]
- [27].Catalona WJ, Partin AW, Slawin KM, et al. Use of the percentage of free prostate-specific antigen to enhance differentiation of prostate cancer from benign prostatic disease: a prospective multicenter clinical trial. JAMA. 1998;279:1542–7. doi: 10.1001/jama.279.19.1542. [DOI] [PubMed] [Google Scholar]
- [28].Stenman UH, Leinonen J, Alfthan H, Rannikko S, Tuhkanen K, Alfthan O. A complex between prostate-specific antigen and alpha 1-antichymotrypsin is the major form of prostate-specific antigen in serum of patients with prostatic cancer: assay of the complex improves clinical sensitivity for cancer. Cancer Res. 1991;51:222–6. [PubMed] [Google Scholar]
- [29].Vickers AJ, Cronin AM, Aus G, et al. A panel of kallikrein markers can reduce unnecessary biopsy for prostate cancer: data from the European Randomized Study of Prostate Cancer Screening in Goteborg, Sweden. BMC Med. 2008;6:19. doi: 10.1186/1741-7015-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].WHO Expert Committee on Biological Standardization . World Health Organ Tech Rep Ser. 1999. p. 889. [PubMed] [Google Scholar]
- [31].Rafferty B, Rigsby P, Rose M, Stamey T, Gaines Das R. Reference reagents for prostate-specific antigen (PSA): establishment of the first international standards for free PSA and PSA (90:10) Clin Chem. 2000;46:1310–7. [PubMed] [Google Scholar]
- [32].Mitrunen K, Pettersson K, Piironen T, Bjork T, Lilja H, Lovgren T. Dual-label one-step immunoassay for simultaneous measurement of free and total prostate-specific antigen concentrations and ratios in serum. Clin Chem. 1995;41:1115–20. [PubMed] [Google Scholar]
- [33].Henderson AR. Chemistry with confidence: should Clinical Chemistry require confidence intervals for analytical and other data? Clin Chem. 1993;39:929–35. [PubMed] [Google Scholar]
- [34].Sottrup-Jensen L. Alpha-macroglobulins: structure, shape, and mechanism of proteinase complex formation. J Biol Chem. 1989;264:11539–42. [PubMed] [Google Scholar]
- [35].Christensson A, Laurell CB, Lilja H. Enzymatic activity of prostate-specific antigen and its reactions with extracellular serine proteinase inhibitors. Eur J Biochem. 1990;194:755–63. doi: 10.1111/j.1432-1033.1990.tb19466.x. [DOI] [PubMed] [Google Scholar]
- [36].Nielsen UB, Cardone MH, Sinskey AJ, MacBeath G, Sorger PK. Profiling receptor tyrosine kinase activation by using Ab microarrays. Proc Natl Acad Sci U S A. 2003;100:9330–5. doi: 10.1073/pnas.1633513100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].MacBeath G. Protein microarrays and proteomics. Nat Genet. 2002;32(Suppl):526–32. doi: 10.1038/ng1037. [DOI] [PubMed] [Google Scholar]
- [38].Zhou HP, Bouwman K, Schotanus M, et al. Two-color, rolling-circle amplification on antibody microarrays for sensitive, multiplexed serum-protein measurements. Genome Biology. 2004;5 doi: 10.1186/gb-2004-5-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Morris KN, Jensen KB, Julin CM, Weil M, Gold L. High affinity ligands from in vitro selection: complex targets. Proc Natl Acad Sci U S A. 1998;95:2902–7. doi: 10.1073/pnas.95.6.2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kim S, Kim Y, Kim P, et al. Improved sensitivity and physical properties of sol-gel protein chips using large-scale material screening and selection. Anal Chem. 2006;78:7392–6. doi: 10.1021/ac0520487. [DOI] [PubMed] [Google Scholar]