

SUMMARY
With the exception of imprinted genes and certain repeats, DNA methylation is globally erased during pre-implantation development. Recent studies have suggested that Tet3-mediated oxidation of 5-methylcytosine (5mC) and DNA replication-dependent dilution both contribute to global paternal DNA demethylation, but demethylation of the maternal genome occurs via replication. Here we present genome-scale DNA methylation maps for both the paternal and maternal genomes of Tet3-depleted and/or DNA replication-inhibited zygotes. In both genomes, we found that inhibition of DNA replication blocks DNA demethylation independently from Tet3 function, and that Tet3 facilitates DNA demethylation by coupling with DNA replication. For both genomes, our data indicate that replication-dependent dilution is the major contributor to demethylation, but Tet3 plays an important role, particularly at certain loci. Our study thus defines the respective functions of Tet3 and DNA replication in paternal DNA demethylation and reveals an unexpected contribution of Tet3 to demethylation of the maternal genome.
INTRODUCTION
DNA methylation at the 5-position of cytosine (5mC) in mammals largely takes place in the context of CpG, and plays important roles in diverse processes including transcription regulation, maintenance of genomic integrity, regulation of genomic imprinting, and X chromosome inactivation (Goldberg et al., 2007; Jaenisch and Bird, 2003; Sasaki and Matsui, 2008). DNA methylation pattern is established and maintained by DNA methyltransferases (DNMTs). Although DNA methylation is originally thought to be a stable modification, recent studies have demonstrated that active DNA demethylation can be achieved through a highly orchestrated enzymatic process involving Ten eleven translocation (TET) protein-mediated iterative oxidation (He et al., 2011; Ito et al., 2010; Ito et al., 2011; Tahiliani et al., 2009), followed by Thymine DNA glycosylase (TDG)-initiated base excision repair (He et al., 2011; Kohli and Zhang, 2013; Maiti and Drohat, 2011). In addition to enzyme-catalyzed active DNA demethylation, DNA methylation can also be lost through replication-dependent dilution when the maintenance DNA methyltransferase DNMT1 is not localized to nucleus or is not functional. Therefore, the steady state level of 5mC is largely determined by a balance between the actions of DNMTs and demethylating enzymes.
Shortly after fertilization, global loss of 5mC takes place in the paternal genome (Mayer et al., 2000; Oswald et al., 2000). This loss of 5mC is explained by rapid conversion of 5mC to 5hmC and its further oxidation products catalyzed by the maternally stored Tet3 proteins (Gu et al., 2011; Inoue et al., 2012; Wossidlo et al., 2011). Immunostaining studies indicated that 5mC derived oxidation products are gradually lost during preimplantation development through a DNA replication-dependent manner (Inoue et al., 2011; Inoue and Zhang, 2011). These observations suggest a simple model by which loss of paternal DNA methylation may be achieved by Tet3-mediated 5mC oxidation followed by replication-dependent dilution of 5hmC/5fC/5caC. However, such model is largely based on immunostaining, which heavily rely on antibody specificity. In addition, because these data are only semi-quantitative, it is not clear to what extent Tet3-mediated oxidation and replication-dependent dilution respectively contribute to the demethylation process. Furthermore, these studies do not provide the resolution that allows pinpointing the genomic location where Tet3-catalyzed 5mC oxidation takes place. Finally, there are also reports that other DNA repair pathways involving deamination may also contribute to zygotic DNA demethylation (Hajkova et al., 2010; Santos et al., 2013). Thus, it is important to analyze zygotic DNA demethylation in greater detail to address the relative contribution of Tet3-mediated 5mC oxidation and DNA replication, and to provide a higher resolution profiling of parental origin-specific 5mC so that the genomic location of Tet3-affected CpGs can be identified.
In this study, we generated a Tet3 conditional knockout mouse model and derived Tet3-null oocytes. By physically isolating paternal and maternal pronuclei from control and Tet3 null zygotes in the presence or absence of a DNA replication inhibitor, we generated genome-scale DNA methylation profiles using reduced representation bisulfite sequencing (RRBS). Comparative analyses of DNA methylation datasets revealed that replication-dependent passive dilution is the major contributing factor for zygotic DNA demethylation, whereas Tet3 facilitates DNA demethylation by coupling with replication. Interestingly, in addition to contributing to paternal DNA demethylation, Tet3 also localizes to maternal pronuclei and promotes maternal DNA demethylation.
RESULTS
Tet3-dependent 5mC oxidation in both paternal and maternal genomes
To better understand to what extent that Tet3 contributes to paternal DNA demethylation in zygotes, we generated a Tet3 conditional knockout (CKO) mouse model in which exons 7–9 encoding the catalytic domain of Tet3 protein are floxed (Figure S1A–C). ZP3-Cre mediated deletion of these exons resulted in generation of oocytes without functional Tet3 proteins. Reverse transcription and polymerase chain reaction (RT-PCR) analyses of mRNAs extracted from CKO oocytes followed by DNA sequencing confirmed that exons 7–9 are indeed absent in the Tet3 mRNA and that this truncation results in a frame-shift, causing premature termination at the 4th amino acid of exon 10 (Figure S1D). Immunostaining with an antibody against the C-terminus of Tet3 protein (Gu et al., 2011) confirmed lack of Tet3 protein in the CKO zygotes (Figure 1A). Similar to previous reports (Gu et al., 2011; Inoue et al., 2012), Tet3 is predominantly localized in the paternal pronucleus. Interestingly, weaker but detectable Tet3 staining signals were also observed in the maternal pronucleus, and this signal was absent in CKO zygotes (Figure 1A). This raised the possibility that Tet3 may also contribute to maternal DNA demethylation. Consistent with the previous observation that Tet3 is responsible for 5hmC generation in the paternal pronucleus (Gu et al., 2011; Inoue et al., 2012; Wossidlo et al., 2011), depletion of Tet3 results in loss of 5hmC in the paternal pronucleus of CKO zygotes (Figure 1B). Importantly, the 5hmC signal detected in the maternal pronucleus at 13 hours post fertilization (hpf) is also lost in the CKO zygotes (Figure 1B). The detected maternal 5hmC seems to be newly generated by Tet3 after fertilization, rather than inherited from oocytes, as no Tet3-dependent 5hmC signal is detected in the maternal pronucleus in earlier pronuclear stage zygotes (8 hpf, Figure S1E). Collectively, these data not only confirm previous observations that Tet3 is responsible for paternal 5mC oxidation, but also suggest that Tet3 contributes to 5mC oxidation in the maternal genome.
Figure 1. Generation and verification of Tet3-KO oocytes and isolation of paternal pronuclei from maternally Tet3-depleted zygotes.

(A) Representative images and quantification verifying depletion of Tet3 protein from conditional Tet3 KO zygotes. Zygotes were stained with anti-Tet3 antibody (red). Maternal pronuclei are marked by immunostaining with an anti-H3K4me3 antibody (green). The relative Tet3 levels are quantified by setting the fluorescent signal of wild-type (WT) maternal pronucleus as 1. ♀, maternal pronucleus; ♂, paternal pronucleus; PB, polar body; CKO, Tet3 conditional knockout. Dotted circles indicate the rim of zygote cytoplasm. Scale bar, 20 μm. *, P<0.01 (student’s t-test). Error bars, s.d.
(B) Representative images and quantification verifying loss of 5hmC in both paternal and maternal pronuclei of CKO zygotes. Zygotes were stained with anti-5hmC (red) and anti-5mC (green) antibodies at 13 hours after fertilization. The graph on the right represents the relative signal intensity of 5hmC with the maternal pronucleus of WT zygotes set as 1.
