

Abstract

RNA oligonucleotides containing a phenyl selenide derivative of 5-methyluridine were chemically synthesized by solid-phase synthesis. The phenyl selenide is rapidly converted to an electrophilic, allylic phenyl seleneate under mild oxidative conditions. The phenyl seleneate yields interstrand cross-links when part of a duplex and is useful for synthesizing oligonucleotide conjugates. Formation of the latter is illustrated by reaction of an oligonucleotide containing the phenyl selenide with amino acids in the presence of mild oxidant. The products formed are analogous to those observed in tRNA that are believed to be formed posttranslationally via a biosynthetic intermediate that is chemically homologous to the phenyl seleneate.
Introduction
The known roles of RNA in biology and biochemistry continue to grow. Its newly discovered features include novel primary and tertiary structures. One way in which organic chemistry can contribute to this important area is by developing methods for synthesizing and analyzing these novel structures. In this report we describe the chemistry that can be used to synthesize RNA-containing C5-substituted uridines and for producing interstrand cross-links in RNA. The latter function may prove useful for detecting tertiary interactions in RNA.
A variety of enzymatic and chemical tools that are used for studying DNA structure are also employed for examining the more complex structure of RNA molecules.1 These tools are being applied in the development of high-throughput methods for RNA structure elucidation.2−4 The tools employed include hydroxyl radical cleavage and chemical reagents that exploit the differential reactivity of nucleotides based upon their solvent exposure.4−7 Dimethyl sulfate and diethyl pyrocarbonate are examples of the latter.1,8,9 In addition, Weeks has pioneered the development of reagents that exploit the accessibility of the 2′-hydroxyl group to extract RNA structural information from differential reactivity.10−13 Each of these reagents provides inferential structural information based on relative reactivity: less-accessible nucleotides that are “buried” within the three-dimensional RNA structure are less reactive.
In contrast, cross-linking agents detect proximal functional groups. A number of cross-linking methods are available, including photolabile nucleotides (e.g., 4-thiouridine, 6-thioguanosine) and exogenous reagents (e.g., 1,4-phenyl diglyoxal).14−16 Researchers have also utilized photochemistry to produce reactive intermediates, such as carbenes and nitrenes, although cross-link yields are often low in these experiments.14 In another approach, cisplatin was elegantly used in conjunction with phosphorothioated RNA to detect short-range interactions.17 Recently, mild methods for cross-linking RNA via modified nucleotides that contain furan or vinyl sulfides have been reported. Oxidation of the furan appended through the 2′-hydroxyl of uridine or introduced as a nonnucleotide spacer in an oligonucleotide produces an electrophilic 1,4-dicarbonyl that reacts with a cytidine in the opposing strand.18 Similarly, oxidation of the vinyl sulfide that is an analogue of adenosine produces a more electrophilic vinyl sulfoxide that reacts with the exocyclic amine of cytidine.19
We previously reported on the mild oxidation of 1 in duplex DNA by a variety of oxidants, such as singlet oxygen, H2O2, and NaIO4 (Scheme 1).20,21 The resulting phenyl selenoxide 2 undergoes rapid [2,3]-sigmatropic rearrangement to an electrophilic species (3) that rearomatizes upon reaction with nucleophiles. The 2′-deoxycytidine analogue 5 undergoes a similar reaction to form 6.22 In duplex DNA electrophiles 3 and 6 react with nucleotides in the opposing strand to produce cross-linked products (e.g., 4). Water slowly traps the electrophiles in the absence of an appropriate nucleophilic partner in DNA, and other nucleophiles such as azide can be used as well. We anticipated that this reactivity pattern could be extended to RNA where it might be useful for probing tertiary interactions, as well as providing a convergent approach for preparing RNA molecules containing C5-functionalized uridines, a common modification in bacterial RNA.23
Scheme 1.

Results and Discussion
Synthesis of RNA Containing a Latent Electrophilic C5-Modified Uridine
Phosphoramidite 12 was synthesized in a manner similar to that previously described for 1 starting from 7 (Scheme 2).24−26 Although the intermediate bromide en route to 8 could be isolated, this proved to be impractical, and it was the bromination step that was responsible for the low yields. The reaction did not proceed to completion, and attempts to push it further resulted in even lower yields. Despite this, the reaction was scalable and 8 was obtained in millimole quantities and carried forward to 10 via ammonolysis product 9. Selective silylation of 10 using Ogilvie’s method yielded only the desired 2′-silyl ether 11, which was then phosphitylated to produce 12.27
Scheme 2.

Oligonucleotides containing 9 (molecules are referred to using the same number whether they are present in a polymer or as a monomer.) were prepared by automated solid-phase synthesis (Figure 1). Standard coupling methods were employed for all phosphoramidites, except for 12, which was manually coupled for 10 min as previously described.28 Coupling yields for 12, as measured using dimethoxytrityl cation detection, varied widely from as low as 40% to almost quantitative. Oligonucleotides were deprotected using a mixture of concentrated aqueous ammonia and methylamine (25 °C, 4 h), followed by desilylation using Et3N·3HF.29,30 Carrying out the deprotection at higher temperatures (50–60 °C) led to decomposition of the phenyl selenide (9). Anhydrous tert-butyl hydroperoxide was substituted for iodine in a mixture of water and pyridine as the oxidant to minimize premature oxidation of the phenyl selenide.31 Despite this safeguard, it was necessary to purify oligonucleotides containing the phenyl selenide 9 by a two-step procedure. Following denaturing polyacrylamide gel electrophoresis, full-length oligonucleotide containing 9 was separated from its more polar product(s) (e.g., 17), believed to result from adventitious oxidation, by reverse-phase HPLC. Purified oligonucleotides were characterized by MALDI-TOF MS. We prepared three oligonucleotides containing 9 for this study (Figure 1). In each instance, a 2′-deoxynucleotide is introduced at the 3′-terminus. This was performed for convenience, so that NaIO4 could be used as an oxidant. However, H2O2 (vide infra) is also a satisfactory oxidant and is compatible with 3′-terminal ribonucleotides.
