

Abstract
The preparation and full characterization of the 4-(nitrophenyl)phenoxyl radical, 2,6-di-tbutyl-4-(4′-nitrophenyl) phenoxyl radical (tBu2NPArO•) is described. This is a rare example of an isolable and crystallographically characterized phenoxyl radical and is the only example in which the parent phenol is also crystallographically well-defined. Analysis of EPR spectra indicates some spin delocalization onto the secondary aromatic ring and nitro group. Equilibrium studies show that the corresponding phenol has an O–H bond dissociation free energy (BDFE) of 77.8 ± 0.5 kcal mol–1 in MeCN (77.5 ± 0.5 kcal mol–1 in toluene). This value is higher than related isolated phenoxyl radicals, making this a useful reagent for hydrogen atom transfer (HAT) studies. Additional thermochemical and spectroscopic parameters are also discussed.
Introduction
Stable and transient phenoxyl radical species are important in chemical processes spanning a large range of applications. Examples include tyrosine/tyrosyl radical mediated enzymatic electron transfers and hydrogen atom transfers,1 food preservation (such as butylated hydroxytoluene or BHT),2 and fundamental studies of proton-coupled electron transfer (PCET)/hydrogen atom transfer (HAT) reactions.3 While most phenoxyl radicals are transient, sufficiently sterically encumbered phenoxyl radicals can be stable in solution under anaerobic conditions.4 The 2,6-di-tert-butyl-4-phenylphenoxyl radical (tBu2PhArO•), for instance, was prepared by Müller and co-workers in 1959, and isolated in 78–88% purity.5 Our laboratory has reported the clean isolation and structural characterization of the 2,4,6-tri-tert-butylphenoxyl radical (tBu3ArO•)6 and the 4,4′ coupled dimer of the 2,6-di-tert-butyl-4-methoxyphenoxyl radical (tBu2MeOArO•).7 The latter has a very weak C–C bond and is primarily dissociated in solution.
The O–H bond dissociation free energies (BDFEs) of the 2,6-di-tert-butyl-6-R-phenols are significantly modulated by the R substituent. The H atom affinities of the corresponding phenoxyl radicals (described by phenolic O–H BDFEs) range from 73.8 kcal mol–1 for the isolable7 R = OMe species to 80.4 kcal mol–1 for the transiently lived8 R = NO2 species (BDFEs in toluene), with the R = tBu and Ph derivatives being the same within error.9a The isolable R = tBu and OMe derivatives have proved to be useful hydrogen atom accepting reagents,10 complementary due to their different hydrogen atom affinities.10a,10b With the goal of preparing an isolable phenoxyl radical with a higher H atom affinity, we report here the preparation, full characterization, and thermochemistry of 2,6-di-tert-butyl-4-(4′-nitrophenyl)phenoxyl radical, or tBu2NPArO•.
Results and Discussion
tBu2NPArO• was prepared by treating a benzene solution of 2,6-di-tert-butyl-4-(4′-nitrophenyl)phenol, tBu2NPArO-H,11 with aqueous 1 M sodium hydroxide and potassium ferricyanide under anaerobic conditions. After 30 min, removal of the solvent under vacuum, extraction of the dark green material with pentane, and crystallization at −30 °C over 24 h yielded black crystals. These were found to be of high purity from elemental analysis and the 1H NMR spectra showed only minor diamagnetic impurities (<5%; see the Supporting Information).
High-quality X-ray crystal structures of tBu2NPArO• and its parent phenol were collected for structural comparison (Figure 1, Table 1). This type of direct structural comparison of a phenoxyl radical/phenol has previously not been possible. The parent phenol of the only previously structurally characterized phenoxyl radical, tBu3ArO-H, was found to be disordered over three positions in its crystals.6
Figure 1.