(C) Representative images of zygotes stained with anti-BrdU antibody indicate that DNA replication is complete at 12 hrs post-fertilization. Zygotes were incubated in media containing BrdU during 7–8 or 12–13 hours after fertilization (hpf).
(D) A schematic representation of pronuclear isolation at 13 hpf at G2 phase. PN, pronucleus.
(E) Confirmation of the successful separation of the paternal and maternal pronuclei as indicated by imprinted genes exhibit expected allele-specific DNA methylation. Base-resolution methylation of paternal and maternal imprinting genes, H19 and Peg10, covered in our RRBS data set is shown.
See also Figure S1.
Loss of Tet3 partially impairs paternal DNA demethylation
Both DNA replication and Tet3-mediated 5mC oxidation can contribute to DNA demethylation. Since Tet3-mediated 5mC oxidation coincides with the onset of DNA replication in zygotes (Inoue et al., 2011; Inoue and Zhang, 2011; Wossidlo et al., 2011), it was crucial to determine the end of the S-phase so that the contribution of replication-dependent dilution can be accurately calculated. To this end, we performed BrdU incorporation experiments following in vitro fertilization (IVF), and found that BrdU incorporation is readily detectable at 7–8 hpf but not at 12–13 hpf (Figure 1C), indicating that DNA synthesis is completed by 12 hpf. To ensure the completion of S-phase at the time of sample collection, we manually collected each paternal and maternal pronucleus at 13 hpf (G2-phase) immediately prior to entering the first mitosis (Figure 1D). Due to the limited amount of samples, we chose to use the RRBS method for DNA methylation analysis. Although this method only covers 5–10% of total CpGs in the genome and is enriched for CpG-rich sequences, it has been proven to be reliable for analyzing small number of cells, and has been used for genome-scale DNA methylation analyses of human and mouse preimplantation embryos at single-base resolution (Guo et al., 2014; Smith et al., 2014; Smith et al., 2012). In parallel to the G2-phase pronuclei, we also performed RRBS analysis of sperm and oocyte DNA (Table S1). As an indication of successful separation of paternal and maternal pronuclei, known germline differentially methylated regions (gDMRs) of paternally imprinted gene H19 and maternally imprinted gene Peg10 exhibited expected allele-specific DNA methylation patterns in our RRBS data set (Figure 1E). In addition, we compared the DNA methylation levels of commonly covered CpG sites of our sperm RRBS data and two published data sets (Smallwood et al., 2011; Smith et al., 2012), and found that the three data sets are highly consistent (data not shown).
Having confirmed successful generation of an RRBS dataset for paternal and maternal DNA, we first focused our analyses on paternal DNA methylation using CpG sites that are commonly covered for at least 10 times in all samples. Consistent with previous reports (Smallwood et al., 2011; Smith et al., 2012), overall DNA methylation in sperm exhibits a bimodal distribution (Figure 2A, left panel). Additionally, we observed an increase of the intermediately methylated CpGs concomitant with a dramatic decrease of highly methylated sites in WT paternal pronuclear DNA (Figure 2A, middle panel). Interestingly, although a dramatic decrease of highly methylated sites was also observed in CKO paternal DNA, the methylation pattern of CKO paternal DNA is significantly different from that of WT paternal DNA (P<2.2 ×10−22, Wilcoxon rank sum test) with many sites showing medium-high DNA methylation, suggesting a partial impairment of DNA demethylation in CKO paternal pronuclei (Figure 2A, right panel). To better examine the degree of demethylation, we calculated DNA methylation levels in 100-bp tiles across the genome and divided the tiles into four groups based on their methylation levels. Compared to sperm, highly methylated tiles (75–100%) are mostly shifted to medium-high (50–75%) and medium-low (25–50%) tiles in both WT and CKO paternal DNA (Figure 2B), indicating the presence of Tet3-independent DNA demethylation. However, when compared to WT, CKO paternal DNA exhibits an increase in the number of medium-high tiles with a concomitant reduction of low and medium-low methylation tiles (Figure 2B), indicating that paternal DNA demethylation is partly impaired in CKO zygotes.
Figure 2. Loss of Tet3 partially impairs paternal DNA demethylation.

(A) Histograms of DNA methylation values across 100-bp tiles in sperm, WT and CKO paternal pronuclei. CpG sites that are at least 10x covered in all samples were examined. Note that common 10x CpG sites are more enriched in CpG-rich sequences and thus show lower overall DNA methylation level compared to that of all detected sites.
(B) Distributions of DNA methylation levels in sperm, WT, and CKO paternal pronuclei. 100-bp tiles of genomic DNA were divided into four groups based on their methylation levels as low (0–25%, purple), medium-low (25–50%, blue), medium-high (50–75%, green), and high (75–100%, red) methylation.
(C) Histograms showing the distributions of RDLSp→♂PN values of 100-bp tiles in WT and CKO paternal DNA. Only tiles that are methylated in sperm (MLSp ≥20%, n=58,101) were examined. RDLSp→♂PN is defined as [(MLSp – ML♂PN)/MLSp]. ML, DNA methylation level. RDL values less than −1 were set to −1.
(D) Boxplot of RDLSp→♂PN values. Red line represents the median. Boxes and whiskers represent for the 25th and 75th, and 2.5th and 97.5th percentiles, respectively.
(E) Distribution of RDLSp→♂PN values in WT and CKO paternal DNA. The RDL values are separated into four groups marked by red (0.7–1), green (0.3–0.7), blue (−0.3–0.3), and purple (<−0.3).
We then restricted our analysis on tiles with methylation level ≥20% in sperm (MLSperm≥20%), and further calculated their relative demethylation levels (RDLs) (Figure 2C). RDLSp→♂PN is defined as [(MLSperm–ML♂ pronucleus)/MLSperm] such that RDL values of 0 and 1 represent maintenance of DNA methylation and complete DNA demethylation (i.e., active DNA demethylation), respectively, and that an RDL value around 0.5 indicates replication-dependent passive dilution. The RDL values exhibit a roughly normal distribution for both WT and CKO zygotes (Figure 2C). In WT zygotes, the distribution is centered at 0.5, suggesting that paternal DNA methylation undergoes passive dilution in general (Figure 2C and 2D). However, the RDL distribution in CKO zygotes shifted to the left and is centered at 0.35 (Figure 2C and D). These results suggest a model where passive dilution plays a major role in paternal DNA demethylation and Tet3-mediated 5mC oxidation facilitates this process. To quantitatively compare the shift of RDL values between WT and CKO paternal DNA, we divided RDLs into four groups that include negative (<−0.3), near-zero (−0.3–0.3, i.e., 0±0.3), near-half (0.3–0.7, i.e., 0.5±0.2), and high RDL (>0.7), and calculated the percentage of total sperm-methylated tiles in each group (Figure 2E). In WT zygotes, 24.9% of tiles are in the high-RDL group suggesting potential active demethylation of these tiles; 39.7% are in the near-half-RDL group indicating replication-dependent dilution; 33.7% are in the near-zero-RDL group which represents maintenance of DNA methylation at these tiles; and finally only 1.6% are in the negative-RDL group, which is expected and shows the robustness of our data set. Interestingly, the percentage of tiles in the high-RDL group decreased from 24.9% in WT zygotes to 7.5% in CKO zygotes, suggesting a role of Tet3 in active DNA demethylation.
