

Abstract
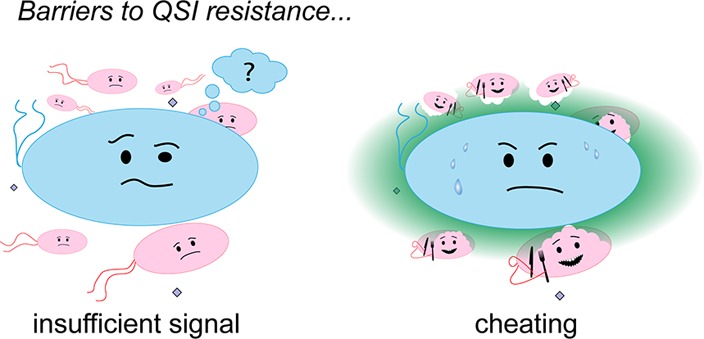
The growing threat of antibiotic resistance necessitates the development of novel antimicrobial therapies. Antivirulence agents that target group-beneficial traits in microorganisms (i.e., phenotypes that help the cells surrounding the producer cell instead of selfishly benefiting only the producer cell) represent a new antimicrobial approach that may be robust against the spread of resistant mutants. One prominent group-beneficial antivirulence target in bacteria is quorum sensing (QS). While scientists are producing new QS inhibitors (QSIs) at an increasing pace for use as research tools and potential therapeutic leads, substantial work remains in empirically demonstrating a robustness against resistance. Herein we report the results of in vitro competition studies in Pseudomonas aeruginosa that explicitly confirm that two separate barriers can impede the spread of resistance to QSIs: (1) insufficient native QS signal levels prevent rare QSI-resistant bacteria from expressing their QS regulon, and (2) group-beneficial QS-regulated phenotypes produced by resistant bacteria are susceptible to cheating by QSI-sensitive neighbors, even when grown on a solid substrate with limited mixing to mimic infected tissue. These results underscore the promise of QSIs and other antivirulence molecules that target group beneficial traits as resistance-robust antimicrobial treatments and provide support for their further development.
Microbes play essential roles in our world. However, these organisms can also have devastating effects on human health and productivity via pathogenic infection. The spread of antibiotic resistance is gradually disarming our society and causing increased morbidity, mortality, and costs associated with infection. In the U.S. alone, antibiotic-resistant hospital-acquired infections kill an estimated 50000–100000 annually1 and cost the U.S. society approximately $35 billion per year.2 This current toll, coupled with the increasing trajectory of resistance development, makes the spread of antibiotic resistance one of the world’s greatest health concerns.3
In view of these challenges, both chemists and biologists have become interested in developing “evolution-proof” drugs that are robust against the development of resistance by microbes.4,5 This represents a substantial undertaking, however. Traditional antibiotics are inherently highly susceptible to resistance, evidenced by the fact that resistance often appears in clinics only a few years after the first therapeutic use of a new antibiotic.5 Antibiotics prevent the growth of all drug-sensitive bacteria but allow resistant bacteria to grow. This scenario presents an extraordinarily strong selective pressure for a single resistant bacterium to propagate through the entire population. In the past decade, “antivirulence” drugs that do not directly kill bacteria but instead prevent pathogens from expressing their detrimental phenotypes have garnered increased attention.4,5 In particular, antivirulence agents that specifically target group-beneficial virulence traits (i.e., phenotypes that, when expressed, help not only the bacterium expressing them, but all of its neighbors as well) may allow for a fundamentally decreased rate of resistance spread within infections. This “resistance robustness” is possible because a resistant bacterium that arises would not selfishly benefit from its resistance but instead would help its antivirulence-drug-sensitive peers at a cost to itself.6−8 Examining resistance spread to such antivirulence approaches was the broad goal of the current study.
Our research group and others have focused on bacterial quorum sensing (QS), a widespread cooperative trait, as one such group-beneficial antivirulence target.9−11 QS is an intercellular chemical signaling mechanism that bacteria use to monitor their local cell densities.12,13 In QS, bacteria constitutively produce low levels of diffusible signal molecules. As the bacteria multiply in a confined space, more signal accumulates until it reaches a threshold concentration that both activates an autoinduction loop for increased signal production and also induces the expression of a set of genes that are beneficial for growth in a cell-dense environment. Viable QS inhibition strategies include competitive inhibition of signal binding to receptor proteins,11,14−19 inhibition of signal synthesis,20 and sequestration and degradation of signals.10,21 We note that the first strategy, competitive inhibition of signal binding, should also inhibit signal production due to the interruption of the signal-synthase autoinduction loop. Several small molecule- and macromolecule-based approaches to QS modulation are shown in Figure 1. Small molecule QS inhibitors (QSIs) targeting QS receptors have arguably seen the most intense study so far.
Figure 1.

Common QS inhibition strategies. (top) Representative competitive inhibitors of signal binding to QS receptor proteins are C-30,19 V-06-018,18 C14,17 itc-13,16 mBTL,15 and AIP-III D4A14. (bottom) Inhibitors of signal synthesis20 and availability10,21 have also been explored.
QS is a compelling resistance-robust antivirulence target because it involves two levels of group behavior that could lead to two barriers to the spread of resistance. First, each individual cell depends on the other cells in the population to produce signal molecules in order for sufficient signal to accumulate to induce QS gene expression.22 Second, many of the genes that are activated by QS are themselves group-beneficial (e.g., biofilm formation and secreted diffusible factors like proteases, siderophores, and toxins).23−25 These group-beneficial genes could render a bacterium that expresses its QS regulon susceptible to “cheating” by neighboring bacteria that lack functioning QS systems. The cheaters benefit from the production of these common goods by QS-active bacteria, but they do not reciprocate by producing the goods themselves. The existence of QS cheaters in bacterial populations has been extensively demonstrated both in vitro(24,26−28) and in vivo,29−31 but studies on the implications for resistance to QSIs are few and yield conflicting results.31−33 For example, one study by Wood and co-workers found that resistance to the QSI furanone C-30 (shown in Figure 1) via increased drug efflux can spontaneously arise and spread when the opportunistic pathogen Pseudomonas aeruginosa is grown in a minimal medium with adenosine as the sole carbon source.32 However, another study by Mellbye and Schuster suggested that QSI resistance does not spread when P. aeruginosa is grown in a minimal medium with bovine serum albumin (BSA) as the only carbon source,33 a competitive growth condition that has shown reasonable correlation with competition studies in animal infections and clinical isolates.26,27,29,30 The authors did not use a small molecule QSI in this latter study but rather used a pair of P. aeruginosa mutants that mimic QSI resistance.33 Lastly, a recent study showed that resistance failed to spread to the inhibition of a QS-regulated trait (siderophore activity in P. aeruginosa).34
We sought to reconcile these previous results in order to guide future QSI and antivirulence research in general. In the current study, we explicitly evaluated two unique obstacles that could preclude the spread of resistance to inhibitors of QS receptors. First, we hypothesized that a few resistant bacteria that spontaneously arise in a population of QSI-sensitive bacteria would not produce sufficient signal molecules to turn on QS and therefore would have no fitness advantage over sensitive bacteria. Second, in a situation where a few resistant bacteria might overcome the first obstacle and express their QS-regulated genes, we hypothesized that those resistant bacteria would be outcompeted by QSI-sensitive bacteria cheating off of the common goods produced by the resistant bacteria,22 even in a physically structured environment that models tissue infections. We explicitly examined these two barriers to resistance in isolation from each other using QS mutants of P. aeruginosa. The results of the competition studies described herein confirmed our hypotheses and provide strong support for QS inhibition as a potential resistance-robust approach to antimicrobial therapy.
