

Abstract
The ebolaviruses cause severe and rapidly progressing hemorrhagic fever. There are five ebolavirus species; although much is known about Zaire ebolavirus (EBOV) and its neutralization by antibodies, little is known about Sudan ebolavirus (SUDV), which is emerging with increasing frequency. Here we describe monoclonal antibodies containing a human framework that potently inhibit infection by SUDV and protect mice from lethal challenge. The murine antibody 16F6, which binds the SUDV envelope glycoprotein (GP), served as the starting point for design. Sequence and structural alignment revealed similarities between 16F6 and YADS1, a synthetic antibody with a humanized scaffold. A focused phage library was constructed and screened to impart 16F6-like recognition properties onto the YADS1 scaffold. A panel of 17 antibodies were characterized and found to have a range of neutralization potentials against a pseudotype virus infection model. Neutralization correlated with GP binding as determined by ELISA. Two of these clones, E10 and F4, potently inhibited authentic SUDV and conferred protection and memory immunity in mice from lethal SUDV challenge. E10 and F4 were further shown to bind to the same epitope on GP as 16F6 with comparable affinities. These antibodies represent strong immunotherapeutic candidates for treatment of SUDV infection.
The ebolaviruses and Marburg virus (MARV) comprise the family Filoviridae of enveloped negative-sense RNA viruses that cause severe hemorrhagic fever.1−4 Based on nucleotide sequence and outbreak location, isolates of Ebola virus are classified into five species: Zaire (EBOV), Tai Forest (TAFV), Sudan (SUDV), Reston (RESTV), and Bundibugyo (BDBV). There are two MARV variants (Marburg and Ravn). Severe human disease, Ebola or Marburg Viral Disease, is associated with EBOV, SUDV, BDBV, and MARV with 30–90% case fatality rates in large outbreaks.2 EBOV and SUDV are the most pathogenic among the ebolaviruses, and both have been associated with recurring outbreaks.5 Among the 13 documented EBOV outbreaks and the six SUDV outbreaks from 1976 to 2012, the average human case fatality rates are 70% and 52%, respectively. Together, EBOV and SUDV have accounted for over 95% of Ebola virus-related deaths;5 these statistics do not include the ongoing large outbreak in West Africa, which is of unprecedented scope and geographic distribution.1,6 Many studies have been directed at understanding EBOV entry and inhibition of virus entry with antibodies and other agents;3,4,7−9 however, considerably less is known about MARV and SUDV despite the increasing prevalence of these two species.
Currently there are no FDA-approved therapies or vaccines to treat any filovirus infection. A number of therapeutic strategies have been proposed, including vaccines, small molecules, and modified oligonucleotides.9−12 Passive immunotherapy has been gaining attention as a therapeutic approach since filovirus-specific polyclonal IgG or cocktails of monoclonal antibodies (mAbs) can provide post-exposure protection against lethal challenge from both EBOV and MARV in mice and non-human primates (NHPs).13−16 The envelope glycoprotein (GP) is the expected primary neutralization target for mAbs and consists of two subunits, GP1 (surface subunit) and GP2 (transmembrane subunit). The mature filovirus GP spike is a trimer of three disulfide-linked GP1–GP2 heterodimers, generated in the producer cell by endoproteolytic cleavage of the GP precursor polypeptide by furin.4,17 GP1 mediates viral adhesion to host cells and regulates the activity of the transmembrane subunit GP2, which mediates fusion of viral and cellular membranes during cell entry. The prefusion EBOV GP1–GP2 spike has a “chalice-and-bowl” morphology. The three GP2 subunits form the chalice at the base of the spike, and the three GP1 molecules form a bowl that sits atop the GP2 chalice.17−19 Together with GP2, the base and head subdomains of GP1 form the conserved structural core of the GP1–GP2 spike. Antibodies that bind both GP1 and GP2 have neutralization potential.
Here we describe the isolation and characterization of protective SUDV-specific mAbs with a human framework using a synthetic antibody approach. A number of mAbs have been described for EBOV, but few have been characterized in detail for SUDV. One of the most potent SUDV mAbs is a murine antibody known as 16F6 that binds to GP from SUDV at the base of the GP chalice-and-bowl.19 While 16F6 exhibits potent neutralization and in vivo protective ability, the murine scaffold presents a potential limitation to its therapeutic use in humans. Serendipitously, we observed that 16F6 has high sequence and structural homology to a commonly used synthetic human antibody framework.20,21 We used a structure-guided approach to design and screen an antibody library that would endow 16F6-like recognition properties onto the human scaffold. The resulting antibodies were characterized for their neutralization potential and ability to confer in vivo protection from lethal SUDV challenge.
Results and Discussion
Library Design and Selection
Sequence and structural alignment of 16F6 (murine scaffold) with the vascular endothelial growth factor (VEGF)-specific synthetic antibody YADS1 (humanized scaffold) revealed marked similarity in the framework regions (Figure 1a and b).19,20 At the amino acid level, there is 77% identity and 87% similarity in noncomplementarity-determining region (CDR) segments, and examination of the structural alignment between the two antigen-binding fragments (Fabs) revealed strong homology of framework segments leading into the CDR loops. Superimposing the frameworks of the two Fabs and variable domains (Fvs) excluding CDR loops revealed RMSDs of 2.6 and 1.3 Å over 381 and 182 Cα atoms, respectively. Although there is marked variability in the loop conformations themselves, we surmised that appropriate positioning of the beginning and end of the CDR loops would allow productive conformations in a humanized antibody containing the 16F6 CDR segments. YADS1 shares a framework with many synthetic antibodies and is derived from the VH3-23 germline segment that has favorable characteristics such as high expression, stability, and mutability.20−22 This analysis suggested that incorporation of 16F6-like recognition onto the YADS1 scaffold might provide a successful strategy for creating SUDV antibodies bearing a humanized framework that are suitable for immunotherapy.
Figure 1.