5fC/5caC partially contributes to the observed active DNA demethylation
It is worth noting that Tet3 converts paternal 5mC not only to 5hmC but also to 5fC and 5caC (Inoue et al., 2011). Since both 5fC and 5caC are interpreted as unmodified cytosines in RRBS analysis, it is possible that a fraction of the observed active demethylation in the high-RDL group results from the conversion of 5mC/5hmC to 5fC/5caC. This possibility is supported by the observation that the percentage of high-RDL group decreased from 24.9% in WT to 7.5% in CKO zygotes (Figure 2E). To further examine the contribution of 5fC/5caC in the observed active DNA demethylation, we sought to distinguish 5fC/5caC sites in our analysis. Because currently available technologies do not allow profiling 5fC/5caC with very limited samples such as zygotes (Plongthongkum et al., 2014), we instead performed further analysis by digging into our RRBS data set (Figure S2). The RRBS method utilizes the MspI restriction enzyme that recognizes CCGG sequences. However, this enzyme cuts very inefficiently when the inner C of the CCGG sequences is replaced by 5fC/5caC (Ito et al., 2011). Therefore, the 5′-end of the RRBS reads, which is generated by MspI digestion during library construction, should only contain less-than-expected amount of 5fC/5caC, while the remaining 3′-end of the reads should contain normal levels of 5fC/5caC as the bulk genome. Indeed, we observed less high-RDL tiles in the MspI sites (16.8%, Figure S2A and S2C) compared to the rest of the reads (26.3%, Figure S2B and S2C). This result supports the notion that 5fC/5caC contributed to the observed active demethylation. However, RDL analyses of MspI sites and the rest of the reads generally yield similar results (Figure S2A and B), which are also similar to those using whole RRBS reads for analysis (Figure 2C, WT), suggesting that the contribution of 5fC/5caC to global DNA demethylation is limited. Thus, we cannot exclude the possibility that some loci in the high-RDL group are restored to unmodified cytosines by unidentified pathways.
Replication-dependent dilution is the major contributor of paternal DNA demethylation
To investigate the contribution of DNA replication to the zygotic DNA demethylation, we analyzed the methylation status of zygotic DNA in the presence of the DNA replication inhibitor aphidicolin. Successful inhibition of DNA replication by aphidicolin treatment is supported by the lack of BrdU incorporation (Figure 3A). Similar to a previous report (Wossidlo et al., 2011), inhibition of DNA replication does not affect Tet3-mediated 5mC oxidation as 5hmC, 5fC and 5caC are robustly detected in paternal pronuclei (Figure 3B and Figure S3A and B), and it also does not significantly change the intensity of the total 5mC signal (Figure 3B). We then performed RRBS analysis after isolation of paternal and maternal pronuclei from aphidicolin-treated WT and CKO 13 hpf zygotes (Figure 1D). We found that, in the presence of aphidicolin, the mean methylation level of the paternal DNA is highly similar to that of sperm DNA regardless of Tet3 functionality (Figure 3C), which is consistent with unsupervised clustering of global methylation patterns (Figure 3D), as well as some representative genomic loci (Figure 3E). Furthermore, for tiles with methylation level ≥20% in sperm (MLSperm≥20%), the RDL values peak at zero in both aphidicolin-treated WT and CKO zygotes (Figure 3F), in contrast to the roughly normal distribution centered at 0.5 or 0.35 in the untreated WT or CKO zygotes, respectively (Figure 2C). Taken together, our study provides high-resolution genome-scale evidence supporting the idea that DNA replication is the major contributor of paternal DNA demethylation regardless of the presence of Tet3 proteins. It is important to note that RRBS detects the relative amount of methylation and thus is affected by the dilution that occurs as a result of replication, whereas immunostaining measures the absolute amount of DNA methylation present and is therefore less strongly affected.
Figure 3. DNA replication is essential for paternal DNA demethylation.

(A) Representative images of zygotes stained with anti-BrdU antibody. Zygotes were incubated in the media containing BrdU in the presence (Aphi) or absence (Mock) of aphidicolin during 7–8 hpf and fixed at 8 hpf. Dotted circles indicate the rim of zygote cytoplasm. Scale bar, 20 μm.
(B) Representative images of control and aphidicolin-treated zygotes stained with anti-5hmC (red) and anti-5mC (green) antibodies. Scale bar, 20 μm. The graphs at right indicate relative signal intensity of 5hmC or 5mC. The value of the maternal pronucleus in WT zygotes was set as 1. The numbers of zygotes quantified are indicated below the bars. Error bars, s.d.
(C) Mean DNA methylation levels of sperm and paternal DNA of both WT and CKO zygotes with or without aphidicolin treatment.
(D) Cluster analysis of global DNA methylation landscape (CpGs with ≥ 10x coverage) of sperm and paternal pronuclear samples. DNA methylation pattern in paternal pronuclei of aphidicolin-treated CKO zygotes is most similar to that in sperm.
(E) Representative genomic loci showing the distinctive roles of Tet3 and DNA replication in paternal DNA demethylation.
(F) Histogram showing the distribution of RDLSp→♂PN values of 100-bp tiles in WT and CKO paternal DNA in the presence of aphidicolin. Only tiles that are methylated in sperm (MLSp≥20%, n=58,101) were examined. RDL values less than −1 were set to −1.
See also Figure S3.
Tet3 knockout impairs DNA demethylation of both genic and repetitive sequences
We next investigated the Tet3 dependency of DNA demethylation for various genetic elements. To this end, we first plotted the mean DNA methylation levels of different genic regions (Figure 4A). Most of these genic regions exhibit loss of DNA methylation from sperm to pronucleus transition, which was partially impaired by Tet3 knockout similar to the global analysis (Figure 3C). The only exception is the 5′UTR, which is already extremely hypomethylated in the sperm genome. We then examined the methylation of various repeats and found that Tet3 knockout resulted in increases in DNA methylation of all repeats that are methylated in sperm (Figure 4A). Interestingly, the contribution of Tet3 to long terminal repeat (LTR) retrotransposons was less prominent when compared to other repeats (Figure 4A). Further analysis of different LTR families showed that Tet3 has very little effect on the methylation of endogenous retrovirus-1 (ERV1) family and intracisternal A-particles (IAPs) (Figure 4A). Taken together, we conclude that Tet3 generally contributes to DNA demethylation of both genic and repetitive regions in the paternal genome.
Figure 4. Tet3 knockout impairs DNA demethylation of both genic and repetitive sequences.

(A) Mean DNA methylation levels of sperm and paternal pronuclear samples (both WT and CKO zygotes with or without aphidicolin treatment) in various genic and repetitive sequences, and in subcategories of LTR repeats.
(B) Heatmap of 37,548 100-bp tiles that are methylated in sperm (MLSp≥20%) and demethylated after fertilization (RDLWT ≥ 0.3). These tiles can be divided into two groups. Tet3 KO affects demethylation of Group 1 tiles (RDLCKO/RDLWT≤0.6, n=20,745) but not Group 2 tiles (RDLCKO/RDLWT >0.6, n=16,803, including 1,082 tiles with RDLCKO/RDLWT >1.7).
(C) Box plot of methylation levels in sperm, paternal pronuclei of both WT and CKO zygotes for Group 1 and Group 2 tiles defined in (B).
See also Figure S4.