Results and Discussion
Development of Experimental Conditions
We chose the Gram-negative pathogen P. aeruginosa for study because of its prevalence in antibiotic-resistant infections3 and its well-characterized QS system. This pathogen uses primarily LuxR/LuxI-type circuits for QS (i.e., the las and rhl systems),13 with which it controls the production of an arsenal of virulence factors and growth into impermeable biofilms in infections.23,35P. aeruginosa infection models in mice have demonstrated that QS is important for abundant growth in infections, presumably due to the QS-regulated production of secreted proteases and siderophores and defenses against the immune system.36−38 As such, common nutrient-rich growth media that do not require QS for growth are poor models of in vivo infection growth. We therefore utilized QS-selective growth media for the current study, in which the supplied carbon sources can only be utilized by bacteria after digestion by QS-regulated enzymes.26−28,32,33,39 Since P. aeruginosa regulates many phenotypes by QS, some of which are “selfish” (i.e., primarily aid growth of the individual expressing the phenotype) and many of which are “group-beneficial” (i.e., significantly aid growth of neighboring bacteria as well as the individual expressing the phenotype),23 both selfish and group-beneficial QS-selective media were used.26,27
The selfish QS-selective medium contained adenosine as the only carbon source because adenosine metabolism in P. aeruginosa requires the production of nucleoside hydrolase (Nuh), a periplasmic protein that is under QS control. In turn, the group-beneficial QS-selective medium contained the protein BSA as the main carbon source. Metabolism of BSA requires the presence of an extracellular protease to liberate simple peptide nutrients, which can be used not only by the bacterium that secreted the protease but also by cheating neighbors that do not secrete proteases. A predominant secreted protease in P. aeruginosa is elastase B (LasB), which is produced under QS control. We found that both media were QS selective, since wild-type P. aeruginosa PAO1 grew substantially better (i.e., reached maximal cell density over 5 days faster) than a QS mutant strain (ΔlasR, ΔrhlR) that lacked functional LuxR-type receptors in both media (Figure 2A,B; see Methods for details of strains).
Figure 2.

Demonstration of selective media and experimental setup. (A) Growth curves for monoculture QS+ (R) and QS– (S) strains grown in QSM + 0.1% adenosine (selfish QS-selective medium). (B) Growth curves for R and S in QSM + 0.1% CAA + 1% BSA (group-beneficial QS-selective medium). In both QS-selective media, monoculture R grew substantially better than monoculture S. (C) Schematic of the competition studies performed herein. A QSI-resistant mimic (PAO1::mini-Tn7-GFP-GmR) and a QSI-sensitive mimic (ΔlasR, ΔrhlR, TcR) were mixed and grown in QS-selective media. Initial and final ratios of resistant/sensitive bacteria were calculated by counting colony forming units (CFUs) of resistant mimics on gentamicin-containing plates and CFUs of sensitive mimics on tetracycline-containing plates. GmR = gentamicin resistant; TcR = tetracycline resistant.
At the outset, we considered treating P. aeruginosa with small molecule QSIs in these QS-selective media and monitoring whether resistance arose and spread over time.32 However, the QSIs in P. aeruginosa that are free of off-target growth affects do not sufficiently inhibit QS. For example, two of the most potent LasR receptor inhibitors, V-06-018 reported by Greenberg and co-workers18 and N-(3-nitrophenylacetanoyl)-l-homoserine lactone (C14) reported by our laboratory17 (Figure 1), still allow for at least 40% of the native level of LasB protease production in P. aeruginosa. This partial inhibition would lead to a weak QS-based selective pressure that would preclude a definitive selection experiment in our media. In addition, while furanone C-30 (Figure 1) is a widely studied QSI and has been shown to inhibit some QS-regulated behaviors in P. aeruginosa by more than 90%,19 we observed significant non-QS-based growth inhibition by this compound at the concentrations necessary for QS inhibition (see Supplementary Text and Supplementary Figure 1, Supporting Information). As highlighted above, Wood and co-workers used this QSI in their recent study to demonstrate that resistance can quickly arise to QSIs.32 However, we believe that C-30’s off-target growth effects impose non-QS-based selective pressures for the spread of resistance. In fact, such off-target growth effects are a feature that we and other research groups explicitly avoid in the ongoing design of improved QSIs.11
Thus, to test resistance to a future ideal QSI in the current study, we instead utilized a pair of P. aeruginosa strains to mimic a QSI-sensitive bacterium and a QSI-resistant bacterium (Figure 3).33 To mimic a QSI-sensitive bacterium (termed S hereafter) having its QS system chemically knocked down nearly 100% by a potent and selective QS receptor inhibitor, we used the P. aeruginosa QS mutant strain (ΔlasR, ΔrhlR). To mimic a QSI-resistant bacterium (termed R hereafter) having a functioning QS system in the presence of a QSI, we used wild-type P. aeruginosa PAO1. The QS mutant had no observable growth defects (see Supplementary Text and Supplementary Figure 2B, Supporting Information), except in QS-selective media (Figure 2A,B). These two strains therefore model sensitivity and resistance to an ideal QSI that completely inhibits QS with no off-target effects (Figure 3).
Figure 3.