Library design and selection. Sequence (A) and structural (B) alignment of light and heavy chain variable domains (VL and VH, respectively) in YADS1 and 16F6. The library design is shown in panel A, below the 16F6 sequence. Positions of randomization are indicated with an ‘X’ and an asterisk. The randomization scheme was as follows: VL position 24 (MRG codon that encodes K/R/Q); positions 53 and 56 (NNK, all 20 amino acids); position 54 (CKT, L/R); and position 55 (YAT, H/Y). VH positions 29, 34, and 100b (HTK, F/I/L/M), 35 (YWT, F/H/L/Y), 52a (YCG, P/S), 60 (SCT, A/P), 93 (KCT, A/S), and 100a (KYT, F/V/S/A). In the structural alignment, the top-down view shows structural variability in the CDR loops; the spheres represent positions that were randomized. (C) Distribution of residues in the ELISA-positive populations resulting from functional (GPSUDV) or display (M2) selection. On the x-axis, the position numbering is followed by the order of residue identities beginning with the most frequently observed (1st) to the 4th in descending order. In cases where more than four residues were permitted or observed, the rest were binned into a 5th class (“other”). At positions 55 and 100b, spurious additional mutations (Tyr in both cases) beyond the encoded diversity were observed in the selected pool.
We designed and constructed a 16F6 humanization library based on a chimeric template where CDR segments from 16F6 were grafted onto the YADS1 scaffold (Figure 1a). Restricted diversification was permitted at positions near the CDR segments that did not involve direct antigen contacts at the analogous sites in 16F6 but had differing residue identity between 16F6 and YADS1. For example, CDR-H1 position 29 (Ile in YADS1 but Phe in 16F6) was allowed to vary among the four residues Phe/Ile/Leu/Met, thus encoding both the 16F6 and YADS1 residues as well as two others (Leu and Met) with similar properties. This randomization strategy was applied to several positions in CDR-H1, H2, and H3, as well as CDR-L1 and L3. In addition, two residues in CDR-L2 (53 and 56) differed among YADS1 (Tyr and Ser, respectively) and 16F6 (both Thr), but the corresponding residues made partial contacts to the antigen in 16F6. Therefore, these two positions were diversified with an NNK (N = A/T/C/G; K = G/T) codon allowing all 20 possible amino acids. Full details for the randomization strategy are discussed in the Figure 1 caption. In total, 13 positions were randomized with a theoretical diversity 4.5 × 108 at the DNA level and 4 × 107 at the protein level; the library was produced with ∼1010 unique members, allowing exhaustive sampling. The library was expressed as a bivalent Fab fusion to the pIII gene as has been previously described for other synthetic antibodies.20,21 This bivalent display format was chosen to more accurately mimic the nature of an IgG, which would be the preferred downstream therapeutic format.
Six rounds of selection were performed against a soluble, recombinant SUDV GP (Boniface variant) that encompasses residues 33–649 (GPSUDV). This protein includes all major domains on GP1 and GP2 except the transmembrane domain and C-terminal tail and is mostly trimeric in solution (data not shown). Previous work has demonstrated that GP constructs expressed in mammalian cells but lacking the transmembrane segment provide a suitable mimic of the prefusion spike for antibody selection, binding, and other structural studies.7,8,18,19 Several hundred clones were screened for binding to GPSUDV from output populations of rounds 3–6 by monoclonal phage ELISA to identify 38 clones representing 17 unique sequences (see Supporting Information). The distribution of amino acids among these clones is represented in Figure 1c. To compare requirements for recognition and stability, a parallel selection to determine display preferences was performed against anti-FLAG antibody M2; a FLAG epitope was included at the C-terminus of the phage displayed light chain, and therefore any preferences for display will be evident with this analysis.23,24 Comparison of amino acid composition among ELISA-positive clones from the functional selection (GPSUDV) and the display selection (M2) distribution revealed moderate preferences for GPSUDV recognition at many positions but strong preferences for the 16F6 residues at positions 29, 100a, and 100b of the heavy chain. Inspection of the 16F6 crystal structure suggests that F29 and F100b likely support loop conformations, as these residues are buried and participate in internal hydrophobic interactions (see Supporting Information).19 Residue F100a participates in the structural paratope, and therefore this residue is likely critical for the intermolecular interface. In contrast, the 16F6 residue Ser at position 52a of CDR-H2 is less preferred compared with the YADS1 cognate residue Pro. This preference is more evident in clones showing stronger neutralization efficacy (below). Since Pro has a strong effect on loop conformations, the Pro at this position may serve to orient the CDR-H2 loop toward favorable GP recognition in the human framework.
Antibody Binding and Neutralization
The panel of 17 unique antibodies were produced as IgG1 molecules and characterized. As a preliminary screen, the mAbs were tested for their ability to inhibit infection of a vesicular stomatitis virus pseudotyped with the SUDV glycoprotein (VSV-GPSUDV) at 20 nM and 100 nM (Figure 2a and 2b). Murine 16F6 and the human EBOV-specific antibody KZ52 were included as positive and negative controls, respectively.25 Overall, the humanized mAbs had a range of activity against VSV-GPSUDV, with the most potent inhibiting at levels on-par with murine 16F6 (∼90% inhibition at 20 nM and ≥95% at 100 nM). Notably, KZ52, an EBOV-specific human antibody, had no activity even at 100 nM, consistent with previous reports of its strain specificity.25,26 All 17 mAbs were also tested against VSV-GPEBOV but had no activity at 100 nM, and thus the humanized variants maintained the specificity profile of the murine 16F6 parent. This orthogonality in neutralization activity for 16F6 and its humanized counterparts (monospecific for SUDV) and KZ52 (monospecific for EBOV) is notable since the independent cocrystal structure complexes of 16F6 Fab-GPSUDV and KZ52 Fab-GPEBOV revealed that both antibodies bind at similar locations on the glycoprotein.7,18,19 In fact, the structural epitopes of the two Fabs overlap by 10 residues, suggesting this region is a shared neutralization epitope among the ebolaviruses.19 However, no broadly neutralizing antibody (bNAb) against the filoviruses has yet been described. As a point of comparison, bNAbs against HIV-1 gp120 and influenza HA target conserved regions, and in these cases a compact structural epitope that focuses on conserved residues is likely critical for broad potency.27−29
Figure 2.