Tet3 is required for efficient demethylation of certain genomic loci
We next asked whether Tet3-mediated DNA demethylation is targeted to specific genomic loci. In the 37,548 tiles (24% of total 10x commonly covered tiles) that undergo dramatic DNA demethylation during sperm to pronucleus transition (MLsperm≥20% and RDLWT≥0.3), 55% tiles show relatively lower RDL values in CKO zygotes than in WT zygotes (RDLCKO/RDLWT≤0.6, n=20,745), suggesting that demethylation of these loci is partially dependent on Tet3 (Group 1); DNA demethylation of the rest 45% of the tiles (RDLCKO/RDLWT >0.6, n=16,803, including 1,082 tiles with RDLCKO/RDLWT >1.7) is independent of Tet3 (Group 2) (Figure 4B). It is worth noting that demethylation of Group 1 tiles is not completely abolished in the absence of Tet3 (Figure 4C) but is totally blocked by aphidicolin treatment (Figure 3C and 3F), indicating that Tet3 only facilitates DNA demethylation by coupling with replication. This implies that, in the absence of Tet3, paternal DNA methylation of Group 1 loci may has a high chance of being maintained during the first round of DNA replication and that Tet3-mediated oxidation may ensure passive demethylation of these loci. Despite identification of these Tet3-dependent demethylated loci, we were not able to identify obvious features that can distinguish Group 1 and Group 2 loci with regard to their GC content (Figure S4A), CpG density (Figure S4B), enrichment for any genomic elements (Figure S4C and D), or dynamic DNA methylation change during preimplantation development (Figure S4E). Further studies are required to reveal the specificity of Tet3 dependency.
Maternal depletion of Tet3 does not affect zygotic gene activation and preimplantation development
It is generally believed that the hypomethylation state of early embryos may functionally contribute to the dynamic change of gene expression during preimplantation development, including zygotic gene activation (ZGA) at 2-cell embryos (Schultz, 2002). Thus, we analyzed the potential effect of partial impairment of DNA demethylation caused by Tet3 KO on ZGA. RNA-Seq analysis (Table S2) of protein coding genes at 2-cell embryos revealed that transcriptomes in WT and CKO embryos are highly similar (R=0.96) (Figure 5A). With a 2-fold cutoff (FDR<5%), we only detected 33 upregulated and 59 downregulated genes, which have no clear GO term enrichment. In addition, the differentially expressed genes do not show enrichment for Group 1 tiles (Figure 5A), which are hypermethylated in the paternal DNA of CKO zygotes. Since several types of transposable elements start expression at the onset of ZGA (Peaston et al., 2004), we compared their expression in WT and CKO 2-cell embryos and found that they are generally not affected by Tet3 depletion (Figure 5B). Furthermore, RNA-Seq analysis at the blastocyst stage revealed that neither protein coding genes, nor repeat sequences exhibit significant alteration in CKO embryos compared to WT embryos (Figure 5C and D). This was supported by the normal development of CKO embryos up to the blastocyst-stage (Figure 5E). Thus, we conclude that Tet3-mediated DNA demethylation is neither important for global gene expression, nor for development of preimplantation embryos.
Figure 5. Tet3-mediated DNA demethylation does not affect zygotic gene activation and preimplantation development.

(A) Scatter plot comparing RNA-Seq results of protein coding genes of WT and CKO 2-cell embryos. Genes containing the Group 1 tiles described in Figure 4B in their promoter or gene body are highlighted in blue.
(B) Scatter plot comparing RNA-Seq results of various repetitive elements in WT and CKO 2-cell embryos.
(C) Scatter plot comparing RNA-Seq results of protein coding genes of WT and CKO blastocysts. Genes containing the Group 1 tiles described in Figure 4B in their promoter or gene body are highlighted in red.
(D) Scatter plot comparing RNA-Seq results of various repetitive elements in WT and CKO blastocysts.
(E) Percentages of fertilized 1-cell embryos developed to the indicated stages. The experiments were repeated three times, with each experiment using more than18 embryos. A total of 121 and 63 of WT and CKO embryos, respectively, were examined. Error bars, s.d.
See also Table S2.
Tet3 contributes to maternal DNA demethylation
The presence of Tet3 protein and 5hmC signal in maternal pronuclei (Figure 1A and B) prompted us to ask whether Tet3 may also contribute to maternal DNA demethylation. It is generally accepted that maternal DNA demethylation is solely mediated by replication-dependent dilution (Seisenberger et al., 2013; Wu and Zhang, 2014). Consistent with this notion, the global DNA methylation profile of the maternal pronuclei in aphidicolin-treated zygotes resembles that of oocytes (Figure 6A–C). In untreated zygotes, the percentage of highly methylated tiles detected in oocytes is dramatically decreased during oocyte to pronucleus transition regardless of the presence of Tet3 (Figure 6A and C). However, similar to observations in paternal pronuclei, there are more intermediately methylated 100-bp tiles in the maternal pronuclei of CKO zygotes (Figure 6C), suggesting a role of Tet3 in facilitating maternal DNA demethylation. We also examined the methylation levels at various genic and repetitive regions and found that the role of Tet3 in facilitating maternal DNA demethylation is widespread throughout the genome (Figure 6D), similar to that of paternal DNA. These data suggest that Tet3 also contributes to maternal DNA demethylation in zygotes, although to a much less extent when compared to that of paternal DNA.
Figure 6. Tet3 contributes to maternal DNA demethylation.

(A) Histograms of methylation values across 100-bp tiles in oocytes, WT and CKO maternal pronuclei with or without aphidicolin treatment. CpG sites that are at least 10x covered in all samples are represented.
(B) Mean DNA methylation levels of oocytes and maternal pronuclei of both WT and CKO zygotes with or without aphidicolin treatment.
(C) Distribution of DNA methylation levels in oocytes, WT, and CKO maternal pronuclei. 100-bp tiles of genomic DNAs are divided into four groups based on their methylation levels as low (0–25%, purple), medium-low (25–50%, blue), medium-high (50–75%, green), and high (75–100%, red) methylation.
(D) Mean DNA methylation levels of oocyte and maternal pronuclear samples (both WT and CKO zygotes with or without aphidicolin treatment) in various genetic elements.
DISCUSSION
In this study, we performed genome-scale DNA methylation analysis of manually isolated paternal and maternal pronuclei of WT and Tet3 CKO zygotes with or without the treatment of the DNA replication inhibitor aphidicolin. Comparative analysis allowed us to pinpoint the specific contribution of replication-dependent dilution and Tet3-mediated oxidation to both paternal and maternal genomes. Our data suggest that Tet3 partly contributes to the paternal DNA demethylation including both genic and repetitive sequences without apparent preference for any specific genome features (Figure 4 and S4). Tet3-mediated demethylation is greatly impaired when DNA replication is blocked, indicating that DNA replication is the predominant driving force for paternal DNA demethylation and that Tet3-dependent demethylation is highly associated with DNA replication (Figure 3). Our analysis also revealed a surprising contribution of Tet3 to maternal DNA demethylation (Figure 6) although the extent of Tet3 contribution to maternal demethylation is much less pronounced compared to that for paternal DNA (Figure 3C and 6B). Inhibition of DNA replication also prevents maternal DNA demethylation (Figure 6A–C). Collectively, our study reveals a combinatorial effect of Tet3 and DNA replication to the loss of both paternal and maternal DNA methylation in mouse zygotes.