Figure 1.

Sequences of oligonucleotides containing 9. Capital letters indicate a 2′-deoxyribonucleotide and small letters a ribonucleotide.
Rapid Formation of an Electrophile from 9 upon Mild Oxidation
Mild oxidation of phenyl selenides 1 and 5 rapidly produces the corresponding allylic phenyl seleneates (3, 6) via the respective selenoxides (e.g., 2). Sodium periodate was the oxidant of choice for the 2′-deoxyribonucleosides, but this reagent is incompatible with the vicinal diol in ribonucleosides. The oxidation of ribonucleosides with NaIO4 is so facile that this reagent is used in conjunction with reductive amination to produce 3′-oligonucleotide conjugates.32 Consequently, H2O2 was used. 1H NMR analysis (with water suppression) of the oxidation carried out in deuterated phosphate buffer (50 mM, pD 7.4) showed that 9 behaves in a very similar manner to 1 and 5 (Figure 2).20,22 The phenyl selenide 9 was consumed within minutes, giving rise to a diastereomeric mixture of the allylic phenyl seleneate (16, Figure 2B). The corresponding selenoxide was not detected. In the absence of a nucleophile, 16 reacted slowly with H2O to form 17, which was only a minor product after 18 h at 25 °C (Figure 2C). Phenyl seleneate 16 reacts more slowly with H2O in buffer than does 3 or 6, both of which showed complete conversion to their respective hydroxymethyl species (e.g., 17) after 18 h.20,22
Figure 2.
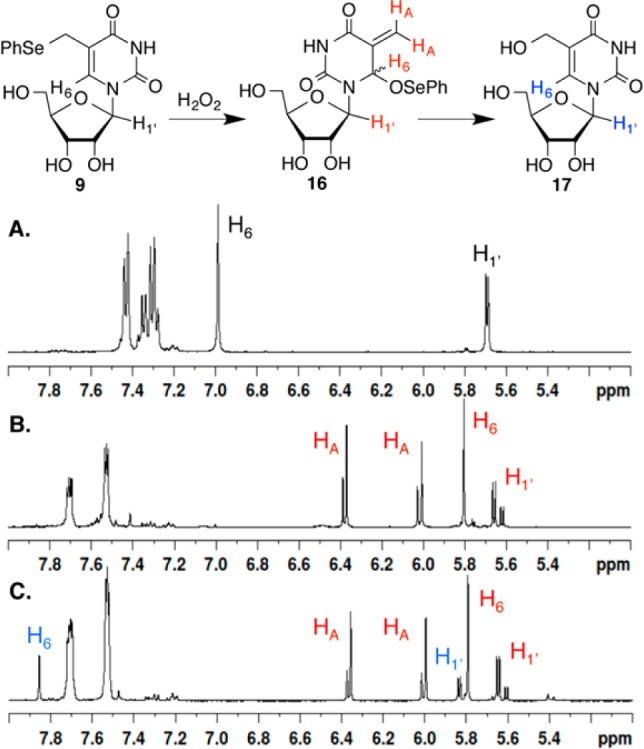
1H NMR analysis of H2O2 (50 mM) oxidation of 9 (50 mM) in deuterated phosphate buffer (50 mM, pD 7.4) at 25 °C. A. Prior to H2O2 addition. B. Fifteen minutes after H2O2 addition. C. Eighteen hours after H2O2 addition.
Interstrand Cross-Link Formation from 9 upon Mild Oxidation
UV-melting experiments indicated that 9 did not significantly destabilize duplex RNA. The Tm (10 mM phosphate pH 7.4, 100 mM KCl, 2 mM MgCl2) for 19a containing 9 (56.3 ± 0.4
°C) was just slightly lower than that of the analogous duplex
containing uridine in place of the phenyl selenide (19b, 58.7 ± 0.3 °C).33 Stable hybridization
is a requirement for ICL formation. Initial cross-linking experiments
were carried out on 9 in a duplex composed of 2′-deoxynucleotides
(18, Figure 3). Treatment of 5′-32P-18 with NaIO4 (5 mM) yielded 51%
interstrand cross-linked (ICL) product in 3 h at 25 °C and more
than 45% in 30 min. This is comparable to and even higher than the
ICL yields obtained from 1 and 5 under the
same conditions.20,22 In contrast, oxidation of 5′-32P-19a reliably produced less than 15% of the
cross-linked product at 25 °C and only 30 ± 4% at 37 °C
in 5 h. Similarly, oxidation of 5′-32P-19a by H2O2 (10 mM) at 37 °C produced the
cross-linked product in 26 ± 1% yield. Hydroxyl radical cleavage
indicated that the A opposite 9 (A24) was
the major cross-linking site, but a small amount of cross-linking
also occurred at A25.33,34 Mass spectrometry
of the ICL confirmed that the product corresponded to the mass of
the duplex following the loss of benzeneselenol. Upon the basis of
these observations and the precedent established by 1, we propose that 23 corresponds to the major ICL product
from oxidation of 9 in 19a.21 Hence, 16 (produced from oxidation of 9) reacts predominantly with the opposing adenine, producing
the cross-link product analogous to 4 (Scheme 1).