Neutralization of GPSUDV-mediated cell entry. (A and B) Single point neutralization assays using the VSV-GPSUDV pseudotype virus at 20 nM (A) or 100 nM (B) antibody concentrations. The y-axis is plotted on logarithmic scale to illustrate the full dynamic range of the assay. Eleven clones exhibited high neutralization potential (∼95% or higher) at 100 nM. (C and D) Dose–response curves for neutralization of VSV-GPSUDV pseudotype (C) or authentic SUDV (D) virus by E10 and F4. The authentic virus neutralization assays were performed alone and in the presence of 5% guinea pig complement. The inset shows the curves with the x-axis in log scale.
Eleven of the clones exhibited strong neutralization potential (∼95% or higher) at 100 nM; among these, E10 and F4 had favorable expression yield, were well-behaved, and thus were carried forward for further studies as representative examples. Dose-dependent inhibition profiles against VSV-GPSUDV revealed IC50 values below 10 nM (Figure 2c). Both mAbs were tested against authentic SUDV under Biosafety level 4 (BSL4) conditions and found to neutralize with IC50s below 4 nM (E10) and 8 nM (F4) (Figure 2d). When the SUDV neutralization assays were performed in the presence of 5% guinea pig complement, IC50s below 4 nM were observed in both cases. These results indicate that E10 and F4 have potent neutralization activity against SUDV GP-mediated cell entry. The fact that IC50s against SUDV were similar in the presence or absence of complement suggests that the mechanism involves binding of GP and inhibition of cell entry, but not opsonization or activation of the complement pathway. We performed competitive ELISA experiments to determine if 16F6, E10, and F4 shared a common epitope. 16F6 was biotinylated (b16F6), and then the capacity of unbiotinylated 16F6 (positive control), E10, and F4 to inhibit b16F6 binding in a dose-specific manner was evaluated. As shown in Figure 3, all three unbiotinylated antibodies competed effectively with b16F6 with IC50 values below 0.2 μM for 16F6 and F4 and ∼0.6 μM for E10. These results indicate that all three antibodies share an epitope and have relatively comparable binding affinities to GPSUDV, with modestly reduced affinity in the case of E10. Since library diversification focused mostly on supporting CDR loop residues, rather than direct contact sites, it is not surprising that affinities of the humanized clones were comparable or slightly decreased relative to the murine 16F6 parent.
Figure 3.
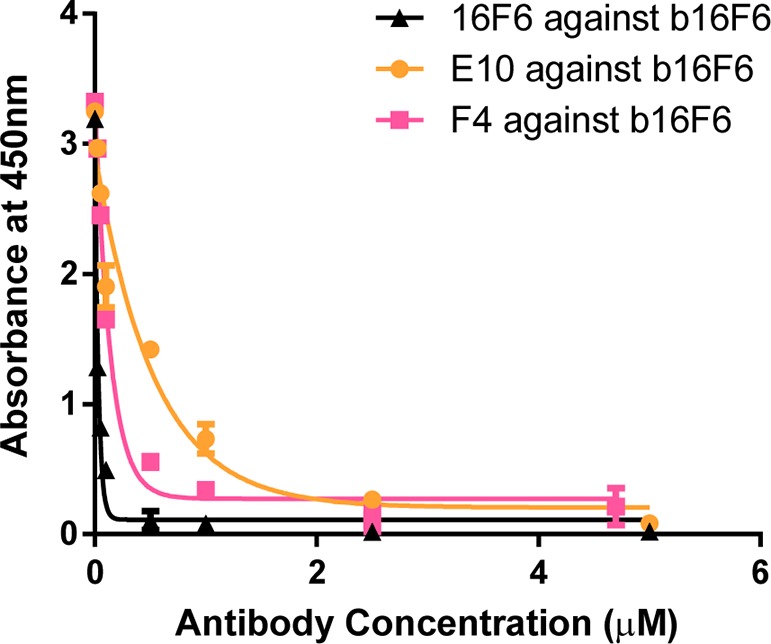
Competition ELISA of 16F6, F4, and E10. Binding of biotinylated 16F6 (b16F6) to immobilized GPSUDV was competed with unbiotinylated 16F6, E10, or F4 yielding IC50s below 0.2 μM for 16F6 and F4 and ∼0.6 μM for E10.
To explore the relationship between neutralization potency and binding capacity, we performed ELISAs with several of the mAbs that had varying neutralization potency (Figure 4). These can be grouped into strong neutralizers (E10, F4, 52D11, 52F2, and 41F10), moderate neutralizers (41C6 and 31F8), and non-neutralizers (35E3 and 51E1). The half-maximal binding titer (EC50) correlated strongly with neutralization efficacy. The strong neutralizers had EC50s ranging from 2.1 nM to 14.8 nM against GPSUDV, while moderate neutralizers 41C6 and 31F8 bound GPSUDV but did not exhibit saturation binding even at micromolar antibody concentrations. The non-neutralizers 35E3 and 51E1 did not have appreciable binding activity. Notably, in all cases where binding activity was observed, it was specific for GPSUDV over wells containing a negative control (2% nonfat dry milk, NFDM). All 17 mAbs were also tested for binding to GPEBOV and found to have no activity, consistent with their monospecific neutralization activity.
Figure 4.

Comparative binding studies of strong, modest, and poor neutralizing antibodies. ELISA binding assays for representative antibodies from the strong, modest, and poor neutralizer categories against GPSUDV and 2% NFDM. The half-maximal binding titer (EC50) is provided. None of the mAbs had activity for GPEBOV; these data are shown for E10 and F4.
In Vivo Protection against Viral Challenge
Antibodies E10 and F4 were assessed for their ability to confer protection from SUDV challenge in mice. Although a mouse-adapted SUDV strain has been developed, it is not lethal to immunocompetent mice. Here, the human-lethal wild-type SUDV virus was used to infect 4-week-old Type 1 interferon α/β receptor knockout mice (Type 1 IFNα/β R–/–), and the ability of the mAbs to confer post-exposure protection was evaluated. The murine 16F6 and an EBOV-specific mAb (Z.6D8, ref (30)) were included as controls. Mice were provided with either three doses (days −1 (pre-exposure), +1 and +4 (post-exposure); Figure 5a and b) or two post-exposure doses (days +1 and +4, Figure 6a and b) of 500 μg mAb via the intraperitoneal route and challenged with a target dose of 1,000 plaque forming units (pfu) of wild-type SUDV (day 0). Mice receiving the SUDV-specific mAbs (16F6, E10, and F4) showed similar levels of protection (80–100% survival, Figures 5a and 6a), whereas those mice treated with an anti-EBOV specific mAb (Z.6D8) showed little or no protection. When mice received one dose of mAb prior to viral challenge (three-dose regime), no significant weight loss, which is a measure of clinical illness, was observed following challenge in the mice that received SUDV-specific antibodies, whereas the Z.6D8-treated control animals exhibited significant weight loss (maximal loss of 25%) (Figure 5b). In fact, a weight gain was observed with SUDV-specific antibody treatment, which is to be expected due to the fact that the mice were 4 weeks old when challenged and still maturing. When initiation of treatment in mice was delayed to day +1 (post-exposure two-dose regime), we observed no lethality when the SUDV-specific mAbs were provided as treatment (Figure 6a). However, with the delay in treatment, we observed a definite weight loss in the antibody-treated mice that was absent in the day −1 (pre-exposure) three-dose treatment scenario (Figure 6b).