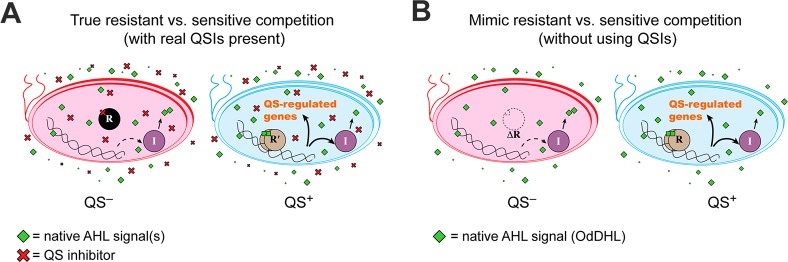
Comparison of a “true” QSI resistance competition to the experimental mimic competition in this study. “R” circles represent QS receptor proteins, and “I” circles represent QS signal synthase proteins. In the true case (A), the wild-type bacteria have chemically knocked down QS (red), and the resistant mutants are still capable of QS even in the presence of the QSI (blue). In the mimic case (B), a P. aeruginosa ΔlasR ΔrhlR mutant has a genetically knocked down QS system (red) to mimic the QSI-sensitive strain, and the resistant mimic is wild-type PAO1 (blue), which is fully capable of QS under the experimental conditions. In both panels, substantial native AHL signals are shown, but if the resistant bacteria are rare, much less signal will actually be present (due to poor signal production by the QS-inhibited strains).
To model a few resistant bacteria arising spontaneously under QSI treatment, we grew populations of the QSI-sensitive mimics (S) seeded with a small number of QSI-resistant mimics (R). We labeled strains R and S with different antibiotic resistance markers, which allowed us to readily count the R/S ratio before and after growth to determine whether the few resistant bacteria were more fit and spread through the population (Figure 2C). In all subsequent experiments, resistance spread was quantified by calculating the relative fitness of R vs S, which is the final ratio of R/S divided by the initial R/S ratio (see Methods).40 Relative fitness values >1 indicated that a spread of resistance had occurred.
First Barrier to Resistance: Nonquorate Signal Levels
We first tested the hypothesis that a population of QSI-sensitive bacteria treated with a QSI would not produce sufficient signal to activate the QS system of a small number of QSI-resistant “infiltrators.” Therefore, if such QSI-resistant mutants were to arise, they would not be more fit than neighboring QSI-sensitive bacteria. An example case is the development of a mutant that effluxes a QSI efficiently. The effective concentration of the QSI would be lower for that cell, so its QS system would no longer be inhibited. However, that cell would still require a quorum level of native QS signal in order to express its QS-regulated genes. Since the other cells in the population are still inhibited, they would not express sufficient QS signal to induce QS in the resistant bacterium (Figure 4A).
Figure 4.

Relative fitness of resistant (R) versus sensitive (S) mimic strains. R and S were grown in coculture with selfish phenotype selection (adenosine carbon source, gray) and group-beneficial phenotype selection (BSA carbon source, black). (A) Schematic demonstrating a nonquorate rare signal-dependent QSI-resistant mutant. (B) Relative fitness of signal-dependent R vs S. (C) Schematic demonstrating a signal-independent QSI-resistant mutant that can express its QS regulon, even when rare. (D) Relative fitness of signal-independent R vs S. Relative fitness values >1 indicate that the resistant mimic is more fit and will spread. Data are represented as box and whisker plots. Each dot is an individual data point. Boxes encompass the inner quartiles, and horizontal lines are median values. Whiskers extend to the furthest data points. The statistical significance of relative fitness deviations from 1 were tested via paired t tests comparing the logarithm of the final R/S ratio to the logarithm of the initial R/S ratio for each sample (****p < 0.0001, ***p < 0.001, **p < 0.01, *p < 0.05, not significant (ns) p > 0.05).
We examined this barrier by competing the resistant mimic (R) with the sensitive mimic (S) in coculture in the “selfish” QS-selective media. By using the selfish media, we excluded any potential fitness effects due to cheating (i.e., the second hypothesized barrier, see below) and therefore explicitly tested the existence of only the first barrier. We mixed the bacteria at three different initial proportions of R (50%, 1%, and 0.01%). In the extreme case that 50% of the population was initially resistant, sufficient native QS signal should be produced for the resistant bacteria to be quorate, as shown by quantifying the amount of native QS signal (N-(3-oxo)-dodecanoyl-l-homoserine lactone, OdDHL) extracted from grown culture (see Supplementary Figure 3, Supporting Information). However, at initial proportions at or below 1% R, the population should remain nonquorate, even at high total bacterial densities (Supplementary Figure 3, Supporting Information). The most relevant case in an infection would be very low levels of resistant bacteria spontaneously arising (≪1%); thus we tested 0.01% R.
In each competitive coculture trial at quorate levels of R (i.e., 1:1 R/S ratio), the fitness of R was greater than S (i.e., relative fitness R/S > 1, Figure 4B). However, when R was seeded as ≤1% of the population (relevant to resistance arising as ≪1% of an infection), the resistant mimics did not consistently outcompete their QSI-sensitive neighbors. These data support the hypothesis that low levels of QSI-resistant bacteria are incapable of expressing their QS genes and therefore have no fitness advantage over QSI-sensitive neighbors. We note that this finding expands on the recent work of Mellbye and Schuster (as introduced above),33 who demonstrated that QSI resistance failed to spread in a group-beneficial selective medium. The authors concluded that resistance failed to spread in their experiment because the phenotypes under QS control (i.e., production and secretion of proteases) were group-beneficial. Our results now demonstrate that even using a selective pressure based on a selfish QS-regulated phenotype, QSI resistance should not spread due to a dependence on signal production. This result is significant, because it argues that QSI resistance could be even less likely to spread than previous work has suggested.
Our data were initially difficult to reconcile with the work of Wood and co-workers using furanone C-30 (vide supra).32 While our results suggest that improved drug efflux mutants should not have a fitness advantage when they are at low levels in a QS-inhibited population, Wood and co-workers showed that improved-efflux mutants (via overexpression of the MexAB-OprM drug efflux pump) were able to readily spread under C-30 treatment. As mentioned above, we believe that the selective pressure present in this previous study is due to furanone C-30 imposing general, non-QS-related growth-inhibitory affects on P. aeruginosa in minimal media and is not due to the QSI activity of C-30. If C-30 were an ideal QSI without off-target effects, we contend that low levels of improved efflux mutants would not spread through a population. Therefore, the Wood study and our study work together to underscore the need to design better QSIs that do not suffer from off-target affects that can select for resistance.