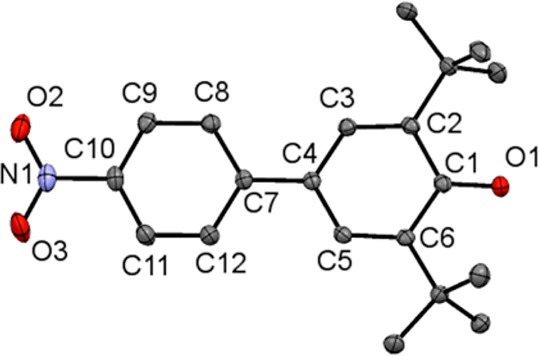
ORTEP drawing of tBu2NPArO• showing 50% probability thermal ellipsoids and labels for select atoms. Hydrogen atoms are omitted for clarity.
Table 1. Select Bond Lengths (Å) and Aryl–Aryl Torsion Angles (deg) of tBu2NPArO• and tBu2NPArO-H.
| tBu2NPArO• | tBu2NPArO-H | difference | |
|---|---|---|---|
| O1–C1 | 1.2509(14) | 1.3794(12) | –0.1285(18) |
| C1–C2 | 1.4699(17) | 1.4100(14) | 0.0599(22) |
| C2–C3 | 1.3696(16) | 1.3944(13) | –0.0248(21) |
| C3–C4 | 1.4194(16) | 1.3928(13) | 0.0266(21) |
| C4–C5 | 1.4228(17) | 1.3964(13) | 0.0264(21) |
| C5–C6 | 1.3711(16) | 1.3941(14) | –0.0230(21) |
| C6–C1 | 1.4751(16) | 1.4120(14) | 0.0631(21) |
| C4–C7 | 1.4754(16) | 1.4829(13) | –0.0075(21) |
| C7–C8 | 1.4069(17) | 1.4008(14) | 0.0061(22) |
| C8–C9 | 1.3833(17) | 1.3861(14) | –0.0028(22) |
| C9–C10 | 1.3873(18) | 1.3839(15) | 0.0034(23) |
| C10–C11 | 1.3828(19) | 1.3842(15) | –0.0014(24) |
| C11–C12 | 1.3842(17) | 1.3853(14) | –0.0011(22) |
| C12–C7 | 1.4114(16) | 1.4023(14) | 0.0091(21) |
| C10–N1 | 1.4272(16) | 1.4672(13) | –0.0400(21) |
| N1–O2 | 1.2262(16) | 1.2312(13) | –0.0050(21) |
| N1–O3 | 1.2289(16) | 1.2286(13) | –0.0003(21) |
| avg Ar–Ar torsion anglea | 17.5(1) | 31.9(1) | –14.4(1) |
Average aryl–aryl torsion angle (deg) refers to the average dihedral angle measured for C5–C4–C7–C12 and C3–C4–C7–C8.
The largest difference between the phenoxyl and phenol structures is in the O1–C1 bond distance, 1.251 vs 1.379 Å. This bond shortening of 0.128 Å is consistent with previous conclusions that phenoxyl radicals have significant ketone character, as suggested by the resonance forms in Scheme 1.4,6,12 The changes in the phenolic aromatic bond lengths support this model, as the C1–C2 and C1–C6 bond lengthen (avg 0.062 Å) more than the C3–C4 and C4–C5 bonds (avg 0.027 Å) while the C2–C3 and C5–C6 bonds shorten (avg −0.024 Å).
Scheme 1. Radical Resonance Forms of tBu2NPArO•.

The aryl–aryl linkage is slightly shorter in the radical, by 0.0075(22) Å, suggesting a small quinomethide component (Scheme 1). This is also suggested by the 0.0400(21) shortening of the C10–N1 bond to the nitro group, and the smaller average aryl–aryl torsion angle,13 of 17.5° in the radical vs 31.9° observed in the phenol.