Figure 5.

Protection of mice with three doses of mAb at days −1, +1, and +4. Survival (A and C) and weight change (B and D) curves for initial challenge experiment (A and B) and rechallenge of surviving mice (C and D). Each weight change data point represents the mean of the mice remaining in the group for a given time point. For the rechallenge experiment, 4-week old mice were used for the Z.6D8-treated control group. For survival curves, statistically significant differences from the Z.6D8 control group are indicated with asterisks: (*, p < 0.05; **, p < 0.01; ***, p < 0.0001). The p value for 16F6- vs F4-treated rechallenged mice is indicated.
Figure 6.

Protection of mice with two doeses of mAb at days +1 and +4. Survival (A and C) and weight change (B and D) curves for initial challenge experiment (A and B) and rechallenge of the surviving mice (C and D). The one Z.6D8-treated control mouse that survived the initial challenge also survived the rechallenge. On the rechallenge plots (C and D), data are also shown for challenge of nine-week old naïve mice, which do not completely succumb to infection but do show signs of illness in the form of weight loss. For survival curves, statistically significant differences from the Z.6D8 control group are indicated with asterisks: (**, p < 0.01).
Thirty-five days following the initial challenge, surviving mice (those treated with SUDV-specific antibodies previously) were rechallenged with 1,000 pfu wild-type SUDV with no additional antibody treatment to assess if memory cell immunity was generated after the first exposure and treatment (Figure 5c and d for the three-dose group, and Figure 6c and d for the post-exposure two-dose group). Mice were 9 weeks old when they were challenged unless otherwise specified. Surviving mice from the three-dose group that received E10 and F4 mAbs had a nonstatistically significant trend of better rechallenge protection than mice receiving 16F6 (p < 0.094, Figure 5c). As a control to demonstrate that virus injected in rechallenge studies was capable of inducing morbidity/mortality, we included a naïve cohort of 4-week-old mice that received the anti-EBOV specific Z.6D8 antibody. As expected there was again little or no protection noted with this treatment. Weight changes were not as apparent with the rechallenged mice in general because they were 9 weeks old and thus more developed at the time of the second infection (Figures 5d and 6d). When surviving mice from the two-dose treatment were rechallenged at 9 weeks of age, all were protected with little or no observable weight loss. During the initial challenge experiment from this two-dose post-exposure treatment group, one of the control Z.6D8 mice survived; this mouse also survived the rechallenge. To gain additional insight, a group of naïve, untreated 9-week-old mice were also included in the rechallenge experiment in Figure 6c. SUDV was not completely lethal to 9-week mice (survival was 80%), but the animals showed clinical signs of disease such as weight loss. When compared to 9-week mice that survived the initial infection either with SUDV mAb treatment (16F6, E10, F4) or without (Z.6D8), it is apparent that the surviving mice have memory immunity and show no weight loss as a result of infection (Figure 6d).
Conclusions
Here we describe the in vitro and in vivo activity of synthetic SUDV-specific antibodies containing a human framework. These mAbs show potent neutralization activity against both pseudotyped and authentic viruses and confer protection and memory immunity from infection in mice. Although therapeutic, antibody-mediated protection against SUDV in larger animals has yet to be demonstrated, the studies showing EBOV protection of NHPs,13−16 in many cases following post-exposure treatments, suggest that SUDV antibody therapies should be explored and the mAbs reported herein represent viable candidates. Antibodies have advantages of favorable serum half-life and are generally well-tolerated especially if they contain human scaffolds; though there is only one FDA-approved antibody for antiviral infection (against respiratory syncytial virus, RSV),31 a number of mAb treatments are being considered for various viral diseases.32−34
The SUDV GP sequence is 56% identical to that of EBOV GP at the amino acid level but differs in a number of structural and functional aspects. The SUDV GP appears to be more dynamic, is more susceptible to proteolytic degradation, and electrostatically has more negative surface charge character than does EBOV GP.17,19 Given these differences, it is not surprising that antibody-mediated neutralization requirements also differ, exemplified by the fact that KZ52 and 16F6 bind to similar overall positions on the prefusion GP spikes but have strain-specific activity (KZ52 for EBOV and 16F6 for SUDV); here we report that our humanized analogues retain this strain selectivity. SUDV has been responsible for numerous recent Ebola virus outbreaks, including one of the largest (Gulu district of Uganda in 2000 with 425 reported cases and 224 deaths), and the only recorded outbreaks in 2012. Development of SUDV-specific mAbs is therefore important for both diagnostic and research purposes as well as for potential immunotherapies.