An insightful conclusion from the work of Wood and co-workers is that many P. aeruginosa strains in chronic infections are already resistant to QSIs via overexpression of the MexAB-OprM pump.32 This mode of small molecule resistance is common and presumably arises due to the selective pressure imposed by previous treatment with antibiotics. Consequently, we believe that ideal resistance-robust QSIs should (1) not have nonselective growth inhibitory effects and (2) not be susceptible to the same resistance mutations as traditional antibiotics that are used before (or in conjunction with) the QSI. Our findings suggest that if such QSIs are developed, they will be robust against the spread of signal-dependent resistance mechanisms, regardless of the selfishness of the QS-regulated phenotypes needed for growth. In this context, recent studies in our laboratory have shown that certain QSI scaffolds can evade active efflux in P. aeruginosavia the MexAB-OprM pump,41 which serves to leverage these compounds for future development.
Second Barrier to Resistance: Group-Beneficial QS-Regulated Genes
Although signal-dependent mechanisms of resistance would be thwarted by the first barrier described above (e.g., efflux pump overexpression, QSI degradation, or target protein modification to become immune), additional mechanisms of resistance are conceivable that would not require quorate levels of native signal in order to express the QS regulon. Examples of “signal-independent” resistance mechanisms include mutations that lead to constitutive expression of the QS regulon or cause the QS receptor protein to respond to the QSI as an agonist instead of an antagonist. The latter mechanism has been observed with mutations of the QS receptors CviR42 and LuxR17,43 in Chromobacterium violaceum and Vibrio fischeri, respectively. These resistance mechanisms would enable rare resistant bacteria to express different genes than their QSI-sensitive neighbors, which could give them a fitness advantage (see Figure 4C). In these cases, we reasoned that a second barrier would arise to inhibit the spread of resistance: cheating by QSI-sensitive bacteria off the group-beneficial phenotypes expressed by the QSI-resistant bacteria. The concept of social cheating has been well studied for the past 50 years,44,45 and in particular, QS-based cheating has been demonstrated in vitro(24,26,27) and in infections in vivo.29,30 These past studies have explored whether low levels of cheaters could invade a population of cooperative bacteria. In contrast, we sought to address the opposite question in the current study, by investigating whether low levels of QS-cooperators can outcompete QS-cheaters. This question is directly relevant to the situation of QSI-resistant cooperators arising in a population of QSI-sensitive cheaters. To our knowledge, the only other study that investigated this situation is that of Mellbye and Schuster (vide supra);33 however, the authors did not test very low initial levels of resistance (≪1% of the cells), which should be the most relevant condition to initial stages of resistance spread. Furthermore, this past work mimicked signal-dependent resistance mechanisms and therefore could have underestimated the ability of signal-independent mechanisms to spread. We therefore explicitly tested the existence of this second barrier by performing new competition experiments with low initial frequencies of signal-independent QSI-resistant cooperators to test whether QSI-resistant bacteria could be more fit and spread.
To imitate signal-independent resistance mechanisms, we developed experimental protocols where the resistant mimics were artificially coerced to express their QS-regulated genes even when they were rare in the population. For selection based on a selfish phenotype, the native P. aeruginosa QS signal OdDHL was added to the selection medium to induce expression of nuh. To induce production of LasB in the group-beneficial selection, the resistant mimic was engineered to constitutively express lasB (see Supplementary Text and Supplementary Figures 4 and 5, Supporting Information). Using these procedures, we found that the signal-independent resistant mimics were substantially more fit than the sensitive mimics (i.e., relative fitness R/S > 1) under selfish selection, even when the signal-independent resistant mimics initiate at low levels in the population (see Figure 4D). In line with our initial hypothesis, however, signal-independent resistant mimics were not more fit than sensitive mimics when growth was dependent on the group-beneficial QS-regulated production of LasB (relative fitness R/S ≤ 1; Figure 4D). In total, these results demonstrate, for the first time, that even signal-independent resistant bacteria are incapable of spreading when the QS-regulated selective pressures at work are group-beneficial, which provides a second barrier to the spread of QSI resistance.
Although previous research has suggested that selfish QS-regulated traits could be relevant in infections and could drive the spread of resistance to QSIs,28,32 recent experiments with mouse infection models instead match these results in protease-selective media very well.29,46 A study of cystic fibrosis clinical isolates30 also suggests that group-beneficial selective pressures are significant in P. aeruginosa infections in humans. Furthermore, a very recent study demonstrated that resistance did not develop to a virulence inhibitor targeting the agr system of Staphylococcus aureus in a mouse infection model, presumably due to social cheating.47 Therefore, we believe that this hurdle of social cheating will present an obstacle to the spread of QSI resistance in infections as well as in vitro.
Effect of Local Population Structure on Resistance Spread
The competition studies above, as well as related experiments,32,33 were all performed in well-mixed liquid culture. In reality, many infections have a more spatially structured, biofilm appearance.48,49 As Figure 5A illustrates, structured populations on solid matrices can keep secreted goods closer to the bacteria that produce them.40,50 As such, the impact of population structure on QS resistance spread could be significant. To examine this phenomenon, we converted the group-beneficial selective medium described above to a solid growth medium50 and repeated the competition studies with initial ratios of 1:100 R/S. Multiple degrees of population structure were tested by altering the plating technique to obtain interspersed monoclonal patches of R and S that were each 0.1 mm, 1 mm, or 1 cm diameter (Figures 5B–D). Larger monoclonal patches should provide greater sharing of goods among the resistant mimics because the resistant cells are on average closer to other resistant cells than sensitive cells. We reasoned that this cooperativity should provide the resistant mimics with a greater advantage over the sensitive mimics. Our results showed that only when the patches were very large (1 cm diameter) did the signal-independent resistant mimics have an advantage (Figure 5E).
Figure 5.

