

Abstract
Multiple myeloma (MM) remains incurable with current therapy, indicating the need for continued development of novel therapeutic agents. We evaluated the activity of a novel phenylbutyrate-derived histone deacetylase inhibitor, AR-42, in primary human myeloma cells and cell lines. AR-42 was cytotoxic to MM cells at a mean LC50 of 0.18 ± 0.06 μmol/l at 48 hr and induced apoptosis with cleavage of caspases 8, 9 and 3, with cell death largely prevented by caspase inhibition. AR-42 downregulated the expression of gp130 and inhibited activation of STAT3, with minimal effects on the PI3K/Akt and MAPK pathways, indicating a predominant effect on the gp130/STAT-3 pathway. AR-42 also inhibited interleukin (IL)-6-induced STAT3 activation, which could not be overcome by exogenous IL-6. AR-42 also downregulated the expression of STAT3-regulated targets, including Bcl-xL and cyclin D1. Overexpression of Bcl-xL by a lentivirus construct partly protected against cell death induced by AR-42. The cyclin dependent kinase inhibitors, p16 and p21, were also significantly induced by AR-42, which together with a decrease in cyclin D1, resulted in G1 and G2 cell cycle arrest. In conclusion, AR-42 has potent cytotoxicity against MM cells mainly through gp130/STAT-3 pathway. The results provide rationale for clinical investigation of AR-42 in MM.
Keywords: multiple myeloma, apoptosis, cell cycle
Multiple myeloma (MM) is a clonal disorder of terminally differentiated B cells characterized by accumulation of slowly proliferating plasma cells with an incidence of 3–4 per 100,000 in the United States.1 While advances in treatment, including the use of high-dose chemotherapy and novel drugs such as thalidomide, lenalidomide and bortezomib have improved patient outcomes,1 relapses invariably occur, indicating a need for continued investigation of novel agents.
Intracellular signaling through the JAK/STAT, phosphatidylinositol 3-kinase (PI3K)/Akt pathway, and Ras/Raf/MEK/extracellular signal-regulated kinase pathways contributes to the survival, growth, proliferation and chemoresistance of MM cells.2,3 Constitutive activation of STAT3 has been detected in MM as well as a number of other cancer types.4 Stat3 signaling participates in oncogenesis through the upregulation of genes encoding apoptosis inhibitors (e.g., Bcl-xL, Mcl-1 and survivin) and cell cycle regulators (e.g., cyclin D1 and c-Myc).5
Histone deacetylases (HDAC) are enzymes that determine the acetylation status of histones, affecting chromatin structure and gene expression, and have emerged as a potential therapeutic target. Initially, inhibitors of HDAC (HDACi) were developed as agents that affect epigenetic processes by inducing histone hyperacetylation, leading to chromatin remodeling and reactivated expression of transcriptionally repressed genes.6 However, in addition to histone acetylation dependent-modulation of transcription, HDACi may also exert their anticancer activity through action on nonhistone substrates with pivotal roles in transformed cells.7,8 It is now known that HDACi can modulate a wide variety of cellular functions, including transcriptional reactivation of dormant tumor suppressor genes as well as modulating the expression of genes and proteins critical to cell proliferation, cell cycle progression, apoptosis, cytoskeleton modifications and angiogenesis.6,7
The investigation of HDACi on MM cells has been limited to studies using sodium butyrate and trichostatin A,9 valproic acid,10 LBH589,11 NVP-LAQ82412 and vorinostat.13–15 However, it is likely that significant differences exist between different HDACi with respect to potency and cellular activity AR-42 (formerly known as (S)-HDAC-42) is a novel orally bioavailable, phenylbutyrate-based HDAC inhibitor with a low-nanomolar IC50 for HDAC inhibition and is currently planned for clinical evaluation as a therapeutic agent in cancer (Arno Therapeutics, Parsippany, NJ). Significant antitumor activity, with higher potency compared to vorinostat, has been reported with AR-42 against prostate cancer cells.8,16 In PC-3 cells, AR-42 decreased the protein levels of phosphorylated (p)-Akt, Bcl-xL and survivin.8 In this study, we evaluate the activity of AR-42 against MM cells and investigate its potential mechanisms of action in this disease. AR-42 suppressed gp130 expression, and both constitutive and inducible STAT3 activation. This correlated with downregulation of STAT3 downstream cell survival and proliferation factors, Bcl-xL and cyclin D1, leading to induction of apoptosis and G1 and G2 cell cycle arrest in MM cells.