Figure 3.

Sequences of oligonucleotide duplexes containing 9. Capital letters indicate a 2′-deoxyribonucleotide and small letters a ribonucleotide.
Kinetic analysis of the cross-linking of 19a correlated with the observed lower yields (Figure 4). The first-order rate constant was 1.0 ± 0.2 × 10–4 s–1 (t1/2 = 116 min). This is more than 11 times slower than the rate constant for cross-linking from 1 in a comparable DNA duplex.35 The slower reactivity of 16 with water observed by 1H NMR is not the source of decreased cross-linking rate constant. If it were, then 9 would also have reacted more slowly in the DNA duplex (18). Subsequent cross-linking experiments were typically carried out overnight at 37 °C to ensure complete reaction of 9 (or more accurately 16). In search of an explanation for why the ICL yield from 19a was so much lower than in 18, we focused on possible differences in the rotational barrier of the glycosidic bond in 16 as a function of oligonucleotide sequence, because ICL formation requires occupation of the syn-conformation in which the allylic phenyl seleneate is oriented toward the opposing strand in the duplex (Scheme 3). This implicitly assumes that the rate-limiting step in cross-linking is adoption of the syn-conformation. This is unknown for 16 but has been shown to be the case in related studies in which ICLs are produced upon photolysis of 1.21,36
Figure 4.

Interstrand cross-link growth from 5′-32P-19a as a function of time at 37 °C following oxidation by NaIO4 (5 mM).
Scheme 3.

We initially examined the effects of mismatches at the adjacent base pair (20a, 20b). Duplex destabilization in the vicinity of the 9-adenosine base pair might facilitate adoption of the syn-conformation by weakening base pairing. However, in side-by-side reactions the ICL yields from 19a (22 ± 2%), 20a (21 ± 2%), and 20b (22 ± 5%) were within experimental error of one another. Consequently, we must rely upon related literature to rationalize the slower cross-linking by 16 than by 3 and the assumption. It is well established that the barrier for rotation about the glycosidic bond in pyrimidines is lower when the nucleoside is in the C2′-endo conformation than when it populates the C3′-endo conformation (Scheme 3).37 2′-Deoxynucleotides adopt the C2′-endo conformation in B-form DNA whereas RNA duplexes typically adopt an A-form structure in which the ribonucleotides are in the C3′-endo conformation. Furthermore, recent calculations predict that 2′-deoxy pyrimidine nucleosides populate the syn-conformational isomer that is required for ICL formation more readily than do the comparable ribonucleosides.38 Although ribonucleotides typically adopt the C3′-endo conformation in duplex RNA (e.g., 19a), structurally related 2′-fluorothymidine is believed to populate the C2′-endo isomer when only one molecule of it is present in a DNA duplex.39 We hypothesize that the surrounding duplex environment influences the sugar pucker of 9 (16) much the same way that the conformation of 2′-fluorothymidine is affected. Lower ICL yields and slower cross-linking are observed from 19a because 9 (16) exists predominantly in the C3′-endo conformation, whereas the C2′-endo conformation of the phenyl selenide should be relatively preferred in the DNA duplex (18).
Cross-linking in duplexes containing 9 treated with NaIO4 was general and in many ways paralleled the reactivity of 1.21 For instance, the ICL yield declined to 14 ± 2% when 9 was flanked by adenosines (22) that provide greater π-stacking from 22 ± 2% in 19a. We also observed that ICLs form more efficiently when 9 is opposite adenosine and cytidine than when the phenyl selenide is opposed by guanosine or uridine (Figure 5). This could be the result of the positioning of the electrophile in syn-16 with the relatively more nucleophilic N1 and N3 of adenine (19a) and cytosine (21b), respectively, compared to the N1 (21a) and N3 (21c) of guanine and uracil. As was the case with 19a, hydroxyl radical cleavage of the isolated ICLs indicated that the nucleotide opposite 9 (16) is the major site of cross-linking.33 Salt content also had an effect on ICL yields in 19a (Figure 6). Addition of MgCl2 to NaIO4 oxidations of 19a significantly lowered the cross-link yields, and to a smaller extent so too did increasing the concentration of NaCl from 100 mM to 250 mM. One possible explanation for the reduced ICL yields with either increasing MgCl2 or NaCl concentration is that the duplex is more stable in higher salt, resulting in an increase in the barrier for rotation about the glycosidic bond in 16 (Scheme 3). The larger decrease observed by the addition of MgCl2 may also be explained by the report that magnesium rigidifies the RNA.40 This too could increase the barrier to forming the syn-conformation of 16 that is required for cross-linking.
Figure 5.

Interstrand cross-link yield from 9 as a function of opposing nucleotide at 37 °C following oxidation by NaIO4 (5 mM).
Figure 6.

Interstrand cross-link yield from 9 in 19a as a function of salt at 37 °C following oxidation by NaIO4 (5 mM). Note: [NaCl] = 100 mM as [MgCl2] is varied (left), and [MgCl2] = 0 as [NaCl] is varied (right).
Synthesis of Amino Acid Conjugates from 9
Oligonucleotide conjugates have useful applications in biotechnology and as potential therapeutic agents.41−47 In addition, RNA is often posttranslationally modified. Some amino acid modifications at the C5-position of uridine are proposed to proceed via electrophilic intermediates analogous to 16 that result from cysteine addition to the C6-position.23 Consequently, we carried out conjugation reactions between 14 and amino acids to demonstrate the utility of phenyl selenide 9 (Scheme 4). The respective conjugates of 14 (10 μM) with glycine (24a, 59%) and phenylalanine (24b, 43%) were isolated by reverse-phase HPLC and characterized by MALDI-TOF MS, following reaction in the presence of the amino acid (10 mM) and NaIO4 (5 mM) at 37 °C for 3 h.