The X-band CW EPR spectrum of tBu2NPArO• in toluene displays a multiline pattern centered at g = 2.007(2) that is well modeled by simulation (Figure 2). Hyperfine coupling constants were assigned by comparison to previously reported phenoxyl radical data9 and from the structural changes observed in the crystal structure: a3,5(2H) = 1.80 G, a8,12(2H) = 1.61 G, a9,11(2H) = 0.74 G and aNO2(1N) = 0.50 G.14 The 14N hyperfine coupling indicates spin density on the nitro group, as depicted in the bottom of Scheme 1. The observed spin density onto the nitro group suggests that the thermochemistry of tBu2NPArO• should be perturbed from that of the unsubstitued tBu2PhArO•.
Figure 2.
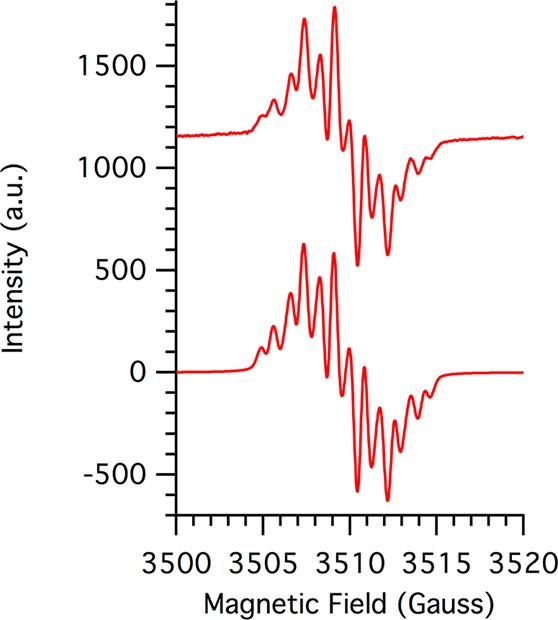
X-band EPR spectrum of 1 mM tBu2NPArO• in toluene recorded at 25 °C (top) and simulation (bottom).
The O–H BDFE of tBu2NPArO-H, was determined by equilibration with the thermochemically well-established3atBu3ArO• radical. In either acetonitrile-d3 or toluene-d8, a known concentration of tBu2NPArO-H was combined with several different concentrations of tBu3ArO• (eq 1).
| 1 |
Integration of the 1H NMR signals of these solutions gave equilibrium concentrations from which equilibrium constants were determined: Keq(acetonitrile) = 0.25 ± 0.03, Keq(toluene) = 0.26 ± 0.03. Thus, the O–H bond in tBu2NPArO-H is 0.8 ± 0.1 kcal mol–1 stronger than that in tBu3ArO-H in both acetonitrile and toluene. Using the known BDFE values of tBu3ArO-H3 and eq 2 gives BDFE(tBu2NPArO-HMeCN) = 77.8 ± 0.5 kcal mol–1 and BDFE(tBu2NPArO-Htol) = 77.5 ± 0.5 kcal mol–1. While this is a small increase, tBu2NPArO• is to our knowledge the thermodynamically strongest isolable, reagent quality organic hydrogen atom abstractor available.
| 2 |
Pedulli and co-workers have previously reported an empirical correlation between the O–H bond strengths of 2,6-tert-butyl-substituted phenols with the EPR hyperfine coupling constants, a3,5, of the corresponding phenoxyl radicals.9a Figure 3 shows a slightly modified version of this correlation using revised BDFE values.15 The values for tBu2NPArO• follow this correlation very closely.
Figure 3.
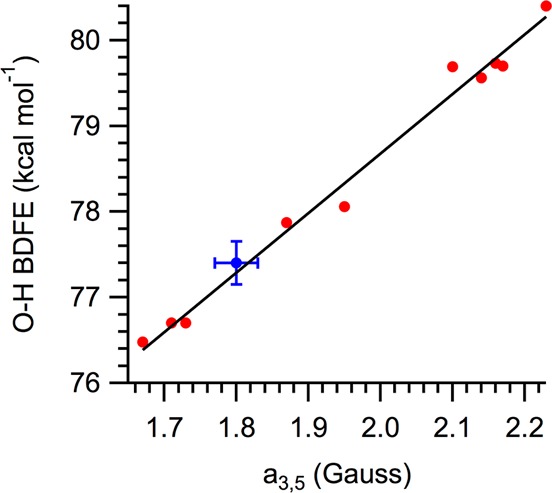
2,6-tBu2-4-X-ArO–H bond dissociation free energies (BDFEs) vs 3,5 hyperfine coupling constant for 2,6-tBu2-4-X-ArO• radicals. Data in red are part of the correlation reported in ref (9a) (revised to use updated BDFE values15); blue data point is tBu2NPArO(-H).