DNA replication and Tet3-mediated DNA demethylation predominantly account for paternal DNA demethylation
Based on our results, and combined with previous studies, we envision that 5mC in sperm genome has at least five possible destinations following fertilization (Figure 7). First, certain 5mCs (#1 in Figure 7), including those in imprinting control regions and some families of repetitive sequences, are maintained during DNA replication (Guo et al., 2014; Smith et al., 2014; Smith et al., 2012; Wang et al., 2014). Such maintenance of DNA methylation seems to be achieved by a somatic form of DNA methyltransferase 1 that is localized in nuclei of preimplantation embryos (Hirasawa et al., 2008; Kurihara et al., 2008). Second, certain 5mCs (#2 in Figure 7), represented by Group 2 tiles in Figure 4B, are lost in a Tet3-independent but replication-dependent manner. This implicates that these 5mCs go through passive dilution without being oxidized, which has been believed to take place only in the maternal genome. Third (#3 in Figure 7), 5mCs are oxidized to 5hmC by Tet3 and then passively diluted. When not oxidized (i.e., in Tet3 CKO zygotes), these 5mCs have a higher chance to be maintained during DNA replication, which causes the apparent hypermethylation as represented in Group 1 in Figure 4B. Thus, Tet3 may ensure these methylation sites take a passive dilution route by means of oxidation. Fourth, 5mCs are converted to 5fC and 5caC (#4 in Figure 7) and then diluted through DNA replication (Inoue et al., 2011). Because demethylation of these Cs is Tet3-dependent, these 5mCs are also represented in Group 1 of Figure 4B. The amount of these 5mCs are, however, likely to be very small, given that RRBS analyses detected very minor ‘demethylation’ (Figure 3C) despite their definitive presence based on immunostaining (Figure S3). Lastly, a small portion of 5mCs could be demethylated in a Tet3- and DNA replication-independent manner (#5 in Figure 7). The existence of such bases is suggested by the observation of a significant number of tiles within high RDL (0.7–1.0) in the Tet3 CKO paternal genome (Figure 2C, 2E, 3F). This notion is consistent with a recent study using WGBS analysis that suggested the existence of DNA demethylation mechanism that is independent of passive dilution of 5mC or 5mC oxidative products (Wang et al., 2014).
Figure 7. Schematic model for paternal DNA demethylation.
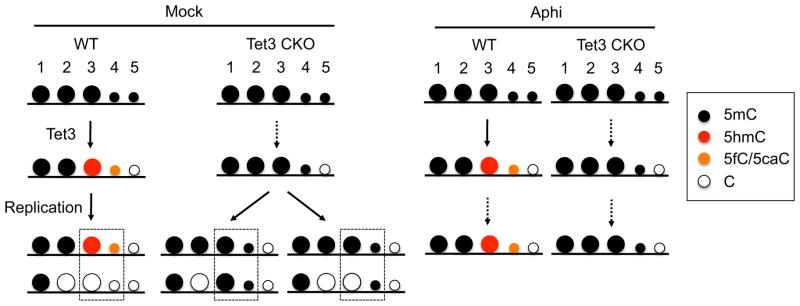
Detailed explanation is described in the discussion. Because the amounts of 5mCs taking the 4th or 5th paths are relatively small, these are indicated as smaller circles. Dotted arrows indicate blocked pathways. Dotted squires indicate Tet3-dependent demethylation loci as represented in the Group 1 of Figure 4B.
Despite the existence of replication- and Tet3-independent demethylation pathway, our RRBS analysis of carefully timed and hand-picked pronuclei do not support the previous conclusion that the majority of sperm methylation undergoes active demethylation independent of Tet3-mediated oxidation and DNA replication (Wang et al., 2014). One likely cause of this difference is that the 2-cell embryos assumed to be G1 phase in the previous study might have actually been in S phase, as these were collected at 30–32 h after hCG injection followed by natural mating. Because the cell cycle is not synchronized when embryos are collected from mice after natural mating and the duration of G1-phase at the 2-cell stage is brief (only 3 h) (Kang et al., 2014), careful timing of the end of the first mitotic cleavage is required for the collection of G1-phase 2-cell embryos (Kang et al., 2014). If S-phase 2-cell embryos contaminated the assumed G1-phase 2-cell embryos analyzed in the previous study, the level of demethylation would be overestimated due to the second round of passive dilution. Alternatively, since RRBS analysis covers a relative small portion of CpGs (5–10%), we cannot exclude the possibility that the results of RRBS analysis may not be representative of bulk genome and that some regions where RRBS does not cover have unusually high levels of Tet3/DNA replication-independent demethylation. Despite the lower coverage of RRBS compared to WGBS, genomic regions covered by RRBS are enriched in CpG-dense regions including promoters where DNA methylation is important for gene regulation. Thus, our RRBS analysis indicating that DNA replication and Tet3-dependent demethylation accounts for almost all DNA demethylation is highly valuable.
Tet3-mediated maternal DNA demethylation
Since initial observation of asymmetric DNA demethylation (Mayer et al., 2000; Oswald et al., 2000), it has been long thought that only the paternal genome, but not the maternal genome, is subject to active demethylation. Consistent with this notion, several recent studies have shown that only the paternal genome undergoes Tet3-mediated oxidation (Gu et al., 2011; Inoue et al., 2012; Inoue et al., 2011; Inoue and Zhang, 2011; Iqbal et al., 2011; Nakamura et al., 2012; Wossidlo et al., 2011). This conclusion was largely based on immunostaining analysis showing that Tet3 and 5hmC are predominantly localized in the paternal pronucleus of zygotes (Gu et al., 2011; Inoue et al., 2012; Iqbal et al., 2011) and that Stella/PGC7 prevents Tet3 from localizing to the maternal pronucleus (Nakamura et al., 2012; Wossidlo et al., 2011). In this study, we provide evidence demonstrating that not only the paternal genome, but also the maternal genome is subject to Tet3-mediated DNA demethylation (Figure 6). We show that both Tet3 and 5hmC are present in the WT maternal pronuclei, but not in the CKO zygotes (Figure 1A, B). Maternal DNA demethylation facilitated by Tet3 is distributed throughout the genome without detectable enrichment in specific genomic loci (Figure 6D). This result is consistent with a recent study that reports the detection of 5hmC and 5fC in the maternal genome of 2-cell embryos (Wang et al., 2014). Although both paternal and maternal genomes go through Tet3-mediated demethylation, the extent of Tet3-mediated maternal demethylation is much less pronounced when compared to that of the paternal genome (Figures. 3C, 6B), consistent with the immunostaining analysis (Figures. 1A, B). This can be explained by the combined effect that Stella/PGC7 not only prevents the localization of Tet3 to the maternal pronuclei but also suppresses the activity of Tet3 (Bian and Yu, 2013; Nakamura et al., 2006; Nakamura et al., 2012). Future study may reveal how, where, and to what extent that Stella may protect the maternal genome from Tet3-mediated active DNA demethylation.
Tet3-mediated demethylation and zygotic gene activation
Dynamic change of transcription accompanies preimplantation development (Hamatani et al., 2004; Peaston et al., 2004). For example, the first zygotic transcription starts at the middle of 1-cell stage and the major wave of zygotic gene activation begins at the 2-cell stage (Schultz, 2002). Given that promoter DNA methylation plays an important role in the repression of gene expression, it is believed that global demethylation during preimplantation development contributes to the dynamic transcriptional change. Recent studies indicated that promoter methylation of coding genes and certain LTR subfamily is indeed negatively correlated with their transcription (Guo et al., 2014; Smith et al., 2014). However, it is unclear whether Tet3-mediated demethylation plays a role in transcriptional regulation. Although our previous studies using siRNA-mediated Tet3 knockdown revealed no significant effect on the activation of representative transposable elements in 1- and 2-cell embryos (Inoue et al., 2012), analysis of global transcriptional effect of Tet3 during preimplantation is still lacking. In this study, we analyzed the transcriptome of Tet3 CKO 2-cell and blastocyst embryos and found that, in contrast to the presumed effect on zygotic gene activation, both global gene expression profiles and the expression of genes whose demethylation is impaired by Tet3-depletion are not significantly altered in Tet3 CKO embryos (Figure 5). These results suggest that Tet3-mediated demethylation is dispensable for transcriptional regulation during preimplantation development. Given that Tet3 CKO embryos show sub-lethality in post-implantation development (Gu et al., 2011), the hyper-methylation caused by loss of Tet3 function may have a long-term effect during later developmental stages. Future studies should reveal the biological function of Tet3-mediated DNA demethylation.