Methods
Phage Display
The synthetic gene for a template for construction of the 16F6 humanization library was cloned as a bivalent Fab into phagemid for pIII display.35 The template consisted of DNA encoding a chimera between the YADS1 framework with 16F6 CDR segments and with 16F6 residue identity at the intended positions of randomization. Oligonucleotide-directed mutagenesis was used to incorporate diversity elements.36 The library DNA was electroporated into SS320 E. coli and library phage amplified according to standard protocols.36
Library sorting was performed according to modified published protocols.8,35 Recombinant GPSUDV (residues 1–649, with the first 32 residues comprising the signal sequence that is removed during maturation) that was expressed in HEK293 cells served as the selection target was purchased from the Protein Expression Laboratory, Frederick National Laboratory for Cancer Research. Briefly, 0.5 μg/well of GPSUDV was immobilized on 96-well Maxisorp immunoplates (Fisher Scientific, Nepean, ON, Canada) in phosphate buffered saline (PBS) pH 8.0 for 14–16 h at 4 °C. Wells were blocked with 5% NFDM (1.5 h, RT). Phage pools from the 16F6 humanization library were cycled through six rounds of binding selection using the immobilized GPSUDV as a capture target and 5% NFDM as a negative control. Stringency was increased throughout the selection by decreasing the number of wells containing antigen and antigen loading concentrations with progressive rounds of selection. The output phage population from between rounds was amplified in E. coli XL1-Blue cells in 2xYT broth supplemented with 5 μg/mL tetracycline. Output phage population was added to 5 mL cultures at OD600 of ∼0.6 and shaken at 37 °C for 1 h. M13-K07 helper phage were added (1010 infectious units (iu)/mL), and the culture was shaken at 37 °C for an additional 1 h. The 5 mL culture was transferred to a 25 mL culture of 2xYT broth supplemented with 100 μg/mL carbenicillin and 50 μg/mL kanamycin and allowed to shake at 37 °C overnight (14–16 h). The cells were removed by centrifugation, and the amplified phage was precipitated by the addition of 3% (w/v) NaCl and 4% (w/v) PEG 8000. The phage was pelleted by centrifugation and suspended in PBS + 0.05% Tween 20 containing 0.5% BSA (PBT).
Output populations were screened for binding using monoclonal phage ELISA. GPSUDV or M2 (0.5 μg/well) was immobilized on wells, and wells were blocked with 5% NFDM as described above. After the sixth round of selection, clones from rounds 3–6 were grown overnight in 96-well deep well plates with 2xYT broth supplemented with carbenicillin and M13K07 helper phage (1010 iu/mL). The culture supernatants were applied directly to ELISA wells containing antigen (1 h, RT) to identify binding clones targeting GPSUDV. The phage solutions were decanted, and the wells were washed 4–6 times with PBS + 0.05% Tween (PBST). To score binding, a solution containing 1/1000 dilution of anti-M13/horseradish peroxidase (HRP) conjugate (GE Heathcare, Piscataway, NJ) was added and allowed to bind for 45–60 min. The wells were again washed 4–6 times with PBST and developed using 3,3′,5,5′-tetramethylbenzidine substrate (Sigma-Aldrich, St. Louis, MO). The ELISA signal was measured after quenching the signal with 0.5 M sulfuric acid and determining the absorbance at 450 nm. Phage clones exhibiting phage ELISA signals toward GPSUDV of at least 2-fold higher than toward 5% NFDM were subjected to DNA sequence analysis.
Expression and Purification of IgGs
The variable domain DNA for each phage clone was amplified by PCR, digested, and subcloned into pMAZ-IgL and pMAZ-IgH vectors.37 Vectors for the heavy and light chain were transfected into HEK293F cells (Invitrogen, Grand Island, NY) using 2 μg/mL linear polyethylenimine (PEI), molecular weight 25,000 Da according to the manufacturer’s instructions (Polysciences, Warrington, PA). Cell cultures were incubated at 37 °C for 5–6 days post-transfection. The cell cultures were centrifuged, and the supernatants were applied to a protein-A affinity column (∼2 mL packed beads per 600 mL culture) (Pierce, ThermoScientific, Rockford, IL). IgG proteins were eluted with 100 mM glycine, pH 2.0 and neutralized with 2 M Tris, pH 7.5. The eluent was dialyzed into PBS, pH 7.4 and the IgG protein was concentrated.
Neutralization Assays with VSV-GPSUDV and VSV-GPEBOV
Neutralization assays were performed using vesicular stomatitis virus pseudotyped to display the GP from either SUDV or EBOV in place of its native G glycoprotein (VSV-GPSUDV or VSV-GPEBOV, respectively). The viral genome encodes an enhanced green fluorescent protein (eGFP), and infection is scored by counting fluorescent cells after infection. The protocol for VSV-GP production has been described elsewhere.38 Briefly, the virus-containing supernatants were harvested and concentrated by pelleting through a 10% sucrose cushion. Virus stocks were titered by infecting African Green Monkey kidney (Vero) cells with serial dilutions and counting eGFP-positive cells by fluorescence microscopy. VSV-GP was used to infect Vero cells at approximate multiplicities of infection of 0.1–1.0 in Dulbecco’s modified Eagle medium (DMEM) containing 2% fetal bovine serum (FBS; Thermo Scientific, Waltham, MA), such that 20–200 cells were infected per well. Vero cell monolayers consisting of ∼7.5 × 104 cells/well in a 48-well plate were incubated for 14–16 h with pseudotyped virus that had been preincubated with dilutions of the IgG. Infection was scored by manually counting eGFP-positive cells under a fluorescence microscope, 14–16 h after initial exposure.
Plaque Reduction Neutralization Assays with Authentic SUDV
Dilutions of the antibody of interest were made in a sterile 96-well plate (Costar/Corning Incorporated, Corning, NY) in Eagle Minimum Essential Media (EMEM) (Sigma-Aldrich, St. Louis, MO) supplemented with 5% FBS. In a sterile 6-well plate (Costar/Corning Incorporated, Corning, NY), 125 μL of authentic EBOV or SUDV diluted to 1200 pfu/mL was added to each well, and the plates were incubated at 37 °C for 1 h. Virus was added to a well containing media alone (no antibody) as a control for 100% infection. Vero-E6 cells were exposed to 100 μL of the virus/antibody mixture and incubated at 37 °C for an additional 1 h. During this time, the plates were gently rocked every 15 min to ensure homogeneity and prevent drying. After 1 h of incubation, 2 mL of primary overlay (EMEM with 10% FBS and 1% Gentamicin (Sigma-Aldrich, St. Louis, MO) with 1% SeaKem ME agarose (Lonza, Cohasset, MN)) was added to each well, and the plates were incubated at 37 °C for 6 days. On Day 7 post-exposure to virus, neutral red solution (EMEM with 10% FBS and 1% gentamicin with 5% neutral red (Gibco/Invitrogen, Grand Island, NY) was added to all cell-containing wells, and cells were incubated at 37 °C overnight. Infection was scored by counting the number of plaques per well, using the number of plaques on the control well (no antibody) as 100% infection.