Scheme 4.

Summary
Phenyl selenide 9 was incorporated into chemically synthesized oligonucleotides. When part of a duplex, 9 yields ICLs predominantly with the opposing nucleotide upon mild oxidation by H2O2 or NaIO4 via the allylic phenyl seleneate (16). The allylic phenyl seleneate (16) is also useful for forming oligonucleotide conjugates, as demonstrated by reacting the single stranded oligonucleotide 14 with amino acids. Conjugation with amino acids is particularly relevant to the biosynthesis of tRNA.23 These reactions suggest that 9 may be a useful tool for producing modified RNA molecules in a convergent manner, providing novel platforms for the delivery of nucleic acids to cells. This chemistry may also prove valuable as a structural probe for detecting tertiary interactions in RNA such as kissing loops and pseudoknots.48−52 If 9 can be incorporated into RNA via its respective nucleotide triphosphate, as the respective 2′-deoxynucleotide phenyl selenide is, it may also be useful for randomly probing RNA folding.2,3,53
Experimental Section
General Methods
Solvents used in reactions were purified and dried (using CaH2 or Na/benzophenone) by distillation before use. Reagents were purchased from commercial sources and were used without further purification. Reactions were carried out under a positive pressure of argon atmosphere and monitored by TLC on silica gel G-25 UV254 (0.25 mm). Spots were detected using UV light and/or by charring with a solution of either ammonium molybdate, ceric ammonium sulfate in water and H2SO4, or p-anisaldehyde in ethanol and H2SO4. Flash chromatography was performed on silica gel 60 (40–60 μm). The ratio between silica gel and crude product ranged from 100:1 to 20:1 (w/w).
Oligonucleotides were synthesized on an Applied Biosystems Incorporated 394 oligonucleotide synthesizer. Phenyl selenide oligonucleotides were synthesized using 2′-O-TOM RNA phosphoramidites commercially available from Glen Research. Phosphoramidite 12 was coupled manually (10 min), as previously described.28 Commercially available THF/pyridine/acetic anhydride was used as a capping reagent and 1 M tert-butyl hydroperoxide in toluene was used as an oxidizing reagent. Oligonucleotides were deprotected using concentrated AMA [50% NH4OH, 50% methylamine (40% in H2O)] at room temperature for 4 h, desilylated in a 1.8 M TEA·3HF solution (2:1:1.5 N-methylpyrrolidinone:TEA:TEA·3HF), purified by 20% denaturing PAGE, isolated by the crush and soak method, desalted using C-18-Sep-Pak cartridges (Waters), and characterized by MALDI-TOF.29,30 Oligonucleotides containing the phenyl selenide modification (9) were subjected to additional purification by HPLC on a RP-C18 column, with monitoring carried out at 260 nm. The peak of interest was collected at 20.1 min using the following gradient conditions: 0–18 min 10–20% B in A, 18–23 min 20–80% B in A, 23–28 min, 80% B in A, 28–30 min 80–10% B in A, 30–50 min 10% B in A, at a flow rate of 1.0 mL/min [A: 0.05 M TEAA (pH 7.0)/MeCN 95:5; B: 0.05 M TEAA (pH 7.0)/MeCN 50:50]. Oligonucleotides were 5′-32P-labeled by T4 polynucleotide kinase (New England Biolabs) and γ-32P-ATP (PerkinElmer) using standard protocols.54 Experiments involving radiolabeled oligonucleotides were analyzed following PAGE using a Storm 840 phosphorimager and Imagequant TL software. Radiolabeled oligonucleotides were hybridized with 5 equiv of complementary oligonucleotides in 10 mM potassium phosphate (pH 7.2) and 100 mM NaCl at 65 °C for 15 min, slowly cooled to room temperature, and then stored at 4 °C overnight.
Preparation of 8
N-Bromosuccinimide (NBS) was freshly recrystallized from water and dried under vacuum overnight. A solution of 7 (2 g, 3.5 mmol), NBS (934 mg, 5.25 mmol), and AIBN (93 mg, 0.57 mmol) was suspended in dry benzene (30 mL). The solution was a slight yellow color, and the solid did not completely dissolve. The solution was heated to reflux for 6 h, whereupon the solution turned from dark red to brown. The solvent was evaporated in vacuo to yield a brown solid. Diphenyl diselenide (2.18 g, 7 mmol) was dissolved in dry DMF (15 mL) to create an orange solution. NaBH4 (530 mg, 14 mmol) was added slowly to the solution over 10 min. Upon each addition of NaBH4, the solution bubbled fiercely and faded from an orange color to colorless. The brown residue from above was dissolved in dry DMF (15 mL) and added to the solution. The reaction was stirred overnight, yielding a light brown mixture. The reaction was quenched by the addition of H2O (50 mL), whereupon the mixture bubbled violently. The mixture was extracted with EtOAc (4 × 50 mL). The organic layers were combined and split in half. Each half was washed with NaHCO3 (60 mL) and brine (60 mL) and dried over NaSO4. The solvent was evaporated to yield a yellow oil (3.25 g) which was then purified by flash chromatography (EtOAc/DCM 2%–10%) to give 8 as a white foam (805 mg, 32%): 1H NMR (CDCl3) δ 3.27 (d, 1H, J = 12.4 Hz), 3.54 (d, 1H, J = 12.4 Hz), 4.61 (m, 3H), 5.49 (t, 1H, J = 6 Hz), 5.75 (m, 1H), 6.30 (d, 1H, J = 6 Hz), 6.84 (s, 1H), 7.26 (m, 4H), 7.38 (m, 6H), 7.57 (m, 5H), 7.97 (m, 4H), 8.07 (dd, 2H, J = 6.8 Hz), 9.82 (bd s, 1H); 13C NMR (CDCl3), 14.4, 21.2, 23.3, 60.6, 64.1, 71.4, 73.8, 80.7, 87.3, 113.7, 128.3, 128.6, 128.75, 128.84, 129.0, 129.3, 129.48, 129.51, 129.8, 130.0, 130.1, 133.87, 133.94, 134.0, 135.27, 135.33, 150.0, 161.8, 165.4, 165.5, 166.1; IR (KBr): 3060, 2923, 1726, 1601, 1452, 1378, 1315, 1266, 1178, 1070, 1025 cm–1; HRMS (ESI/APCI-TOF) m/z calculated for [M + H]+ C37H31N2O9Se 727.1195, found 727.1184.