EXPERIMENTAL PROCEDURES
Collection of sperm, oocytes, and pronuclei of zygotes
All animal studies were performed in accordance with guidelines of the Institutional Animal Care and Use Committee at Harvard Medical School. Sperm genomic DNA was extracted as described previously (Weyrich, 2012). MII oocytes were collected from 7–10 week-old C57BL/6J females superovulated by injecting 7.5 I.U of PSMG (Millipore) and hCG (Millipore). They were briefly incubated in M2 media containing 0.3 mg/ml hyaluronidase (Millipore) for removing cumulus cells.
When collecting pronuclei of zygotes, 7–10 week-old CKO (Zp3-Cre, f/-) or WT (f/f) females were superovulated. MII oocytes were collected from the mice, transferred into HTF medium supplemented with 10 mg/ml bovine serum albumin (BSA; Sigma-Aldrich), and inseminated with activated spermatozoa obtained from the caudal epididymides of adult C57BL/6J male mice. Spermatozoa capacitation was attained by 1 h incubation in HTF medium. Zygotes were cultured in a humidified atmosphere of 5% CO2/95% air at 37.8°C. Five hours after fertilization, zygotes were cultured in KSOM (Millipore). Twelve hours post-fertilization (hpf), zygotes were transferred into M2 media containinig 5 μM cytochalasin B. Zona pellucidae were cut by a Piezo impact-driven micromanipulator (Prime Tech Ltd., Ibaraki, Japan) and pronuclei of the zygotes were isolated. The parental pronuclei were distinguished by (1) the distance from the second polar body and (2) the pronuclear size. The isolated paternal and maternal pronuclei were frozen at 13 hpf in −80°C.
Aphidicolin treatment and BrdU incorporation
At 5 hpf, zygotes were transferred into KSOM containing 3 μg/ml aphidicolin (Sigma-Aldrich). When labeling ongoing replication by BrdU incorporation, zygotes were incubated with KSOM containing 100 μM BrdU for 1 h. Following the incubation, zygotes were briefly washed with M2 media and then fixed.
RRBS library preparation
To prepare RRBS libraries from limited number of pronuclei and MII oocytes, we adopted and improved a single-tube method that has recently been successfully used for DNA methylation analysis of single mouse cells and human early embryos (Guo et al., 2013; Smallwood et al., 2011). Briefly, 5 μl of lysis buffer (20 mM Tris-EDTA [pH 8.0], 20 mM KCl, and 0.3% Triton X-100, 1 mg/ml QIAGEN Protease) was directly added to the frozen samples (in < 1μl PBS) followed by 3 hours of incubation at 50 °C and 30 minutes of inactivation at 75 °C. Then to the same tube, 13 μl of MspI digestion master mix containing 10 units of MspI (Thermo Scientific) and 1.8 μl 10x Buffer Tango (Thermo Scientific) was added followed by 3 hours of incubation at 37 °C and 20 minutes of inactivation at 80 °C. The digested DNA was then filled-in and tailed with an extra A to the 3′ end by adding 2 μl of end-preparation master mix containing 5 units of Klenow Fragment (exo-; Thermo Scientific), 1.8 μl of 10x Buffer Tango, 0.4 mM dATP, and 0.04 mM each of dGTP and dCTP (Thermo Scientific), followed by 40 minutes of incubation at 37 °C and 15 minutes of inactivation at 75 °C. Adaptor ligation was then performed by adding 5 μl of ligation master mix, which contains 30 Weiss Units of T4 DNA Ligase (HC, Thermo Scientific), 0.5 μl of 10x Buffer Tango, 5mM ATP (Thermo Scientific), and 150 nM of methylated custom adaptor (Forward: 5′-ACACTCTTTCCCTACACGACGCTCTTCCGATC*T-3′, Reverse: 5′-/5Phos/GATCGGAAGAGCACACGTCTGAACTCCAGTC-3′, where * indicates phosphorothioate bond). After three incubation steps (16 °C for 30 min, 4 °C overnight, and 65 °C for 20 min), the reaction mix was subjected to EpiTect Fast Bisulfite Conversion Kit (QIAGEN) following manufacturer’s low-concentration protocol. The bisulfite converted DNA was immediately subjected to PCR amplification using KAPA HiFi HotStart Uracil+ ReadyMix (Kapa Biosystems) and NEBNext Primers for Illumina (New England Biolabs). Typically, 21 PCR cycles were required when starting with ~20 pronuclei. After amplification, final libraries were obtained by size selection of 150–500 bp DNA fragments using SPRIselect beads (Beckman Coulter). RRBS libraries were mix with 20% PhiX Control (Illumina) or other balanced libraries, and single-end 50 bp sequencing was performed on a HiSeq 2500 sequencer (Illumina).
Calculating methylation levels and relative demethylation levels (RDLs)
RRBS sequencing reads were mapped to the mouse genome (mm9) using Bismark v0.10.1 (Babraham Bioinformatics) after adapter trimming by Trim Galore (Babraham Bioinformatics). The methylation level of each covered cytosine in CpG context was calculated as the number of reported C divided by the total number of reported C and T. Only CpG sites that were covered by at least ten reads were considered for the subsequent analyses. When comparing different samples, only CpG sites that were commonly 10x covered in all samples were used. For 100-bp tile analysis, we binned the genome into consecutive 100-bp tiles, and only keep those tiles covering at least two 10x covered CpG sites. The average methylation level of all CpG sites within each tile was considered as the methylation level of that 100-bp tile. The RDLSperm→♂PN was calculated as [(MethylationSperm–Methylation♂ pronucleus)/MethylationSperm] such that RDL values of 0 and 1 represent the maintenance of DNA methylation and complete DNA demethylation (i.e., active DNA demethylation), respectively, and a value around 0.5 indicates replication-dependent dilution of methylation. Only those tiles with ≥20% DNA methylation in sperm were considered for RDL analysis. Statistical analyses were implemented with R (http://www.r-project.org/).
Whole mount immunostaining
Embryos were fixed in 3.7% paraformaldehyde (PFA) in PBS containing 0.2% Triton X-100 for 20 min and then washed with PBS containing 10 mg/mL BSA (PBS/BSA). After blocking in PBS/BSA for overnight, anti-Tet3 rabbit polyclonal (1/4000, a gift from GL Xu) and anti-H3K4me3 mouse monoclonal antibodies (1/500, Millipore #05-1339) were incubated for 2 h at room temperature. When zygotes were stained with anti-5mC, -5hmC, -5fC, and -5caC antibodies, they were fixed with 3.7%PFA for 20 min, permeabilized with 0.5% Triton X-100 for 15 min, and treated with 4N HCl for 15 min. When stained with anti-BrdU antibody, 4N HCl was treat for 30 min. After the acid treatment, the zygotes were neutralized with 100 mM Tris-HCl (pH8.0) for 15 min and then incubated with PBS/BSA overnight. Anti-5mC (1/100, Eurogentec #BI-MECY-0500) and anti-5hmC (1/500, Active Motif), anti-5fC [1/2000: (Inoue et al., 2011)], or anti-5caC [1/2000: (Inoue et al., 2011)] antibodies or anti-BrdU (1/200: Roche Diagnostic) antibody diluted in PBS/BSA were treated for 1 h at room temperature. After washing with PBS/BSA for 1 h, they were incubated with a 1:250 dilution of fluorescein isothiocyanate-conjugated anti-mouse IgG (Jackson Immuno-Research, West Grove, PA) and Alexa Flour 546 donkey anti-rabbit IgG (Life technologies) for 1 h. The zygotes were then mounted on a glass slide in Vectashield anti-bleaching solution with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA). Fluorescence was detected under a laser-scanning confocal microscope with a spinning disk (CSU-10, Yokogawa) and an EM-CCD camera (ImagEM, Hamamatsu). All images were acquired and analyzed using Axiovision software (Carl Zeiss). The fluorescent signal intensity was quantified with Axiovision. The quantification data were analyzed by two-tailed Student’s t-test. A value of P<0.01 was considered statistically significant.