PRNT assays with complement were performed using Low-Tox Guniea pig complement (Cedarlane). The lyophilized complement was resuspended with 1 mL of ice-cold water and then diluted 1:18 in ice-cold PBS and filter sterilized. This complement mixture was incubated in a 1:1 ratio with 65 μL of 2,400 pfu/mL virus stock 37 °C for 1 h (this gave a final viral titer of 1,200 pfu/mL). Vero-E6 cells were exposed to 100 μL of the virus/antibody/complement mixture at 37 °C for 1 h with gentle rocking every 15 min. The remaining steps were identical to the PRNT assay without complement as described above.
Binding ELISAs
The target proteins were directly immobilized onto 96-well Maxisorp plates (GPSUDV and GPEBOV = 0.5 μg/well) by incubating in PBS pH 8.0 for 14–16 h at 4 °C. PBS, pH 7.4, containing 5% NFDM was used to block the wells after target immobilization (incubation for 60–90 min at RT). Negative control plates were coated with 5% NFDM only. IgGs were diluted into PBT, applied to the wells, and incubated at RT for 1 h. The plates were washed with PBST and incubated for 45–60 min with Protein A/HRP antibody conjugate (1:1000 dilution in PBT). The wells were washed 4–6 times with PBST and developed as described above. The absorbance at 450 nm was determined. The data were fit to standard four-parameter logistic equations using Graphpad Prism (GraphPad Software, La Jolla, CA). The half-maximal binding titers (EC50) were obtained from the inflection point in the curves.
Competition ELISAs were performed as previously described.8 16F6 was biotinylated (b16F6) using a NHS-PEG4-BIOTIN labeling kit (Thermo Scientific, Rockford, IL) according to the manufacturer’s instructions. GPSUDV was immobilized on 96-well Maxisorp plates (Corning Incorporated, Corning, NY) at a concentration of 0.5 μg/well. b16F6 was diluted to 12 μg/mL in PBST buffer with or without varying amounts of three unbiotinylated competitors, 16F6, F4, and E10. The mixtures of b16F6 with or without competitors were applied to the wells and incubated at RT for 1 h. The plates were washed with PBST, and then horseradish peroxidase/streptavidin conjugate (1:1000 dilution in PBST buffer) was added and incubated for 45 min. The plates were washed with PBST, developed with TMB substrate, and quenched with 0.5 M H2SO4. Absorbance at 450 nm was measured.
Mouse Protection Studies
Male and female Type 1 IFN α/β receptor knockout mice (Type 1 IFNα/β R–/–) purchased from Jackson Laboratory (4–14 weeks of age) were utilized in these experiments. Mice were treated via the intraperitoneal route (ip) with 500 μg of indicated mAb at either day −1, +1, and +4 or 500 μg of indicated mAb at day +1 and +4 and challenged ip with a target dose of 1,000 plaque forming units (pfu) of wild-type SUDV. Surviving mice were rechallenge IP with target dose 1000 pfu of wild-type SUDV with no treatment provided. Following all challenges mice were monitored daily for morbidity and mortality.
Acknowledgments
This work was supported by the NIH (R01-AI090249 (J.R.L.), R01-AI088027 (K.C.), and U19-AI09762), the Canadian Institutes for Health Research Grant MOP-93725 (S.S.S.), and JSTO-CBD Defense Threat Reduction Agency CB3947 (J.M.D.). J.F.K was supported in part by NIH Medical Scientist Training Program T32-GM007288, and J.C.F. by NIH Cellular and Molecular Biology and Genetics Training Program T32-GM007491. Opinions, conclusions, interpretations, and recommendations are those of the authors and are not necessarily endorsed by the U.S. Army. The mention of trade names or commercial products does not constitute endorsement or recommendation for use by the Department of the Army or the Department of Defense.
Glossary
Abbreviations
- MARV
Marburg virus
- EBOV
Zaire ebolavirus
- SUDV
Sudan ebolavirus
- NHP
non-human primate
- GP
glycoprotein
- CDR
complementarity-determining region
- Fab
antigen binding fragment
- VSV-GP
vesicular stomatitis virus pseudotyped with GP
- BSL4
Biosafety level 4
- bNAb
broadly neutralizing antibodies
- NFDM
nonfat dry milk
- pfu
plaque-forming units
- PBS
phosphate-buffered saline
- FBS
fetal bovine serum
- EMEM
Eagle minimal Essential Media
- VL
light chain variable domain
- VH
heavy chain variable domain
Supporting Information Available
Sequences of the 17 mAbs characterized in neutralization and binding studies. Figure S1 showing potential roles of 16F6 combining site residues in the interaction with GPSUDV. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
# These authors contributed equally to this work.
The authors declare the following competing financial interest(s): The United States Army Medical Research Institute of Infectious Disease (USAMRIID) has filed a patent application on the murine mAb 16F6 (WO2011071574 A2) entitled “Monoclonal Antibodies against Glycoprotein of Ebola Sudan Boniface Virus” with J.M.D. as a coinventor. The Albert Einstein College of Medicine has filed a patent application on the humanized mAbs discussed herein (U.S. Provisional Patent Application No. 61/830,325 and U.S. Utility Application No. 14/291,608) entitled “Therapy for Filovirus Infection” with G.C., J.F.K., S.E.Z., J.C.F., K.C., J.M.D., S.S.S., and J.R.L. as coinventors.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Feldmann H. (2014) Ebola—A Growing Threat?. N. Engl. J. Med. 10.1056/NEJMp1405314. [DOI] [PubMed] [Google Scholar]
- Feldmann H.; Geisbert T. W. (2011) Ebola Haemorrhagic Fever. Lancet 377, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saphire E. O. (2013) An Update on the Use of Antibodies against the Filoviruses. Immunotherapy 5, 1221–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E. H.; Chandran K. (2012) Filovirus Entry into Cells—New Insights. Curr. Opin. Virol 2, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- http://www.cdc.gov/ncidod/dvrd/spb/mnpages/dispages/ebola.htm.