Material and Methods
Myeloma cells, culture conditions and reagents
The MM cell lines U266, H929, RPMI 8226, ARH-77 and IM-9 cell lines were purchased from American Type Culture Collection (Manassas, VA). Cell lines were cultured in RPMI 1640 media (Gibco, Invitrogen Company, Grand Island, NY) and supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Invitrogen Company, Grand Island, NY), 100 units/ml penicillin, 10 μg/ml streptomycin and 2 mM L-glutamine (Gibco). Primary MM cells were purified from bone marrow aspirates obtained after informed consent from patients at the time of diagnostic aspiration. Approval was obtained from the Institutional Review Board of Indiana University. CD138+ cells from bone marrow aspirates were separated using an LS+ column and a magnetic separator according to the manufacturer’s instructions (Miltenyi Biotech, Auburn, CA) with resulting purity of >90% in all cases. Cell viability as assessed by trypan blue exclusion was consistently >95%. CD138+ cells were cultured in RPMI 1640 containing 10% FBS under the same condition as cell lines. The caspase inhibitor, Q-VD-OPH, was purchased from Calbiochem, EMD Biosciences (La Jolla, CA). (S)-HDAC-42 (AR-42) was synthesized in Dr. Ching-Shih Chen’s laboratory at the Ohio State University, with purity exceeding 99% as shown by nuclear magnetic resonance spectroscopy. The drug was diluted to an initial stock solution of 100 mmol/l in dimethyl sulfoxide (DMSO), and aliquots were made of the stocks and stored at −80°C, avoiding multiple freeze–thaw cycles.
Cell viability assay
Cell viability was analyzed by the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI). Myeloma cells were plated in 96-well flat-bottomed plates in a 100 μl total volume at a density of 2 × 104 cells per well. Triplicate wells were treated with 10% FBS-supplemented RPMI 1640 media containing 0, 0.1, 0.25, 0.5, 0.75, 1.0, 2.5 and 5.0 μmol/l AR-42, or vehicle. The plates were incubated at 37°C in 5% CO2 for 24 or 48 hr. During the last 4 hr of the cell culture, the combined 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium inner salt/electron coupling reagent phenazine methosulfate (MTS/PMS) solution was added into each well of the 96-well assay plate containing 100 μl of cells in culture medium according to the manufacturer’s instructions. Then the cells were incubated at 37°C for 4 hr and absorbance was measured at 490 nm in a Victor 3 plate reader (PerkinElmer Life and Analytical Sciences, Shelton, CT). Cell viability under drug treatment is reported as a percentage of control.
Apoptosis assay
Following incubation with drug, cells were stained with annexin V–fluorescein isothiocyanate (FITC) and propidium iodide (PI) according to the manufacturer’s directions (BD Pharmingen, San Diego, CA) and analyzed by flow cytometry using a Beckman-Coulter FC500 cytometer (Beckman-Coulter, Miami, FL). Fluorophores were excited at 488 nm, and fluorescence was measured using channel FL1 for annexin V-FITC, channel FL3 for PI. Data were analyzed with the CXP software package (Beckman-Coulter). At least 10,000 cells were counted for each treated sample. In experiments assessing caspase-dependent apoptosis, 20 μmol/l Q-VD-OPH was added 60 min before the addition of AR-42, and flow cytometric analysis performed. In each case, at least three independent experiments were preformed and the reported data represent the mean of the replicates.