Structured environments retain susceptibility to cheating. (A) Schematic showing that a well-mixed culture would quickly distribute goods away from the producers and also prevent producers from making monoclonal patches of high goods concentrations. (B–D) Images demonstrating the increasing degrees of population structure that were tested. In panels B and C, fluorescence microscopy was used to visualize 1:10 ratios of R producing GFP (green) and S producing mCherry (red). In panel D, a 1:100 ratio of R vs S was visualized without magnification: the macrocolony marked with a red dot is R, and the 99 other spots on the plate are S. (E) Relative fitness of signal-independent R vs S in liquid group-beneficial medium and in solid group-beneficial medium with different degrees of population structure. Data analysis was analogous to that described in Figure 4. (F) Image visualizing the diffusion of protease-digested goods after 1 day of growth of a 1:100 ratio of R vs S. The bright halo around the single R colony (circled) is diffused protease-digested fluorogenic substrate. The substrate diffusion indicates the distance over which QSI-sensitive bacteria can cheat off neighboring QSI-resistant bacteria.
We hypothesized that the necessary patch size for a resistance advantage was related to the distance that goods diffuse from the producer. To test the diffusion distance of the protease-digested goods, we added a LasB substrate to the plate that fluoresces once cleaved by LasB and then repeated the competition experiment above with 1 cm colony spacing. After only 1 day of incubation (before substantial selective growth has occurred), the digested product had diffused approximately 1 cm past the edges of the resistant colony (see Figure 5F). A similar degree of diffusion has previously been reported for siderophores through agar.50 Since the 1 cm population structure was on the same size scale as the goods diffusion (as in the right image of Figure 5A), the goods stayed mostly near the large protease-producing resistant colony, making the resistant bacteria more fit. However, when the patches were smaller, the diffused goods benefitted hundreds to tens of thousands of surrounding QSI-sensitive patches, which enabled substantial cheating. Together, these results indicate that resistance should not spread within a microbial population under group-beneficial selection, even if grown in a viscous or spatially structured environment, as long as the colony-to-colony distance is smaller than the diffusion distance of secreted goods. Although diffusion rates through 1.5% agar are unlikely to directly correlate to diffusion in infections in vivo, we note that the 1 cm separation distance needed for the spread of resistance is more than 100× larger than the monoclonal colony separation observed within reported images of biofilm infection biopsies (≤0.1 mm).48
Conclusions and Outlook
We expect QSI-resistant mutants to arise in nature.32,42 However, at the outset of this study, we hypothesized that the QSI-resistant mutants would struggle to overtake their population relative to traditional antibiotic resistant mutants. The competition studies reported herein provide the first empirical evidence that (1) QS-signal-dependence is sufficient to impede the spread of many mechanisms of QSI resistance, (2) cheating is sufficient to impede the spread of even signal-independent mechanisms of QSI resistance under in vivo-relevant group-beneficial selection, and (3) reasonable degrees of population structure on a solid matrix still do not enable signal-independent QSI-resistant bacteria to spread. While our experiments were designed to directly mimic resistance to QSIs that block QS receptor function, we believe that the results apply broadly to the other QS inhibition strategies (i.e., inhibition of signal synthesis and sequestration and degradation of signal molecules; Figure 1). Because previous research has shown good correlation between protease-based in vitro selective pressures and in vivo selective pressures in mouse infections and human cystic fibrosis lungs,26,27,29,30 we are optimistic that these barriers to the spread of resistance will be relevant in infections. Ongoing research in our laboratory is focused on studying the impact of multispecies cultures51,52 and population expansion53 on the spread of QSI resistance, because these have recently been shown to affect microbial competition.
We close by highlighting an additional potential advantage of QS inhibition (or other antivirulence approaches) compared with traditional antibiotics; namely, these approaches should not affect the growth of nonpathogenic bacteria in natural environments. A major cause of the prevalence of resistance in pathogens is that antibiotics select for resistance in the harmless bacteria in human guts and in the environment.54,55 As resistance genes become more abundant in environmental bacteria, the genes have an increased likelihood of transferring to neighboring pathogens.56 Since QSIs and other antivirulence approaches are likely narrow spectrum and only affect fitness in specific settings, they should not broadly increase the prevalence of resistance genes in the environment.5 When this feature is coupled with the results described herein, which indicate that two unique barriers impede QSI-resistant pathogens from outcompeting their QSI-sensitive neighbors, we conclude that QS inhibition and other antivirulence approaches have substantial promise as resistance-robust therapeutics.
Methods
Strains and Routine Growth Conditions
All strains and plasmids used are listed in Supplementary Table 1, Supporting Information. Detailed strain construction procedures are in the Supplementary Text, Supporting Information. In brief, the QSI-sensitive mimic strain (S) (ΔlasR, ΔrhlR, TcR), the QSI-resistant mimic strain (R) (GFP+, GmR), and the signal-independent resistant mimic (R-lasB) (Ptac-lasB, SmR) were constructed from the same parent P. aeruginosa PAO1 strain. S was constructed by homologous recombination,57 and R and R-lasB were constructed by insertion of mini-Tn7 cassettes.58 For microscopy, pMP760559 was added to S to produce mCherry. All plasmids were conjugated into P. aeruginosa strains by mating with Escherichia coli S17-1::λpir. Luria–Bertani (LB) broth (1% peptone, 0.5% yeast extract, 0.5% NaCl, EMD Millipore) was used for all cloning, E. coli growth, and P. aeruginosa overnight cultures. Bacteria were grown at 37 °C with 200 rpm shaking unless noted otherwise. When needed, antibiotics were used at the following concentrations: 15 μg/mL gentamicin, 20 μg/mL tetracycline, 500 μg/mL streptomycin, 100 μg/mL ampicillin, and 50 μg/mL kanamycin.
Competitive Growth Experiments
Overnight cultures of strains R and S were mixed in different ratios to final volumes of 500 μL. The mixtures were rinsed 2 times with M9 salts (47.9 mM Na2HPO4, 22.0 mM KH2PO4, and 8.56 mM NaCl) to remove products from overnight growth. The mixtures were serially diluted and plated on LB + antibiotic plates (one gentamicin or streptomycin and one tetracycline). R and S colony-forming units (CFUs) were counted on their respective antibiotic plates and used to determine the initial R/S ratios. For liquid competition experiments, 2 μL of a 1:10 dilution of the rinsed mixtures was inoculated into wells of 96-well microtiter plates containing 198 μL of the QS-selective growth media (see Supplementary Text, Supporting Information, for recipe)39 supplemented with either 1% BSA (Sigma-Aldrich) and 0.1% casamino acids (CAA; Acros) for group-beneficial selection or 0.1% adenosine (Sigma-Aldrich) for selfish selection. To mimic signal-independent resistance in the adenosine conditions, 1 μL of the native QS signal OdDHL (Sigma-Aldrich) was added from a 400 μM DMSO stock solution (final concentration of 2 μM, with 0.5% DMSO). Growth was monitored by OD600 measurements using a microplate reader (Synergy 2, BioTek Instruments, Inc., see Supplementary Figure 6, Supporting Information, for representative growth curves). When the cultures reached stationary phase or grew for 150 h (whichever occurred first), they were serially diluted in M9 salts and plated on LB + antibiotic plates. CFUs were counted for calculation of final R/S ratios. In case biofilms formed during the course of extended growth, cultures were thoroughly resuspended and mixed by pipetting up and down and scraping the sides and bottoms of the wells before serial dilution and CFU determination. To avoid complications due to evaporation, only the inner wells of the 96-well plates were inoculated, and the outer wells were filled with sterile water or media. At the end of growth, the inoculated wells still contained >170 μL of liquid.