Preparation of 9
Gaseous ammonia was bubbled through a mixture of 8 (800 mg, 1.1 mmol) in MeOH (8 mL) cooled to 0 °C for 40 min. As the solution became saturated with ammonia, the solid 8 slowly dissolved and the liquid became yellow. After the solution was saturated with ammonia, the reaction flask was sealed and the mixture was stirred overnight. After 30 min of reaction, all solid had dissolved into solution and the mixture was a clear yellow color. To quench the reaction, argon was bubbled through the solution for 30 min. The solvent was evaporated to a yellow oil (700 mg). The resulting yellow oil, which was insoluble in the chromatography solvent, was dissolved in MeOH. Silica (3 g) was added to the mixture and the solvent was evaporated, yielding yellow, free-floating silica powder. This powder was added to the top of a flash column, and the compound was purified by flash chromatography (MeOH/DCM 8%–12%), yielding 9 as a white foam (234 mg, 57%): 1H NMR (MeOH-d4) δ 3.72 (d, 1H, J = 2.8 Hz), 3.75 (d, 1H, J = 3.2 Hz), 3.83 (m, 2H), 3.92 (t, 1H, J = 5.2 Hz), 4.04 (m, 2H), 5.91 (d, 1H, J = 4.4 Hz), 7.41 (m, 3H), 7.60 (m, 3H); 13C NMR (MeOH-d4), 14.6, 20.9, 23.8, 24.2, 24.5, 61.7, 63.4, 71.3, 75.9, 86.3, 90.7, 113.2, 109.0, 109.6, 130.5, 130.90, 135.89, 135.94, 136.0, 138.7, 152.3, 164.8, 210.2; IR (KBr): 3388, 2924, 1684, 1559, 1540, 1507, 1275, 1102 cm–1; HRMS (ESI/APCI-TOF) m/z calculated for [M + H]+ C16H19N2O6Se 415.0403, found 415.0402.
Preparation of 10
The triol 9 (452 mg, 1.21 mmol) was azeotropically dried twice from pyridine (2 mL). Dry 9 was dissolved in pyridine (9 mL) and cooled to 0 °C. Vacuum-dried DMTCl (617 mg, 1.82 mmol) and DMAP (29 mg, 0.24 mmol) were added to the cooled solution, which immediately became orange in color. The mixture was allowed warm to room temperature and was stirred under argon for 8 h. The reaction was quenched by the addition of NaHCO3 (15 mL), and the resulting solution was extracted with DCM (2 × 15 mL) and washed with brine (20 mL). The organic layer was concentrated in vacuo to a yellow foam (1.11 g). Flash chromatography (0.5% Et3N to 2% MeOH, 0.5% Et3N in DCM) yielded 10 as a white foam (539 mg, 66%): 1H NMR (CDCl3) δ 3.10 (d, 1H, J = 12 Hz), 3.39 (m, 2H), 4.84 (d, 1H, J = 12 Hz), 3.76 (s, 6H), 4.13 (m, 1H), 4.19 (m, 2H), 5.84 (d, 1H, J = 2.8 Hz), 6.82 (m, 3H), 7.13 (m, 2H), 7.17 (m, 2H), 7.27 (m, 8H), 7.42 (d, 2H, J = 1.6 Hz), 7.46 (s, 1H); 13C NMR (CDCl3), 23.0, 55.5, 62.9, 70.3, 76.6, 77.4, 83.9, 87.0, 90.9, 112.4, 113.6, 127.3, 127.8, 128.3, 128.4, 129.2, 130.0, 130.3, 134.4, 135.5, 135.7, 136.6, 144.6, 150.9, 158.9, 163.1; IR: 3229, 3056, 2925, 1670, 1684, 1653, 1635, 1507, 1457, 1251, 1177, 1086, 1035 cm–1; HRMS (ESI/APCI-TOF) m/z calculated for [M + Na]+ C37H36N2O8NaSe 739.1529, found 739.1528.