- Baize S.; Pannetier D.; Oestereich L.; Rieger T.; Koivogui L.; Magassouba N.; Soropogui B.; Sow M. S.; Keïta S.; De Clerck H.; Tiffany A.; Dominguez G.; Loua M.; Traoré A.; Kolié M.; Malano E. R.; Heleze E.; Bocquin A.; Mély S.; Raoul H.; Caro V.; Cadar D.; Gabriel M.; Pahlmann M.; Tappe D.; Schmidt-Chanasit J.; Impouma B.; Diallo A. K.; Formenty P.; Van Herp M.; Günther S. (2014) Emergence of Zaire Ebola Virus Disease in Guinea—Preliminary Report. N. Engl. J. Med. 10.1056/NEJMoa1404505. [DOI] [PubMed] [Google Scholar]
- Bale S.; Dias J. M.; Fusco M. L.; Hashiguchi T.; Wong A. C.; Liu T.; Keuhne A. I.; Li S.; Woods V. L. Jr.; Chandran K.; Dye J. M.; Saphire E. O. (2012) Structural Basis for Differential Neutralization of Ebolaviruses. Viruses 4, 447–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koellhoffer J. F.; Chen G.; Sandesara R. G.; Bale S.; Saphire E. O.; Chandran K.; Sidhu S. S.; Lai J. R. (2012) Two Synthetic Antibodies That Recognize and Neutralize Distinct Proteolytic Forms of the Ebola Virus Envelope Glycoprotein. ChemBioChem 13, 2549–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M.; Misasi J.; Ren T.; Bruchez A.; Lee K.; Filone C. M.; Hensley L.; Li Q.; Ory D.; Chandran K.; Cunningham J. (2011) Small Molecule Inhibitors Reveal Niemann-Pick C1 Is Essential for Ebola Virus Infection. Nature 477, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisbert T. W.; Feldmann H. (2011) Recombinant Vesicular Stomatitis Virus-Based Vaccines against Ebola and Marburg Virus Infections. J. Infect. Dis. 204(Suppl. 3), S1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warfield K. L.; Swenson D. L.; Olinger G. G.; Nichols D. K.; Pratt W. D.; Blouch R.; Stein D. A.; Aman M. J.; Iversen P. L.; Bavari S. (2006) Gene-specific Countermeasures against Ebola Virus Based on Antisense Phosphorodiamidate Morpholino Oligomers. PLoS Pathol. 2(1), e1 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren T. K.; Wells J.; Panchal R. G.; Stuthman K. S.; Garza N. L.; Van Tongeren S. A.; Dong L.; Retterer C. J.; Eaton B. P.; Pegoraro G.; Honnold S.; Bantia S.; Kotian P.; Chen X.; Taubenheim B. R.; Welch L. S.; Minning D. M.; Babu Y. S.; Sheridan W. P.; Bavari S. (2014) Protection Against Filovirus Diseases by a Novel Broad-Spectrum Nucleoside Analogue BCX4430. Nature 508, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dye J. M.; Herbert A. S.; Kuehne A. I.; Barth J. F.; Muhammad M. A.; Zak S. E.; Ortiz R. A.; Prugar L. I.; Pratt W. D. (2012) Postexposure Antibody Prophylaxis Protects Nonhuman Primates from Filovirus Disease. Proc. Natl. Acad. Sci. U.S.A. 109, 5034–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G.; Richardson J. S.; Pillet S.; Patel A.; Qiu X.; Alimonti J.; Hogan J.; Zhang Y.; Takada A.; Feldmann H.; Kobinger G. P. (2012) Immune Parameters Correlate with Protection against Ebola Virus Infection in Rodents and Nonhuman Primates. Sci. Transl. Med. 4(158), 158ra146 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzi A.; Yoshida R.; Miyamoto H.; Ishijim M.; Suzuki Y.; Higuchi M.; Matsuyama Y.; Igarashi M.; Nakayama E.; Kuroda M.; Saijo M.; Feldmann F.; Brining D.; Feldmann H.; Takada A. (2012) Protective Efficacy of Neutralizing Monoclonal Antibodies in a Nonhuman Primate Model of Ebola Hemorrhagic Fever. PLoS One 7, e36192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olinger G. G. Jr.; Pettitt J.; Kim D.; Working C.; Bohorov O.; Bratcher B.; Hiatt E.; Hume S. D.; Johnson A. K.; Morton J.; Pauly M.; Whaley K. J.; Lear C. M.; Biggins J. E.; Scully C.; Hensley L.; Zeitlin L. (2012) Delayed Treatment of Ebola Virus Infection with Plant-derived Monoclonal Antibodies Provides Protection in Rhesus Macaques. Proc. Natl. Acad. Sci. U.S.A. 109, 18030–18035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Saphire E. O. (2009) Neutralizing Ebolavirus: Structural Insights into the Envelope Glycoprotein and Antibodies Targeted against It. Curr. Opin. Struct. Biol. 19, 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. E.; Fusco M. L.; Hessell A. J.; Oswald W. B.; Burton D. R.; Saphire E. O. (2008) Structure of the Ebola Virus Glycoprotein Bound to an Antibody From a Human Survivor. Nature 454, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J. M.; Kuehne A. I.; Abelson D. M.; Bale S.; Wong A. C.; Halfmann P.; Muhammad M. A.; Fusco M. L.; Zak S. E.; Kang E.; Kawaoka Y.; Chandran K.; Dye J. M.; Saphire E. O. (2011) A Shared Structural Solution for Neutralizing Ebolaviruses. Nat. Struct. Mol. Biol. 18, 1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellouse F. A.; Wiesmann C.; Sidhu S. S. (2004) Synthetic Antibodies from a Four-Amino-Acid Code: A Dominant Role for Tyrosine in Antigen Recognition. Proc. Natl. Acad. Sci. U.S.A. 101, 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellouse F. A.; Li B.; Compaan D. M.; Peden A. A.; Hymowitz S. G.; Sidhu S. S. (2005) Molecular Recognition by a Binary Code. J. Mol. Biol. 348, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Tiller T.; Schuster I.; Deppe D.; Siegers K.; Strohner R.; Herrmann T.; Berenguer M.; Poujol D.; Stehle J.; Stark Y.; Heßling M.; Daubert D.; Felderer K.; Kaden S.; Kölln J.; Enzelberger M.; Urlinger S. (2013) A Fully Synthetic Human Fab Antibody Library Based on Fixed VH/VL Framework Pairings with Favorable Biophysical Properties. MAbs 5, 445–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva G. F.; Harrison J. S.; Lai J. R. (2010) Contribution of Light Chain Residues to High Affinity Binding in an HIV-1 Antibody Explored by Combinatorial Scanning Mutagenesis. Biochemistry 49, 5464–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajdos F. F.; Adams C. W.; Breece T. N.; Presta L. G.; de Vos A. M.; Sidhu S. S. (2002) Comprehensive Functional Maps of the Antigen-Binding Site of an Anti-ErbB2 Antibody Obtained with Shotgun Scanning Mutagenesis. J. Mol. Biol. 320, 415–428. [DOI] [PubMed] [Google Scholar]