For solid competition experiments, the same media recipe was used except 1.5% agar was added, and 0.03% CAA was used instead of 0.1%. Plates were inoculated by three different methods to afford different degrees of population structure (see Supplementary Text, Supporting Information), followed by incubation at 30 °C. When the plates had thick growth and pigment production or had been grown for 12 days (whichever occurred first), cells were resuspended from the plate using 3 × 2 mL rinses with M9 salts and scraping with a bent glass pipet. The resuspensions were serially diluted and plated on LB + antibiotic plates for CFU counting.
For all competition studies, “relative fitness R/S” (v) was calculated by the method of Ross-Gillespie et al.:60v = (x1(1 – x0))/(x0(1 – x1)), where x0 and x1 are the initial and final resistant mimic frequencies, respectively. Values of v > 1 indicate that the resistant mimic outcompeted the sensitive mimic (i.e., resistance is spreading). Values of v ≤ 1 indicate a lack of resistance spread.
Microscopy
Epifluorescence microscopy with GFP and mCherry filters was performed on solid cultures directly through the agar plates after incubation at 30 °C for 6 days (see Supplementary Text, Supporting Information, for details).
Measurement of the Diffusion Distance of LasB-Digested Common Goods
The LasB substrate (2-aminobenzoylalanyl-glycyl-leucyl-alanyl-4-nitrobenzylamide; Peptides International) was added to the QSM + 1% BSA + 0.03% CAA agar mixture from a DMSO stock. The final concentration was 80 μM substrate with 0.03% DMSO. After incubation with bacteria at 30 °C for 24 h, cleaved substrate was imaged with a UV transilluminator (312 nm; TFP-M/WL, Vilber Lourmat) in conjunction with the FOTO/Analyst Apprentice system (Fotodyne, Inc.).
Acknowledgments
Financial support was provided by the NIH (Grants AI063326 and GM109403), the Burroughs Wellcome Foundation, and the Greater Milwaukee Foundation Shaw Scientist Program. J.P.G. was supported in part by the Department of Defense (DoD) Air Force Office of Scientific Research through a National Defense Science & Engineering Graduate (NDSEG) Fellowship (32 CFR 168a) and by the NIH through the UW–Madison Chemistry-Biology Interface Training Grant (NIGMS T32 GM008505). We thank M. Schuster and S. P. Diggle for helpful discussions, B. H. Iglewski, E. P. Greenberg, and T. K. Wood for P. aeruginosa strains, S. Molin for mini-Tn7 transposon strains, E. L. Lagendijk for mCherry plasmids, J. Tommassen for pML27, E. G. Ruby for microscope use, and E. R. Strieter for plasmids and E. coli strains.
Supporting Information Available
Additional methods and experiments in support of the main text, bacterial strains and plasmids used in this study, growth inhibitory effects of brominated furanone C-30 on P. aeruginosa PA14, elastase activity and growth curves of wild-type P. aeruginosa PAO1 compared with mutants, quantification of the native P. aeruginosa QS signal, OdDHL, in mixed R/S cultures, degradation of OdDHL by P. aeruginosa cultures, confirmation of LasB production by the “signal-independent” R-lasB mutant, and representative bacterial growth curves from competition experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Spellberg B.; Blaser M.; Guidos R. J.; Boucher H. W.; Bradley J. S.; Eisenstein B. I.; Gerding D.; Lynfield R.; Reller L. B.; Rex J.; Schwartz D.; Septimus E.; Tenover F. C.; Gilbert D. N. (2011) Combating antimicrobial resistance: Policy recommendations to save lives. Clin. Infect. Dis. 52, S397–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R. R.; Hota B.; Ahmad I.; Scott R. D.; Foster S. D.; Abbasi F.; Schabowski S.; Kampe L. M.; Ciavarella G. G.; Supino M.; Naples J.; Cordell R.; Levy S. B.; Weinstein R. A. (2009) Hospital and societal costs of antimicrobial-resistant infections in a Chicago teaching hospital: Implications for antibiotic stewardship. Clin. Infect. Dis. 49, 1175–1184. [DOI] [PubMed] [Google Scholar]
- Boucher H. W.; Talbot G. H.; Bradley J. S.; Edwards J. E.; Gilbert D.; Rice L. B.; Scheld M.; Spellberg B.; Bartlett J. (2009) Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12. [DOI] [PubMed] [Google Scholar]
- Allen R. C.; Popat R.; Diggle S. P.; Brown S. P. (2014) Targeting virulence: Can we make evolution-proof drugs?. Nat. Rev. Microbiol. 12, 300–308. [DOI] [PubMed] [Google Scholar]
- Clatworthy A. E.; Pierson E.; Hung D. T. (2007) Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 3, 541–548. [DOI] [PubMed] [Google Scholar]
- Andre J.-B.; Godelle B. (2005) Multicellular organization in bacteria as a target for drug therapy. Ecol. Lett. 8, 800–810. [Google Scholar]
- Pepper J. W. (2008) Defeating pathogen drug resistance: Guidance from evolutionary theory. Evolution 62, 3185–3191. [DOI] [PubMed] [Google Scholar]
- Brown S. P.; West S. A.; Diggle S. P.; Griffin A. S. (2009) Social evolution in micro-organisms and a Trojan horse approach to medical intervention strategies. Philos. Trans. R. Soc. London, Ser. B 364, 3157–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praneenararat T.; Palmer A. G.; Blackwell H. E. (2012) Chemical methods to interrogate bacterial quorum sensing pathways. Org. Biomol. Chem. 10, 8189–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara N.; Krom B. P.; Kaufmann G. F.; Meijler M. M. (2011) Macromolecular inhibition of quorum sensing: Enzymes, antibodies, and beyond. Chem. Rev. 111, 195–208. [DOI] [PubMed] [Google Scholar]
- Galloway W. R. J. D.; Hodgkinson J. T.; Bowden S. D.; Welch M.; Spring D. R. (2011) Quorum sensing in Gram-negative bacteria: Small-molecule modulation of AHL and AI-2 quorum sensing pathways. Chem. Rev. 111, 28–67. [DOI] [PubMed] [Google Scholar]