Cyclic voltammetry of tBu2NPArO• in acetonitrile with 0.1 M [nBu4N]PF6 as a supporting electrolyte displayed a reversible couple with E1/2 = −0.436 ± 0.010 V vs Fc+/0. This value is 0.26 V less negative than the related tBu3ArO0/– potential of −0.70 V vs Fc+/0.3a This is much larger than the reported potential difference of only 0.045 V between tBu2PhArO• and tBu3ArO• in 9:1 MeCN/H2O,16 illustrating the effect of the nitro substituent on the phenoxyl/phenol thermochemistry.
The cyclic voltammogram of tBu2NPArO-H displayed an irreversible anodic peak centered at 0.975 ± 0.010 V vs Fc+/0. It is presumably irreversible due to loss of the proton from the highly acidic radical cation.
The reduction potential of tBu2NPArO• and the BDFE(tBu2NPArO-H) imply that in acetonitrile the pKa of tBu2NPArOH is 24 ± 0.4 by Hess’ law (eq 3). Compared to its parent phenylphenol, the (nitrophenyl)phenol has a significantly higher acidity17 and more positive reduction potential. These are both due to the stabilization of the phenoxide anion by the 4-substituted nitrobenzene group. The higher BDFE of tBu2NPArO-H is due to the shifts in pKa and E° not exactly offsetting each other, with the nitro group affecting the pKa less in free energy terms.18
| 3 |
These values can be assembled into a “square scheme” that describes the PCET thermochemistry of tBu2NPArO-H (Scheme 2). We have included the irreversible anodic peak potential, Ea,p, even though it is not a thermochemical value. Using this value to crudely estimate E°(tBu2NPArO-H+/0) ≅ +0.95 V would imply that the pKa of the radical cation is about 9 units lower than that of the phenol.
Scheme 2. Thermochemical “Square Scheme” for tBu2NPArO(-H).
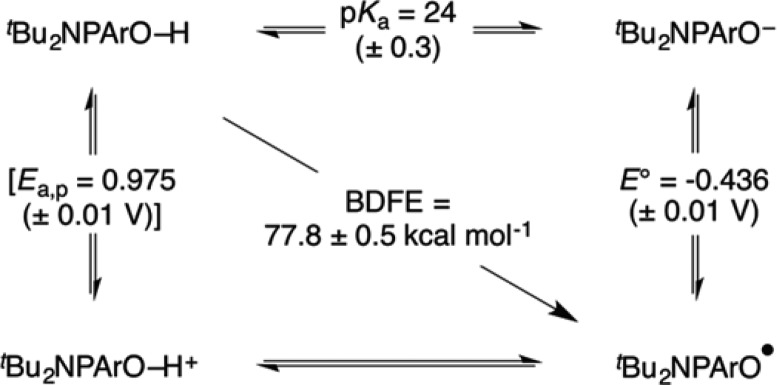
Ea,p is bracketed since it refers to an irreversible anodic peak potential and is not a thermochemical value.
In conclusion, the 4-(nitrophenyl)phenoxyl radical tBu2NPArO• is a previously unreported phenoxyl radical that is easily prepared in high purity and reasonable yield. Equilibrium studies show that the tBu2NPArO-H BDFE is modestly stronger than that of its unsubstituted isolable relative, tBu2PhArO• (ΔBDFEtoluene = 0.8 kcal mol–1). tBu2NPArO-H has the highest reported BDFE of any isolable organic hydrogen atom acceptor: 77.8 ± 0.5 kcal mol–1 in acetonitrile and 77.5 ± 0.5 kcal mol–1 in toluene. The combination of easy isolation of the phenoxyl radical in pure form and its relatively high hydrogen atom affinity should make this a useful reagent for studying hydrogen atom transfer reactions.