Preparation of 11
Tritylated nucleoside 10 (450 mg, 0.66 mmol) was azeotropically dried three times from pyridine (3 × 1.5 mL). Silver nitrate (136 mg, 0.80 mmol) was suspended in a mixture of pyridine (267 μL, 3.32 mmol) and THF (4.5 mL) and sonicated for 30 min. TBDMSCl (121 mg, 0.80 mmol) was added to the contents in the sonicated flask, creating a milky white solution with some undissolved solid. Dried 10 was dissolved in dry THF (1.5 mL) and added to the flask via syringe. No physical change was observed in reaction flask. The reaction was stirred under argon for 5 h. The contents of the reaction flask consisted of a slightly yellow liquid layer and gray solid collected at the bottom of the flask. The heterogeneous mixture was filtered through Celite into a stirring 20% NaHCO3 solution (10 mL), extracted with EtOAc (3 × 10 mL), and concentrated in vacuo to yield a yellow foam (448 mg). Purification by flash chromatography (10% EtOAc in DCM) yielded 11 as a white foam (337 mg, 64%): 1H NMR (CDCl3) δ 0.13 (s, 6H), 0.92 (s, 10H), 2.59 (d, 1H, J = 5.2 Hz), 3.12–3.10 (d, 1H, J = 11.6 Hz), 3.44–3.35 (ddd, 2H, J1 = 19.6, J2 = 10.4 Hz, J3 = 3.2), 3.46–3.43 (d, 1H, 11.6 Hz), 3.77 (s, 6H), 4.10–4.07 (m, 2H), 4.27–4.25 (t, 1H, J = 4.4 Hz), 5.92–5.91 (d, 1H, J = 4.4 Hz), 6.84–6.81 (m, 4H), 7.21–7.15 (m, 4H), 7.34–7.29 (m, 9H), 7.42–7.39 (dd, 2H, J1 = 3.2 Hz, J2 = 1.6 Hz), 7.50 (s, 1H), 8.37 (bd s, 1H); 13C NMR (CDCl3) −4.9, −4.5, 18.2, 22.8, 25.9, 29.9, 55.5, 63.9, 71.1, 76.3, 83.8, 87.3, 88.8, 112,6, 113.6, 127.4, 127.6, 128.3, 128.4, 129.1, 130.3, 130.35, 134.0, 135.7, 136.7, 144.6, 159.0; IR (KBr): 3193, 3056, 2927, 2856, 1700, 1684, 1653, 1507, 1458, 1252, 1176, 1121, 1035 cm–1; HRMS (ESI/APCI-TOF) m/z calculated for [M + Na]+ C43H50N2O8NaSiSe 853.2394, found 853.2395.
Preparation of 12
Silylated nucleoside 11 (101 mg, 0.13 mmol) was azeotropically dried twice over pyridine (1 mL). Dried 11 was dissolved in DCM (0.8 mL), and Hünig’s base was added (87 μL, 63.3 mg, 0.49 mmol). 2-Cyanoethyl N,N-diisopropylchlorophosphoramidite (43 μL, 45 mg, 0.19 mmol) was added to the reaction flask via syringe. The reaction was stirred under argon at room temperature for 2.5 h, yielding a clear yellow solution. The reaction mixture was diluted with DCM (1 mL), quenched with NaHCO3 (2 mL), and extracted with DCM (2 × 2 mL). The organic layer was concentrated to a yellow oil. Flash chromatography (30% EtOAc in hexanes) yielded a mixture of diastereomers of 12 as a white foam (102 mg, 81%): 1H NMR (CDCl3) δ 0.01–0.08 (m, 12H), 0.89 (d, 20H, J = 7.2 Hz), 1.15 (d, 6H, J = 6.8 Hz), 1.18–1.13 (m, 17H), 1.27–1.24 (t, 8H, J = 7.2 Hz), 2.37–2.34 (t, 2H, J = 6.4 Hz), 2.67–2.63 (q, 2H, J = 6.4 Hz), 3.04 (d, 1H, J = 12.4 Hz), 3.09 (d, 1H, J = 12.4 Hz), 3.34–3.29 (dd, 1H, J1 = 16 Hz, J2 = 2.8 Hz), 3.31 (t, 1H, J = 2.8 Hz) 3.41–3.38 (dd, 1H, J1 = 8.4 Hz, J2 = 2.8 Hz), 3.57–3.51 (m, 7H), 3.76–3.74 (m, 12H), 3.89–3.86 (m, 1H), 3.99–3.90 (m, 1H), 4.28–4.21 (m, 3H), 4.35–4.33 (q, 1H, J = 4.4 Hz), 4.40 (t, 1H, J = 4.8 Hz), 4.49–4.46 (dd, 1H, J1 = 6.4 Hz, J2 = 4.8 Hz), 5.95–5.93 (d, 1H, J = 5.6 Hz), 6.05–6.03 (d, 1H, J = 6 Hz), 6.84–6.79 (m, 8H), 7.16–7.12 (m, 6H), 7.33–7.26 (m, 19H), 7.43–7.41 (m, 4H), 7.70 (s, 1H), 7.73 (s, 1H), 8.62 (br, 2H); 31P NMR (CDCl3) 149.1, 150.5; HRMS (ESI/APCI-TOF) m/z calculated for [M + Na]+ C52H67N4O9NaSiPSe 1053.3472, found 1053.3489.
1H NMR Study of Oxidation of 9 by H2O2
Nucleoside 9 was dissolved in D2O (50 mM deuterated phosphate buffer, pD 7.4) to make a 50 mM solution in an NMR tube. The mixture was analyzed via 1H NMR with water suppression, whereupon a 20× solution of H2O2 was added to the tube via pipet. The final concentrations of 9 and H2O2 were 50 mM. The mixture was quickly agitated and placed back in the NMR instrument for kinetic analysis. 1H NMR spectra (8 scans, with water suppression) were taken every minute for 60 min. An additional spectrum was taken of the solution after incubation at room temperature for 18 h.