Western blot analysis
Cells were treated with AR-42 for the indicated hours above, washed with ice-cold PBS and resuspended in lysis buffer containing the phosphatase inhibitors sodium orthovanadate (1 mM) and microcystin LR (1 μmol/l) (both from Sigma, St. Louis, MO). Cell lysates were collected after centrifugation at 13,000 rpm for 10 min. Equivalent amounts of proteins (50 μg) from each lysate were resolved in 4–20% sodium dodecyl sulfate–polyacrilamide gel electrophoresis (SDS-PAGE). Protein was transferred to 0.2 μm nitrocellulose membranes (Schleicher & Schuell, Keene, NH) and blots were probed with primary antibody specific for the following proteins as appropriate at the indicated dilutions: p-Akt-Ser473 (1:200), Akt (1:1,000), caspases 3 (1:1,000), 8 (1:500) and 9 (1:500), poly(ADP-ribose)polymerase (PARP) (1:1,000), XIAP (1:1,000), Bax (1:500), p21 (1:500), p-STAT3 (Tyr705) (1:200), STAT3 (1:200), BID (1:400); BIM (1:500) (Cell Signaling Technology, Beverly, MA), p16 (1:500), Bcl-2 (1:1,000), Bcl-xL (1;500); gp130 (1:200), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:100,000), α-tubulin (1:2,000) (Santa Cruz Biotechnology, Santa Cruz, CA), β-actin (1:2,000) (Novus Biologicals, Littleton, CO), acetyl-Histone H3 (Lys9) (1:1,000) (Millipore Corporation, Billerica, MA) and FLIP (1:1,000) (AXXORA, LLC, San Diego, CA). Following incubation with antibody, the proteins were detected with chemiluminescent substrate (SuperSignal, Pierce, Rockford, IL). Protein bands were quantified by integration of the chemiluminescence signals on Quantity One (Bio-Rad Laboratories, Hercules, CA).
Overexpression of Bcl-xL in MM cell lines
The pCL6IEGwo vector control or a pCL6IEGwo-Bcl-xL lentiviral construct were transduced into U266 cells to generate pCL6IEGwo control and pCL6IEGwo-Bcl-xL cells. The pCL1IEGwo-Bcl-xL construct was made by cloning the full length Bcl-xL cDNA, generated by PCR, into the pCL6IEGwo vector. Western blot analysis confirmed that Bcl-xL was over-expressed in pCL6IEGwo-Bcl-xL cells.
Overexpression of c-FLIP (L) in H929
The pCL1IEG vector control or a pCL1IEG-cFLIP lentiviral construct were transduced into H929 cells to generate H929-pCL1IEG and H929-cFLIP cells. The pCL1IEG-cFLIP construct was made by cloning the full length cFLIP cDNA, generated by PCR, into the pCL1IEG vector. Western blot analysis confirmed that c-FLIP(L) was overexpressed in H929-c-FLIP cells.
Results
AR-42 is a potent antimyeloma agent and induces apoptosis in MM cells
The in vitro activity of AR-42 against MM cells was evaluated after 24–96 hr of exposure to drug. Cells were grown in the absence or the presence of different concentrations (0.1, 0.25, 0.5, 0.75, 1.0, 1.5, 2.5 and 5 μmol/l) of AR-42, and cytotoxicity was measured by the MTS assay. AR-42 effectively induced cell death in all cell lines tested (Fig. 1a). The LC50 of AR-42 after 48 hr of exposure to the drug was 0.25 ± 0.01, 0.15 ± 0.02, 0.25 ± 0.07, 0.11 ± 0.01 and 0.17 ± 0.02 μmol/l, respectively, for U266, H929, RPMI 8226, ARH-77 and IM-9 cells. The cytotoxicity of AR-42 was confirmed in primary MM cells with LC50 of 0.17 ± 0.03 μmol/l at 48 hr. Extending drug exposure to 72 and 96 hr resulted in additional cytotoxicity, indicating that AR-42 also induced the cell death in a time-dependent manner (Fig. 1b; 72 hr result not shown).
Figure 1.