Experimental Section
Materials
Unless otherwise noted, all chemicals were purchased from commercial sources and used without purification. Toluene was dried using a “Grubb’s type” Seca Solvent System. Acetonitrile was purchased from Burdick & Jackson (low-water brand) and stored in an argon-pressurized glovebox plumbed directly into the glovebox. Toluene-d8 and acetonitrile-d3 were dried over NaK and CaH2, respectively, and vacuum distilled. tBu3ArO•6 and tBu2NPArOH19 were prepared following literature methods.
Instrumentation
All NMR spectra were collected on 500 MHz spectrometers and chemical shifts referenced to TMS using residual solvent peaks. The reported EPR spectrum was collected using an X-band spectrometer at room temperature in toluene. Simulation of the spectrum was preformed using the W95EPR program.
Synthesis of tBu2NPArO•
A 100 mL two neck round-bottom flask was charged with 467 mg (1.43 mmol) of tBu2NPArOH dissolved in ∼15 mL of benzene, 5 mL of 1 M NaOH and a stir bar. The flask was fitted with a 180° Schlenk adapter on one neck and a solid addition funnel containing 1.20 g (3.64 mmol) of solid K3Fe(CN)6 on the other neck. The biphasic mixture was degassed by 3 sequential freeze–pump–thaw cycles. After degassed, the mixture was frozen and the K3Fe(CN)6 was added. The frozen mixture was allowed to thaw at room temperature and left to stir. After 1 h of stirring, the solvents were removed in vacuo and extracted with pentane. Crystals were grown from a saturated pentane solution at −30 °C. Yield: 279 mg, 52%. Anal. Calcd for C20H24NO3: C, 73.59; H, 7.41; N, 4.29. Found: C, 73.88; H, 7.60; N, 4.34.
Acknowledgments
We gratefully acknowledge financial support from the US National Institute of Health (2R01GM50422). We thank Professor Stefan Stoll for useful discussions about EPR simulation.
Supporting Information Available
NMR and optical spectra, BDFE calculations, electrochemical data, crystallographic information, and an ORTEP of tBu2NPArOH. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
Department of Chemistry, Yale University, 225 Prospect St, New Haven, CT 06520.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- a Styring S.; Sjöholm J.; Mamedov F. Biochim. Biophys. Acta 2012, 1817, 76. [DOI] [PubMed] [Google Scholar]; b Reece S. Y.; Nocera D. G. Annu. Rev. Biochem. 2009, 78, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]; c McEvoy J. P.; Brudvig G. W. Chem. Rev. 2006, 106, 4455. [DOI] [PubMed] [Google Scholar]; d Whittaker J. W. Chem. Rev. 2003, 103, 2347. [DOI] [PubMed] [Google Scholar]
- Babich H. Environ. Res. 1982, 29, 1. [DOI] [PubMed] [Google Scholar]
- a Warren J. J.; Tronic T. T.; Mayer J. M. Chem. Rev. 2010, 110, 6961. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Weinberg D. R.; Gagliardi C. J.; Hull J. F.; Murphy C. F.; Kent C. A.; Westlake B. C.; Paul A.; Ess D. H.; McCafferty D. G.; Meyer T. J. Chem. Rev. 2012, 112, 4016. [DOI] [PubMed] [Google Scholar]
- Altwicker E. R. Chem. Rev. 1967, 67, 475. [Google Scholar]
- a Müller E.; Schick A.; Scheffler K. Chem. Ber. 1959, 92, 474. [Google Scholar]; b Related 4-tolyl- and 4-anisyl-phenoxyl radicals have been reported but not isolated; see ref (4)