- Maruyama T.; Rodriguez L. L.; Jahrling P. B.; Sanchez A.; Khan A. S.; Nichol S. T.; Peters C. J.; Parren P. W.; Burton D. R. (1999) Ebola Virus Can Be Effectively Neutralized by Antibody Produced in Natural Human Infection. J. Virol. 73, 6024–6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama T.; Parren P. W.; Sanchez A.; Rensink I.; Rodriguez L. L.; Khan A. S.; Peters C. J.; Burton D. R. (1999) Recombinant Human Monoclonal Antibodies to Ebola Virus. J. Infect. Dis. 179(Suppl 1), S235–239. [DOI] [PubMed] [Google Scholar]
- Ekiert D. C.; Bhabha G.; Elsliger M. A.; Friesen R. H.; Jongeneelen M.; Throsby M.; Goudsmit J.; Wilson I. A. (2009) Antibody Recognition of a Highly Conserved Influenza Virus Epitope. Science 324, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X.; Yang Z. Y.; Li Y.; Hogerkorp C. M.; Schief W. R.; Seaman M. S.; Zhou T.; Schmidt S. D.; Wu L.; Xu L.; Longo N. S.; McKee K.; O’Dell S.; Louder M. K.; Wycuff D. L.; Feng Y.; Nason M.; Doria-Rose N.; Connors M.; Kwong P. D.; Roederer M.; Wyatt R. T.; Nabel G. J.; Mascola J. R. (2011) Focused Evolution of HIV-1 Neutralizing Antibodies Revealed by Structures and Deep Sequencing. Science 333, 1592–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T.; Georgiev I.; Wu X.; Yang Z. Y.; Dai K.; Finzi A.; Kwon Y. D.; Scheid J. F.; Shi W.; Xu L.; Yang Y.; Zhu J.; Nussenzweig M. C.; Sodroski J.; Shapiro L.; Nabel G. J.; Mascola J. R.; Kwong P. D. (2010) Structural Basis for Broad and Potent Neutralization of HIV-1 by Antibody VRC01. Science 329, 811–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J. A.; Hevey M.; Bakken R.; Guest S.; Bray M.; Schmaljohn A. L.; Hart M. K. (2000) Epitopes Involved in Antibody-Mediated Protection from Ebola Virus. Science 287, 1664–1666. [DOI] [PubMed] [Google Scholar]
- McLellan J. S.; Chen M.; Leung S.; Graepel K. W.; Du X.; Yang Y.; Zhou T.; Baxa U.; Yasuda E.; Beaumont T.; Kumar A.; Modjarrad K.; Zheng Z.; Zhao M.; Xia N.; Kwong P. D.; Graham B. S. (2013) Structure of RSV Fusion Glycoprotein Trimer Bound to a Prefusion-Specific Neutralizing Antibody. Science 340, 1113–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barouch D. H.; Whitney J. B.; Moldt B.; Klein F.; Oliveira T. Y.; Liu J.; Stephenson K. E.; Chang H. W.; Shekhar K.; Gupta S.; Nkolola J. P.; Seaman M. S.; Smith K. M.; Borducchi E. N.; Cabral C.; Smith J. Y.; Blackmore S.; Sanisetty S.; Perry J. R.; Beck M.; Lewis M. G.; Rinaldi W.; Chakraborty A. K.; Poignard P.; Nussenzweig M. C.; Burton D. R. (2013) Therapeutic Efficacy of Potent Neutralizing HIV-1-Specific Monoclonal Antibodies in SHIV-infected Rhesus Monkeys. Science 503, 224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal P.; Dowd K. A.; Brien J. D.; Edeling M. A.; Gorlatov S.; Johnson S.; Lee I.; Akahata W.; Nabel G. J.; Richter M. K.; Smit J. M.; Fremont D. H.; Pierson T. C.; Heise M. T.; Diamond M. S. (2013) Development of a Highly Protective Combination Monoclonal Antibody Therapy against Chikungunya Virus. PLoS Pathol. 9, e1003312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A.; Thomas W. D.; Guarner J.; Lamirande E. W.; Babcock G. J.; Greenough T. C.; Vogel L.; Hayes N.; Sullivan J. L.; Zaki S.; Subbarao K.; Ambrosino D. M. (2006) Therapy with a Severe Acute Respiratory Syndrome-Associated Coronavirus-Neutralizing Human Monoclonal Antibody Reduces Disease Severity and Viral Burden in Golden Syrian Hamsters. J. Infect. Dis. 193, 685–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson H.; Ye W.; Wernimont A.; Adams J. J.; Koide A.; Koide S.; Lam R.; Sidhu S. S. (2013) CDR-H3 Diversity Is Not Required for Antigen Recognition by Synthetic Antibodies. J. Mol. Biol. 425, 803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu S. S., Weiss G. A.. Constructing phage display libraries by oligonucleotide-directed mutagenesis, in Phage Display: A Practical Approach (Clackson T., and Lowman H. B., Eds.) Chapter 2, pp 27–41, Oxford University Press, New York, NY. [Google Scholar]
- Mazor Y.; Barnea I.; Keydar I.; Benhar I. (2007) Antibody Internalization Studied using a Novel IgG Binding Toxin Fusion. J. Immunol. Methods 321, 41–59. [DOI] [PubMed] [Google Scholar]
- Chandran K.; Sullivan N. J.; Felbor U.; Whelan S. P.; Cunningham J. M. (2005) Endosomal Proteolysis of the Ebola Virus Glycoprotein is Necessary for Infection. Science 308, 1643–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.