Interstrand Cross-Link Formation with Duplex RNA
The 32P-labeled oligonucleotide (0.3 μM) and its complementary sequence (1.5 μM) were dissolved in 100 mM NaCl and 10 mM NaH2PO4 (pH 7.4). The solution was heated to 65 °C and allowed to cool to 4 °C over the course of 1 h. NaIO4 (5 mM) and H2O2 (10 mM) reactions of RNA duplexes (11 nM) were carried out in 10 mM sodium phosphate (pH 7.2) and 100 mM NaCl. Aliquots were taken at prescribed times, immediately quenched with an equal volume of 95% formamide loading buffer, and stored at −20 °C until analysis by 20% denaturing PAGE.
Fe(II)-EDTA Analysis of Cross-Linked DNA
Fe(II)-EDTA reactions were carried out in 500 μM (NH4)2Fe(SO4)2, 500 μM EDTA, 2 mM sodium ascorbate, 100 mM H2O2, 10 mM NaCl, 10 mM potassium phosphate (pH 7.2) for 10 min at room temperature and were quenched with 10 μL of excess thiourea (100 mM). Samples were desalted via C-18 Sep-Pak cartridges (100 mg), lyophilized, suspended in 95% formamide loading buffer, and subjected to electrophoresis on a 20% denaturing polyacrylamide gel. The alkali ladder was generated by treating radiolabeled oligonucleotide with 0.2 M NaOH in 10 mM EDTA at 90 °C for 15 s. The reaction was quenched by addition of 5 μL each of stop buffer (9.5 M urea, 85 mM NaOAc, 1% v/v AcOH) and 95% formamide loading buffer. RNase A sequencing was performed with 1 μU of enzyme in 5 μL of reaction buffer (300 mM NaCl, 5 mM EDTA, and 10 mM Tris-HCl (pH 7.5)) at 37 °C for 20 min. To this reaction was added 5 μL of 95% formamide loading buffer.
Conjugation Reactions with Amino Acids
The phenyl selenide-modified oligonucleotide (14, 10 μM) and the relevant nucleophile (10 mM) were incubated at 37 °C in a mixture of 10 mM potassium phosphate (pH 7.2), 100 mM NaCl, and 5 mM NaIO4 (total volume 10 μL). The resulting solution was diluted with the addition of extra water (15 μL) and filtered through a 0.22 μm filter. The mixture was subjected to UPLC analysis on a RP-C18 HPLC column with monitoring carried out at 260 nm. Peaks were analyzed using the following gradient conditions: 0–18 min 10–20% B in A, 18–23 min 20–80% B in A, 23–28 min, 80% B in A, 28–30 min 80–10% B in A, 30–50 min 10% B in A, at a flow rate of 1.0 mL/min [A: 0.05 M TEAA (pH 7.0)/MeCN 95:5; B: 0.05 M TEAA (pH 7.0)/MeCN 50:50]. The relevant peak of interest was collected from the LC, lyophilized to dryness, and analyzed by MALDI-TOF MS.
Acknowledgments
We are grateful for generous support of this research from the National Institute of General Medical Sciences (GM-054996). We thank Dr. Michael Delaney (Dharmacon Research), Dr. Rakesh Paul (Johns Hopkins University), and Souradyuti Ghosh (Johns Hopkins University) for advice on the manual coupling procedure. We thank Dr. Cathy Moore (Johns Hopkins University) for assistance with the water suppression NMR experiment on 9.
Supporting Information Available
NMR spectra of monomers, mass spectra of oligonucleotides containing nonnative nucleotides, hydroxyl radical cleavage histograms of cross-linked RNA, and full 1H NMR spectra of the oxidation of 9 (Figure 2). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Ehresmann C.; Baudin F.; Mougel M.; Romby P.; Ebel J.-P.; Ehresmann B. Nucleic Acids Res. 1987, 15, 9109–9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero P.; Kladwang W.; VanLang C. C.; Das R. Biochemistry 2012, 51, 7037–7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kladwang W.; Das R. Biochemistry 2010, 49, 7414–7416. [DOI] [PubMed] [Google Scholar]
- Laederach A.; Das R.; Vicens Q.; Pearlman S. M.; Brenowitz M.; Herschlag D.; Altman R. B. Nat. Protoc. 2008, 3, 1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adilakshmi T.; Bellur D. L.; Woodson S. A. Nature 2008, 455, 1268–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston C. Y.; Sclavi B.; Sullivan M.; Deras M. L.; Woodson S. A.; Chance M. R.; Brenowitz M. Methods Enzymol. 2000, 317, 353–368. [DOI] [PubMed] [Google Scholar]
- Tullius T. D.; Greenbaum J. A. Curr. Opin. Chem. Biol. 2005, 9, 127–134. [DOI] [PubMed] [Google Scholar]
- Tijerina P.; Mohr S.; Russell R. Nat. Protocols 2007, 2, 2608–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S.; Zubradt M.; Washietl S.; Kellis M.; Weissman J. S. Nature 2014, 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinnis J. L.; Dunkle J. A.; Cate J. H. D.; Weeks K. M. J. Am. Chem. Soc. 2012, 134, 6617–6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer S. A.; Weeks K. M. J. Am. Chem. Soc. 2007, 129, 4144–4145. [DOI] [PubMed] [Google Scholar]
- Wilkinson K. A.; Merino E. J.; Weeks K. M. J. Am. Chem. Soc. 2005, 127, 4659–4667. [DOI] [PubMed] [Google Scholar]
- Merino E. J.; Wilkinson K. A.; Coughlan J. L.; Weeks K. M. J. Am. Chem. Soc. 2005, 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- Harris M. E.; Christian E. L. Methods Enzymol. 2009, 468, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juzumiene D.; Shapkina T.; Kirillov S.; Wollenzien P. Methods 2001, 25, 333–343. [DOI] [PubMed] [Google Scholar]