- Manner V. W.; Markle T. F.; Freudenthal J. H.; Roth J. P.; Mayer J. M. Chem. Commun. 2008, 256. [DOI] [PubMed] [Google Scholar]
- Wittman J. M.; Hayoun R.; Kaminsky W.; Coggins M. K.; Mayer J. M. J. Am. Chem. Soc. 2013, 135, 12956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook C. D.; Gilmour N. D. J. Org. Chem. 1960, 25, 1429. [Google Scholar]
- a Brigati G.; Lucarini M.; Mugnaini V.; Pedulli G. F. J. Org. Chem. 2002, 67, 4828. [DOI] [PubMed] [Google Scholar]; b Lucarini M.; Pedrielli P.; Pedulli G. F.; Cabiddu S.; Fattuoni C. J. Org. Chem. 1996, 61, 9259. [Google Scholar]; c Lucarini M.; Pedulli G. F. Chem. Soc. Rev. 2010, 39, 2106. [DOI] [PubMed] [Google Scholar]; d Reiker A.; Scheffler K. Justus Liebigs Ann. Chem. 1965, 689, 78. [Google Scholar]
- a Waidmann C. R.; Zhou X.; Tsai E. A.; Kaminsky W.; Hrovat D. A.; Borden W. T.; Mayer J. M. J. Am. Chem. Soc. 2009, 131, 4729. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Warren J. J.; Mayer J. M. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 5282. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Valdez C. N.; Braten M. B.; Soria A.; Gamelin D. R.; Mayer J. M. J. Am. Chem. Soc. 2013, 135, 8492. [DOI] [PubMed] [Google Scholar]; d Schrauben J. N.; Hayoun R.; Valdez C. N.; Braten M. M.; Fridley L.; Mayer J. M. Science 2012, 336, 1298. [DOI] [PubMed] [Google Scholar]; e Manner V. W.; Lindsey A. D.; Mader E. A.; Harvey J. N.; Mayer J. M. Chem. Sci. 2012, 3, 230. [Google Scholar]; f Warren J. J.; Mayer J. M. J. Am. Chem. Soc. 2011, 133, 8544. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Mader E. A.; Manner V. W.; Markle T. F.; Wu A.; Franz J. A.; Mayer J. M. J. Am. Chem. Soc. 2009, 131, 4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chern Y. T.; Ju M. H. Macromolecules 2009, 42, 169. [Google Scholar]
- a O’Malley P. J. J. Phys. Chem. B 2002, 106, 12331. [Google Scholar]; b Xie C.; Lahti P. M.; George C. Org. Lett. 2000, 2, 3417. [DOI] [PubMed] [Google Scholar]
- Average torsion angle refers to the average dihedral angle measured for atoms C3–C4–C7–C8 and C5–C4–C7–C12.
- EPR hyperfine coupling constants reported have an assumed error margin of ∼±0.05 G.
- The correlation in ref (9a) used bond dissociation enthalpies (BDEs), which were determined from equilibration experiments. ΔBDE values were obtained from these equilibrium constants and then converted to BDEs using a gas-phase BDE of tBu3ArO-H. Here, we take their equilibrium constants to be ΔBDFEs and scale them to the reported BDFE of tBu3ArO-H in benzene.3
- a Steuber F. W.; Dimroth K. Chem. Ber. 1966, 99, 258. [Google Scholar]; b Values reported in (a): E1/2(tBu2PhArO•–) = −0.014 V; E1/2(tBu3ArO•–) = −0.059 V (both vs Ag/AgCl; in 9:1 MeCN/H2O; 0.01m [Me4N+][OH–], 0.01m [Me4N+][Cl–])
- The pKa of tBu2PhArO-H is taken to be roughly equal to the pKa of tBu3ArO-H (∼28 in MeCN; ref (3)) since their BDFEs and E° are roughly equivalent.
- CG is a constant that contains the free energy of formation of H•, free energy of solvation of H•, as well as the nature of the electrode. In MeCN, CG = 54.9 kcal mol–1. A more detailed description can be found in ref (3).
- Chern Y. T.; Ju M. H. Macromolecules 2009, 42, 169. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.