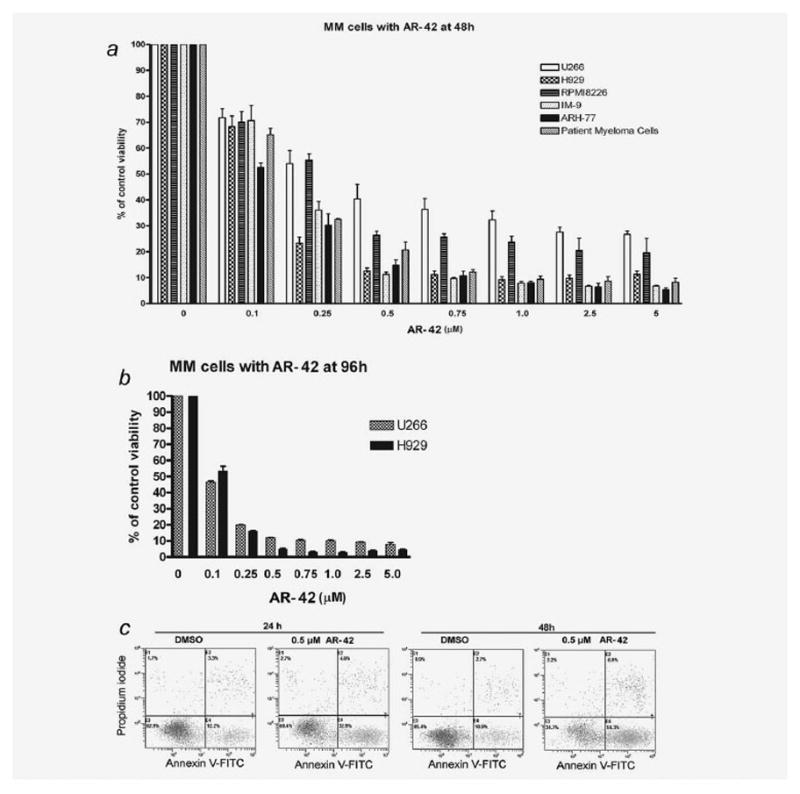
AR-42 is cytotoxic to MM cells in a dose- and time-dependent manner. (a) MTS assay showing percentage of viable MM cells treated with 0, 0.1, 0.25, 0.5, 0.75, 1.0, 2.5 and 5 μmol/l of AR-42 for 48 hr. Results expressed as mean with standard error of three independent experiments in cell lines, and as mean with standard error of two different patient primary MM samples. (b) U266 and H929 MM cells were treated with AR-42 or vehicle for 96 hr, and proliferation measured using MTS assay. (c) Flow cytometry results of annexin V-PI staining of U266 cells after exposure to DMSO or AR-42 (0.5 μmol/l) for 24 or 48 hr. An increase in early apoptotic (A+P−) cells following treatment with AR-42 is shown.
To determine whether MM cell death induced by AR-42 involves apoptosis, flow cytometric analysis with annexin V–PI staining was performed. AR-42 induced apoptosis in a time- and dose-dependent manner in all cell lines tested in the dose range of 0.25–2.5 μmol/l. Figure 1c is a representative example of apoptosis of U266 cell line treated with 0.5 μmol/l of AR-42 at 24 and 48 hr; strong apoptosis was also observed at the other concentrations (data not shown).
AR-42 induces cell death in a caspase-dependent manner by cleavage of caspases 3, 8 and 9
The mechanisms of cell death by different HDACi may vary in different cancer cell types.17–22 We, therefore, explored the effect of AR-42 on caspase-dependent apoptotic pathways. AR-42 induced cleavage of caspases 3, 8 and 9, as well as PARP, in a dose-dependent manner after 24-hr incubation with the drug (Fig. 2a). To determine the dependence of AR-42-induced apoptosis on the caspase pathway, we assessed the ability of the pancaspase inhibitor, Q-VD-OPH to protect against cell death. As shown in Figure 2b, Q-VD-OPH reduces AR-42-induced cell death as determined by annexin V-PI staining and the effect is only partial. We next examined whether Q-VD-OPH was actually inhibiting AR-42 activation of caspase-3 as measured by processing of the proform and downstream cleavage of PARP that is characteristic of caspase-dependent apoptosis. U266 cells were exposed to AR-42 in the presence or absence of Q-VD-OPH and cell lysates made (n = 3). As shown in Figure 2b, Q-VD-OPH greatly diminished processing of cleavage of PARP and caspase-3 as well as preventing cell death. These data together demonstrate that while apoptosis is induced by AR-42 mainly through caspase-dependent mechanisms, noncaspase dependent pathways may also operate.