- Wagner R.; Garrett R. A. Nucleic Acids Res. 1978, 5, 4065–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E. G.; DeRose V. J. J. Am. Chem. Soc. 2011, 134, 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrette L. L. G.; Gyssels E.; Loncke J.; Madder A. Org. Biomol. Chem. 2014, 12, 931–935. [DOI] [PubMed] [Google Scholar]
- Kusano S.; Haruyama T.; Ishiyama S.; Hagihara S.; Nagatsugi F. Chem. Commun. 2014, 50, 3951–3954. [DOI] [PubMed] [Google Scholar]
- Hong I. S.; Greenberg M. M. J. Am. Chem. Soc. 2005, 127, 10510–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H.; Majumdar A.; Tolman J. R.; Greenberg M. M. J. Am. Chem. Soc. 2008, 130, 17981–17987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Hong I. S.; Li H.; Seidman M. M.; Greenberg M. M. J. Am. Chem. Soc. 2008, 130, 10299–10306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M.; Alfonzo J. D. Chem. Biol. 2014, 21, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedballa U.; Vorbruggen H. J. Org. Chem. 1974, 39, 3654–3660. [DOI] [PubMed] [Google Scholar]
- Hong I. S.; Greenberg M. M. Org. Lett. 2004, 6, 5011–5013. [DOI] [PubMed] [Google Scholar]
- Vorbrueggen H.; Bennua B. Chem. Ber. 1981, 114, 1279–1286. [Google Scholar]
- Hakimelahi G. H.; Proba Z. A.; Ogilvie K. K. Can. J. Chem. 1982, 60, 1106–1113. [Google Scholar]
- Ghosh S.; Greenberg M. M. J. Org. Chem. 2014, 79, 5948–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy M. P.; Hanna N. B.; Faroqui F. Tetrahedron Lett. 1994, 35, 4311–4314. [Google Scholar]
- Wincott F.; DiRenzo A.; Shaffer C.; Grimm S.; Tracz D.; Workman C.; Sweedler D.; Gonzalez C.; Scaringe S.; Usman N. Nucleic Acids Res. 1995, 23, 2677–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I. S.; Greenberg M. M. J. Am. Chem. Soc. 2005, 127, 3692–3693. [DOI] [PubMed] [Google Scholar]
- Proudnikov D.; Mirzabekov A. Nucleic Acids Res. 1996, 24, 4535–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information.
- Millard J. T.; Weidner M. F.; Kirchner J. J.; Ribeiro S.; Hopkins P. B. Nucleic Acids Res. 1991, 19, 1885–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X.; Greenberg M. M. Nucleic Acids Res. 2008, 36, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L.; Horvat S. M.; Schiesser C. H.; Greenberg M. M. Org. Lett. 2013, 15, 3618–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haschemeyer A. E. V.; Rich A. J. Mol. Biol. 1967, 27, 369–384. [DOI] [PubMed] [Google Scholar]
- Foloppe N.; Nilsson L. J. Phys. Chem. B 2005, 109, 9119–9131. [DOI] [PubMed] [Google Scholar]
- Williams D. M.; Benseler F.; Eckstein F. Biochemistry 1991, 30, 4001–4009. [DOI] [PubMed] [Google Scholar]
- Fiore J. L.; Holmstrom E. D.; Nesbitt D. J. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2902–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher L.; McGouran J. F.; Kessler B. M.; Schofield C. J.; Davis B. G. Angew. Chem., Int. Ed. 2013, 52, 10553–10558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pourceau G.; Meyer A.; Vasseur J. J.; Morvan F. J. Org. Chem. 2009, 74, 1218–1222. [DOI] [PubMed] [Google Scholar]
- Stetsenko D. A.; Gait M. J.. Methods Mol. Biol. (Totowa, NJ, U.S.) 2005, 288, 205–224. [DOI] [PubMed] [Google Scholar]
- Zaramella S.; Yeheskiely E.; Strömberg R. J. Am. Chem. Soc. 2004, 126, 14029–14035. [DOI] [PubMed] [Google Scholar]
- Seo T. S.; Li Z.; Ruparel H.; Ju J. J. Org. Chem. 2003, 68, 609–612. [DOI] [PubMed] [Google Scholar]
- Birts C. N.; Sanzone A. P.; El-Sagheer A. H.; Blaydes J. P.; Brown T.; Tavassoli A. Angew. Chem., Int. Ed. 2014, 53, 2362–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sagheer A. H.; Brown T. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 15329–15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weixlbaumer A.; Werner A.; Flamm C.; Westhof E.; Schroeder R. e. Nucleic Acids Res. 2004, 32, 5126–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C.; Otabe T.; Matsumoto S.; Dohno C.; Murata A.; Hagihara M.; Nakatani K. Chem.—Eur. J. 2014, 20, 5282–5287. [DOI] [PubMed] [Google Scholar]
- Salim N.; Lamichhane R.; Zhao R.; Banerjee T.; Philip J.; Rueda D.; Feig A. L. Biophys. J. 2012, 102, 1097–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengesbach M.; Kim N.-K.; Feigon J.; Stone M. D. Angew. Chem., Int. Ed. 2012, 51, 5876–5879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.; Ishibe-Murakami S.; Patel D. J.; Serganov A. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14801–14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong I. S.; Ding H.; Greenberg M. M. J. Am. Chem. Soc. 2006, 128, 2230–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J.; Fritsch E. F.; Maniatis T.. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press, Woodbury, NY, 1989. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.