- Miller M.; Bassler B. L. (2001) Quorum sensing in bacteria. Annu. Rev. Microbiol. 55, 165–199. [DOI] [PubMed] [Google Scholar]
- Fuqua C.; Parsek M. R.; Greenberg E. P. (2001) Regulation of gene expression by cell-to-cell communication: Acyl-homoserine lactone quorum sensing. Annu. Rev. Genet. 35, 439–468. [DOI] [PubMed] [Google Scholar]
- Tal-Gan Y.; Stacy D. M.; Foegen M. K.; Koenig D. W.; Blackwell H. E. (2013) Highly potent inhibitors of quorum sensing in Staphylococcus aureus revealed through a systematic synthetic study of the group-III autoinducing peptide. J. Am. Chem. Soc. 135, 7869–7882. [DOI] [PubMed] [Google Scholar]
- O’Loughlin C. T.; Miller L. C.; Siryaporn A.; Drescher K.; Semmelhack M. F.; Bassler B. L. (2013) A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 110, 17981–17986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amara N.; Mashiach R.; Amar D.; Krief P.; Spieser S. A. H.; Bottomley M. J.; Aharoni A.; Meijler M. M. (2009) Covalent inhibition of bacterial quorum sensing. J. Am. Chem. Soc. 131, 10610–10619. [DOI] [PubMed] [Google Scholar]
- Geske G. D.; O’Neill J. C.; Miller D. M.; Mattmann M. E.; Blackwell H. E. (2007) Modulation of bacterial quorum sensing with synthetic ligands: systematic evaluation of N-acylated homoserine lactones in multiple species and new insights into their mechanisms of action. J. Am. Chem. Soc. 129, 13613–13625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müh U.; Schuster M.; Heim R.; Singh A.; Olson E. R.; Greenberg E. P. (2006) Novel Pseudomonas aeruginosa quorum-sensing inhibitors identified in an ultra-high-throughput screen. Antimicrob. Agents Chemother. 50, 3674–3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentzer M.; Wu H.; Andersen J. B.; Riedel K.; Rasmussen T. B.; Bagge N.; Kumar N.; Schembri M.; Song Z.; Kristoffersen P.; Manefield M.; Costerton J.; Molin S.; Eberl L.; Steinberg P.; Kjelleberg S.; Hoiby N.; Givskov M. (2003) Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22, 3803–3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen Q. H.; Grove T. L.; Booker S. J.; Greenberg E. P. (2013) A high-throughput screen for quorum-sensing inhibitors that target acyl-homoserine lactone synthases. Proc. Natl. Acad. Sci. U.S.A. 110, 13815–13820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piletska E. V.; Stavroulakis G.; Larcombe L. D.; Whitcombe M. J.; Sharma A.; Primrose S.; Robinson G. K.; Piletsky S. A. (2011) Passive control of quorum sensing: Prevention of Pseudomonas aeruginosa biofilm formation by imprinted polymers. Biomacromolecules 12, 1067–1071. [DOI] [PubMed] [Google Scholar]
- Swem L. R.; Swem D. L.; O’Loughlin C. T.; Gatmaitan R.; Zhao B.; Ulrich S. M.; Bassler B. L. (2009) A quorum-sensing antagonist targets both membrane-bound and cytoplasmic receptors and controls bacterial pathogenicity. Mol. Cell 35, 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M.; Greenberg E. P. (2006) A network of networks: Quorum-sensing gene regulation in Pseudomonas aeruginosa. Int. J. Med. Microbiol. 296, 73–81. [DOI] [PubMed] [Google Scholar]
- Wilder C. N.; Diggle S. P.; Schuster M. (2011) Cooperation and cheating in Pseudomonas aeruginosa: The roles of the las, rhl and pqs quorum-sensing systems. ISME J. 5, 1332–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popat R.; Crusz S. A.; Messina M.; Williams P.; West S. A.; Diggle S. P. (2012) Quorum-sensing and cheating in bacterial biofilms. Proc. Biol. Sci. 279, 4765–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle S. P.; Griffin A. S.; Campbell G. S.; West S. A. (2007) Cooperation and conflict in quorum-sensing bacterial populations. Nature 450, 411–414. [DOI] [PubMed] [Google Scholar]
- Sandoz K. M.; Mitzimberg S. M.; Schuster M. (2007) Social cheating in Pseudomonas aeruginosa quorum sensing. Proc. Natl. Acad. Sci. U.S.A. 104, 15876–15881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dandekar A. A.; Chugani S.; Greenberg E. P. (2012) Bacterial quorum sensing and metabolic incentives to cooperate. Science 338, 264–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh K. P.; Diggle S. P.; Watters C. M.; Ross-Gillespie A.; Griffin A. S.; West S. A. (2009) Quorum sensing and the social evolution of bacterial virulence. Curr. Biol. 19, 341–345. [DOI] [PubMed] [Google Scholar]
- Köhler T.; Buckling A.; van Delden C. (2009) Cooperation and virulence of clinical Pseudomonas aeruginosa populations. Proc. Natl. Acad. Sci. U.S.A. 106, 6339–6344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler T.; Perron G. G.; Buckling A.; van Delden C. (2010) Quorum sensing inhibition selects for virulence and cooperation in Pseudomonas aeruginosa. PLoS Pathog. 6, e1000883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda T.; García-Contreras R.; Pu M.; Sheng L.; Garcia L. R.; Tomás M.; Wood T. K. (2012) Quorum quenching quandary: Resistance to antivirulence compounds. ISME J. 6, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellbye B.; Schuster M. (2011) The sociomicrobiology of antivirulence drug resistance: A proof of concept. mBio 2, e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Gillespie A.; Weigert M.; Brown S. P.; Kümmerli R. (2014) Gallium-mediated siderophore quenching as an evolutionarily robust antibacterial treatment. Evol. Med. Public Health 2014, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kievit T. R. (2009) Quorum sensing in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 11, 279–288. [DOI] [PubMed] [Google Scholar]