Supplementary Material
Figure S1. Targeted disruption of Tet3 gene
(A) Strategy for targeted disruption of the Tet3 gene. Black boxes indicate the coding exons of Tet3. The asterisk above exon 9 indicates the catalytic domain of Tet3 that has the iron-binding motif essential for its enzymatic activity. Primers used for mouse genotyping are indicated by red arrows.
(B) Representative images of genotyping of mouse tail DNA. The primer set of F3 and R1 was used to detect WT (130 bp) and 2flox (290 bp) alleles. The primer set of F2 and R1 was used to detect 1flox allele (218 bp).
(C and D) Confirmation of exon 7–9 deletion in CKO oocytes. After cDNA preparation by reverse transcription of total RNA from WT or CKO oocytes, PCR amplification using primers amplifying exons 6 and 10 were performed (C). The PCR products were then read by Sanger sequencing, which confirmed that exons 7–9 are indeed deleted in CKO oocytes (D). The amino acids translated from the mRNAs are shown. Note that Tet3 protein is truncated in CKO oocytes.
(E) Representative images of zygotes stained with anti-5hmC (red) and anti-5mC (green) antibodies at 8 hours after fertilization. Scale bar, 20 μm. The graph at right represents relative signal intensity of 5hmC. The value of the maternal pronucleus in WT zygotes was set as 1.0. A total of 23 and 18 of WT and CKO zygotes, respectively, were examined. *P<0.01 by Student’s t-test. Error bars, s.d.
Related to Figure 1
Figure S2. 5fC and 5caC partially contribute to the observed active paternal DNA demethylation.
(A and B) Histograms of RDLSp→♂PN values in WT zygotes using DNA methylation information separately extracted from the MspI site at the 5′-end of RRBS reads (A) or from the rest of the reads (B). Only 100-bp tiles that are methylated in sperm (MLSperm≥20%) were analyzed. Since the 5′ MspI site of an RRBS read is depleted of 5fC/5caC, the decreased percentage of fully demethylated tiles in (A) compared with (B) suggests the contribution of 5fC/5caC in the observed active DNA demethylation. RDLSp→♂PN is defined as [(ML Sp – ML ♂PN)/ML Sp]. RDL values less than −1 were set to −1.
(C) Distribution of RDLSp→♂PN values in (A) and (B). The RDL values are divided into four groups marked by red (0.7–1), green (0.3–0.7), blue (−0.3–0.3), and purple (<−0.3).
See also Figure 2
Figure S3. Inhibition of DNA replication does not affect Tet3-mediated 5mC oxidation.
(A and B) Representative images of zygotes stained with anti-5fC (A) or -5caC (B) and anti-5mC antibodies are shown. Zygotes were incubated in the media with or without Aphidicolin. Scale bar, 20 μm. The graphs on the right indicate relative signal intensity of 5fC or 5caC. The value of the maternal pronucleus in Mock zygotes was set as 1.0. For 5fC staining, both Mock and Aphi groups used a total of 19 zygotes. For 5caC staining, Mock and Aphi groups used a total of 19 and 16 zygotes, respectively. Error bars, s.d.
Related to Figure 3
Figure S4. Group1 and Group2 tiles share similar genomic features and dynamic DNA methylation changes during preimplantation development.
(A and B) Boxplot of GC% (A) and the number of CpG dinucleotides (B) in Group1 and Group2 tiles. No significant difference was found. Red line represents the median, boxes and whiskers represent for the 25th and 75th, and 2.5th and 97.5th percentiles, respectively.
(C) Pie charts showing similar distribution of Group1 and Group2 tiles across the whole genome.
(D) Percentage of Group1 and Group 2 tiles overlapping with various types of repeats. In parentheses are the number of tiles overlapping with the indicated repeat and its percentage in all repeat-overlapping tiles. “Other” stands for all other repeats in the RepeatMasker annotation.
(E) Dynamic methylation levels of Group 1 and Group 2 tiles during embryonic development.
Related to Figure 4
Table S1. Summary of RRBS libraries, Related to Figure 1,3 and 6
Table S2. Summary of RNA-Seq libraries, Related to Figure 5
Acknowledgments
We thank Drs. Hao Wu and Luis M. Tuesta for critical reading of the manuscript, Dr. Guoliang Xu for the Tet3 antibody. Y.Z. is an investigator of the Howard Hughes Medical Institute. The raw data of RRBS and RNA-Seq experiments have been deposited in Gene Expression Omnibus (GEO) under the accession number GSE61331.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bian C, Yu X. PGC7 suppresses TET3 for protecting DNA methylation. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, et al. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature. 2011;477:606–610. doi: 10.1038/nature10443. [DOI] [PubMed] [Google Scholar]
- Guo H, Zhu P, Wu X, Li X, Wen L, Tang F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome research. 2013;23:2126–2135. doi: 10.1101/gr.161679.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Zhu P, Yan L, Li R, Hu B, Lian Y, Yan J, Ren X, Lin S, Li J, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511:606–610. doi: 10.1038/nature13544. [DOI] [PubMed] [Google Scholar]
- Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatani T, Carter M, Sharov A, Ko M. Dynamics of global gene expression changes during mouse preimplantation development. Dev Cell. 2004;6:117–131. doi: 10.1016/s1534-5807(03)00373-3. [DOI] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, Sasaki H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008;22:1607–1616. doi: 10.1101/gad.1667008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Matoba S, Zhang Y. Transcriptional activation of transposable elements in mouse zygotes is independent of Tet3-mediated 5-methylcytosine oxidation. Cell Res. 2012 doi: 10.1038/cr.2012.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21:1670–1676. doi: 10.1038/cr.2011.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334:194. doi: 10.1126/science.1212483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin S, Pfeifer G, Szabó P. Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc Natl Acad Sci U S A. 2011;108:3642–3647. doi: 10.1073/pnas.1014033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, D’Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature genetics. 2003;33(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Kang E, Wu G, Ma H, Li Y, Tippner-Hedges R, Tachibana M, Sparman M, Wolf D, Schöler H, Mitalipov S. Nuclear reprogramming by interphase cytoplasm of two-cell mouse embryos. Nature. 2014 doi: 10.1038/nature13134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502:472–479. doi: 10.1038/nature12750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara Y, Kawamura Y, Uchijima Y, Amamo T, Kobayashi H, Asano T, Kurihara H. Maintenance of genomic methylation patterns during preimplantation development requires the somatic form of DNA methyltransferase 1. Developmental biology. 2008;313:335–346. doi: 10.1016/j.ydbio.2007.10.033. [DOI] [PubMed] [Google Scholar]
- Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. The Journal of biological chemistry. 2011;286:35334–35338. doi: 10.1074/jbc.C111.284620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, Haaf T. Demethylation of the zygotic paternal genome. Nature. 2000;403:501–502. doi: 10.1038/35000656. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Arai Y, Umehara H, Masuhara M, Kimura T, Taniguchi H, Sekimoto T, Ikawa M, Yoneda Y, Okabe M, et al. PGC7/Stella protects against DNA demethylation in early embryogenesis. Nat Cell Biol. 2006;9:64–71. doi: 10.1038/ncb1519. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Liu Y, Nakashima H, Umehara H, Inoue K, Matoba S, Tachibana M, Ogura A, Shinkai Y, Nakano T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature. 2012;486:415–419. doi: 10.1038/nature11093. [DOI] [PubMed] [Google Scholar]
- Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, Dean W, Reik W, Walter J. Active demethylation of the paternal genome in the mouse zygote. Current biology: CB. 2000;10:475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- Peaston A, Evsikov A, Graber J, de Vries W, Holbrook A, Solter D, Knowles B. Retrotransposons regulate host genes in mouse oocytes and preimplantation embryos. Dev Cell. 2004;7:597–606. doi: 10.1016/j.devcel.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nature reviews Genetics. 2014 doi: 10.1038/nrg3772. [DOI] [PubMed] [Google Scholar]
- Santos F, Peat J, Burgess H, Rada C, Reik W, Dean W. Active demethylation in mouse zygotes involves cytosine deamination and base excision repair. Epigenetics & chromatin. 2013;6:39. doi: 10.1186/1756-8935-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nature reviews Genetics. 2008;9:129–140. doi: 10.1038/nrg2295. [DOI] [PubMed] [Google Scholar]
- Schultz R. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum Reprod Update. 2002;8:323–331. doi: 10.1093/humupd/8.4.323. [DOI] [PubMed] [Google Scholar]
- Seisenberger S, Peat JR, Reik W. Conceptual links between DNA methylation reprogramming in the early embryo and primordial germ cells. Curr Opin Cell Biol. 2013;25:281–288. doi: 10.1016/j.ceb.2013.02.013. [DOI] [PubMed] [Google Scholar]
- Smallwood SA, Tomizawa S, Krueger F, Ruf N, Carli N, Segonds-Pichon A, Sato S, Hata K, Andrews SR, Kelsey G. Dynamic CpG island methylation landscape in oocytes and preimplantation embryos. Nature genetics. 2011;43:811–814. doi: 10.1038/ng.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z, Chan M, Humm K, Karnik R, Mekhoubad S, Regev A, Eggan K, Meissner A. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511:611–615. doi: 10.1038/nature13581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Z, Chan M, Mikkelsen T, Gu H, Gnirke A, Regev A, Meissner A. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012 doi: 10.1038/nature10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zhang J, Duan J, Gao X, Zhu W, Lu X, Yang L, Zhang J, Li G, Ci W, et al. Programming and inheritance of parental DNA methylomes in mammals. Cell. 2014;157:979–991. doi: 10.1016/j.cell.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich A. Preparation of genomic DNA from mammalian sperm. In: Ausubel Frederick M, et al., editors. Current protocols in molecular biology. Unit 2 13. Chapter 2. 2012. pp. 11–13. [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, Walter J. 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nature communications. 2011;2:241. doi: 10.1038/ncomms1240. [DOI] [PubMed] [Google Scholar]
- Wu H, Zhang Y. Reversing DNA methylation: mechanisms, genomics, and biological functions. Cell. 2014;156:45–68. doi: 10.1016/j.cell.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Targeted disruption of Tet3 gene
(A) Strategy for targeted disruption of the Tet3 gene. Black boxes indicate the coding exons of Tet3. The asterisk above exon 9 indicates the catalytic domain of Tet3 that has the iron-binding motif essential for its enzymatic activity. Primers used for mouse genotyping are indicated by red arrows.
(B) Representative images of genotyping of mouse tail DNA. The primer set of F3 and R1 was used to detect WT (130 bp) and 2flox (290 bp) alleles. The primer set of F2 and R1 was used to detect 1flox allele (218 bp).
(C and D) Confirmation of exon 7–9 deletion in CKO oocytes. After cDNA preparation by reverse transcription of total RNA from WT or CKO oocytes, PCR amplification using primers amplifying exons 6 and 10 were performed (C). The PCR products were then read by Sanger sequencing, which confirmed that exons 7–9 are indeed deleted in CKO oocytes (D). The amino acids translated from the mRNAs are shown. Note that Tet3 protein is truncated in CKO oocytes.
(E) Representative images of zygotes stained with anti-5hmC (red) and anti-5mC (green) antibodies at 8 hours after fertilization. Scale bar, 20 μm. The graph at right represents relative signal intensity of 5hmC. The value of the maternal pronucleus in WT zygotes was set as 1.0. A total of 23 and 18 of WT and CKO zygotes, respectively, were examined. *P<0.01 by Student’s t-test. Error bars, s.d.
Related to Figure 1
Figure S2. 5fC and 5caC partially contribute to the observed active paternal DNA demethylation.
(A and B) Histograms of RDLSp→♂PN values in WT zygotes using DNA methylation information separately extracted from the MspI site at the 5′-end of RRBS reads (A) or from the rest of the reads (B). Only 100-bp tiles that are methylated in sperm (MLSperm≥20%) were analyzed. Since the 5′ MspI site of an RRBS read is depleted of 5fC/5caC, the decreased percentage of fully demethylated tiles in (A) compared with (B) suggests the contribution of 5fC/5caC in the observed active DNA demethylation. RDLSp→♂PN is defined as [(ML Sp – ML ♂PN)/ML Sp]. RDL values less than −1 were set to −1.
(C) Distribution of RDLSp→♂PN values in (A) and (B). The RDL values are divided into four groups marked by red (0.7–1), green (0.3–0.7), blue (−0.3–0.3), and purple (<−0.3).
See also Figure 2
Figure S3. Inhibition of DNA replication does not affect Tet3-mediated 5mC oxidation.
(A and B) Representative images of zygotes stained with anti-5fC (A) or -5caC (B) and anti-5mC antibodies are shown. Zygotes were incubated in the media with or without Aphidicolin. Scale bar, 20 μm. The graphs on the right indicate relative signal intensity of 5fC or 5caC. The value of the maternal pronucleus in Mock zygotes was set as 1.0. For 5fC staining, both Mock and Aphi groups used a total of 19 zygotes. For 5caC staining, Mock and Aphi groups used a total of 19 and 16 zygotes, respectively. Error bars, s.d.
Related to Figure 3
Figure S4. Group1 and Group2 tiles share similar genomic features and dynamic DNA methylation changes during preimplantation development.
(A and B) Boxplot of GC% (A) and the number of CpG dinucleotides (B) in Group1 and Group2 tiles. No significant difference was found. Red line represents the median, boxes and whiskers represent for the 25th and 75th, and 2.5th and 97.5th percentiles, respectively.
(C) Pie charts showing similar distribution of Group1 and Group2 tiles across the whole genome.
(D) Percentage of Group1 and Group 2 tiles overlapping with various types of repeats. In parentheses are the number of tiles overlapping with the indicated repeat and its percentage in all repeat-overlapping tiles. “Other” stands for all other repeats in the RepeatMasker annotation.
(E) Dynamic methylation levels of Group 1 and Group 2 tiles during embryonic development.
Related to Figure 4
Table S1. Summary of RRBS libraries, Related to Figure 1,3 and 6
Table S2. Summary of RNA-Seq libraries, Related to Figure 5