- Defoirdt T.; Boon N.; Bossier P. (2010) Can bacteria evolve resistance to quorum sensing disruption?. PLoS Pathog. 6, e1000989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh K. P.; Griswold J.; Iglewski B. H.; Hamood A. (1999) Contribution of quorum sensing to the virulence of Pseudomonas aeruginosa in burn wound infections. Infect. Immun. 67, 5854–5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Song Z.; Hentzer M.; Andersen J. B.; Molin S.; Givskov M.; Høiby N. (2004) Synthetic furanones inhibit quorum-sensing and enhance bacterial clearance in Pseudomonas aeruginosa lung infection in mice. J. Antimicrob. Chemother. 53, 1054–1061. [DOI] [PubMed] [Google Scholar]
- Darch S. E.; West S. A.; Winzer K.; Diggle S. P. (2012) Density-dependent fitness benefits in quorum-sensing bacterial populations. Proc. Natl. Acad. Sci. U.S.A. 109, 8259–8263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin A. S.; West S. A.; Buckling A. (2004) Cooperation and competition in pathogenic bacteria. Nature 430, 1024–1027. [DOI] [PubMed] [Google Scholar]
- Moore J. D.; Gerdt J. P.; Eibergen N. R.; Blackwell H. E. (2014) Active efflux influences the potency of quorum sensing inhibitors in Pseudomonas aeruginosa. ChemBioChem. 15, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Swem L. R.; Swem D. L.; Stauff D. L.; O’Loughlin C. T.; Jeffrey P. D.; Bassler B. L.; Hughson F. M. (2011) A strategy for antagonizing quorum sensing. Mol. Cell 42, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C. H.; Arnold F. H.; Leadbetter J. R. (2005) Directed evolution of Vibrio fischeri LuxR for increased sensitivity to a broad spectrum of acyl-homoserine lactones. Mol. Microbiol. 55, 712–723. [DOI] [PubMed] [Google Scholar]
- Hamilton W. D. (1964) The genetical evolution of social behaviour. I. J. Theor. Biol. 7, 1–16. [DOI] [PubMed] [Google Scholar]
- Hamilton W. D. (1964) The genetical evolution of social behaviour. II. J. Theor. Biol. 7, 17–52. [DOI] [PubMed] [Google Scholar]
- Rumbaugh K. P.; Trivedi U.; Watters C.; Burton-Chellew M. N.; Diggle S. P.; West S. A. (2012) Kin selection, quorum sensing and virulence in pathogenic bacteria. Proc. Biol. Sci. 279, 3584–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sully E. K.; Malachowa N.; Elmore B. O.; Alexander S. M.; Femling J. K.; Gray B. M.; Deleo F. R.; Otto M.; Cheung A. L.; Edwards B. S.; Sklar L. A.; Horswill A. R.; Hall P. R.; Gresham H. D. (2014) Selective chemical inhibition of agr quorum sensing in Staphylococcus aureus promotes host defense with minimal impact on resistance. PLoS Pathog. 10, e1004174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmølle M.; Thomsen T. R.; Fazli M.; Dige I.; Christensen L.; Homøe P.; Tvede M.; Nyvad B.; Tolker-Nielsen T.; Givskov M.; Moser C.; Kirketerp-Møller K.; Johansen H. K.; Høiby N.; Jensen P. Ø.; Sørensen S. J.; Bjarnsholt T. (2010) Biofilms in chronic infections - a matter of opportunity - monospecies biofilms in multispecies infections. FEMS Immunol. Med. Microbiol. 59, 324–336. [DOI] [PubMed] [Google Scholar]
- Bjarnsholt T.; Alhede M.; Alhede M.; Eickhardt-Sørensen S. R.; Moser C.; Kühl M.; Jensen P. Ø.; Høiby N. (2013) The in vivo biofilm. Trends Microbiol. 21, 466–474. [DOI] [PubMed] [Google Scholar]
- Kümmerli R.; Griffin A. S.; West S. A.; Buckling A.; Harrison F. (2009) Viscous medium promotes cooperation in the pathogenic bacterium Pseudomonas aeruginosa. Proc. Biol. Sci. 276, 3531–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitri S.; Xavier J. B.; Foster K. R. (2011) Social evolution in multispecies biofilms. Proc. Natl. Acad. Sci. U.S.A. 108, 10839–10846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celiker H.; Gore J. (2012) Competition between species can stabilize public-goods cooperation within a species. Mol. Syst. Biol. 8, 621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyken J. D.; Müller M. J. I.; Mack K. M. L.; Desai M. M. (2013) Spatial population expansion promotes the evolution of cooperation in an experimental prisoner’s dilemma. Curr. Biol. 23, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer M. O. A.; Dantas G. (2011) Antibiotics and the resistant microbiome. Curr. Opin. Microbiol. 14, 556–563. [DOI] [PubMed] [Google Scholar]
- Heuer H.; Schmitt H.; Smalla K. (2011) Antibiotic resistance gene spread due to manure application on agricultural fields. Curr. Opin. Microbiol. 14, 236–243. [DOI] [PubMed] [Google Scholar]
- Bourgeois-Nicolaos N.; Moubareck C.; Mangeney N.; Butel M.-J.; Doucet-Populaire F. (2006) Comparative study of vanA gene transfer from Enterococcus faecium to Enterococcus faecalis and to Enterococcus faecium in the intestine of mice. FEMS Microbiol. Lett. 254, 27–33. [DOI] [PubMed] [Google Scholar]
- Hoang T. T.; Karkhoff-Schweizer R. R.; Kutchma A. J.; Schweizer H. P. (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: Application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212, 77–86. [DOI] [PubMed] [Google Scholar]
- Koch B.; Jensen L.; Nybroe O. (2001) A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. J. Microbiol. Methods 45, 187–195. [DOI] [PubMed] [Google Scholar]
- Lagendijk E. L.; Validov S.; Lamers G. E. M.; De Weert S.; Bloemberg G. V. (2010) Genetic tools for tagging Gram-negative bacteria with mCherry for visualization in vitro and in natural habitats, biofilm and pathogenicity studies. FEMS Microbiol. Lett. 305, 81–90. [DOI] [PubMed] [Google Scholar]
- Ross-Gillespie A.; Gardner A.; West S. A.; Griffin A. S. (2007) Frequency dependence and cooperation: Theory and a test with bacteria. Am. Nat. 170, 331–342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.