Figure 2.
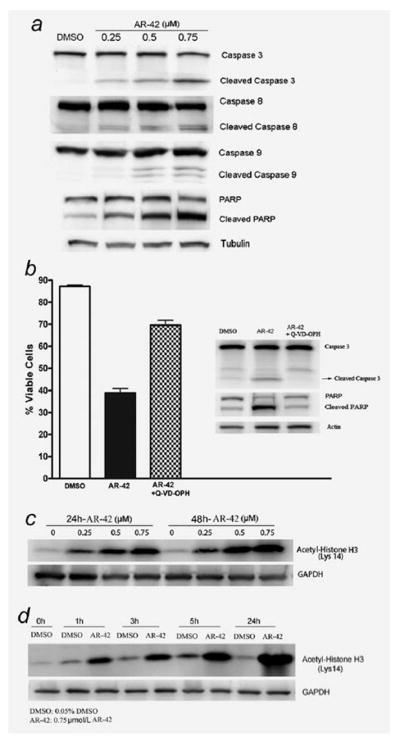
AR-42 induces cell death in a caspase-dependent manner and induces hyperacetylation of histone H3. (a) Caspase activation and PARP cleavage following treatment with AR-42. Lysates from MM cells treated with 0, 0.25, 0.5 and 0.75 μmol/l AR-42 for 24 hr were probed for caspase-3, caspase-8, caspase-9 or PARP by immunoblot. (b) Caspase inhibition protects against AR-42-induced cell death. MM cells were incubated in 0.05% DMSO, 0.5 μmol/l AR-42 or a combination of Q-VD-OPH (20 μmol/l) and 0.5 μmol/l AR-42, followed annexin V-PI staining 48 hr later. Lysates from treated cells were probed for caspase 3 or PARP by immunoblot. Immunoblot is representative of three independent experiments. (c) Immunoblot of cell lysates showing a dose dependent increase in acetylated histone H3 with AR-42 treatment in U266 at 24 and 48 hr. (d) AR-42 (0.75 μmol/l) induced a time dependent increase in histone H3 acetylation in U266 cell lines. Histone H3 acetylation was induced as early as 1 hr and reached a maximum level at 24 hr.
AR-42 induces histone acetylation in MM cells
To verify whether AR-42 induced hyperacetylation of histones, MM cells were treated with AR-42 at different concentrations and time points and histone H3 acetylation analyzed by Western blotting on whole cell lysates. As shown in Figures 2c and 2d for U266 cells as a representative example, AR-42 induced histone H3 acetylation in a time- and dose-dependent manner as early as 1 hr, and reaching maximal effect by 24 hr.
AR-42 decreases gp130 subunit of the interleukin-6 receptor complex levels and inhibits constitutive and inducible STAT3 phosphorylation in MM cells
The proliferation and survival of MM cells are dependent, in large part, on interleukin (IL)-6 and IL-6 receptor stimulation through autocrine and paracrine loops.23,24 IL-6 stimulates three major survival pathways, including the JAK2/STAT3, the Ras/Raf/MEK/MAPK and the PI3K/AKT pathways.25–28 Signaling through the IL-6 receptor is via the gp130 signal transduction subunit, which following dimerization leads to phosphorylation of STAT3 at tyrosine residue 705 leads to activation of the JAK2/STAT3 pathway.29 In the human myeloma cell line U266, STAT3 is constitutively activated through an IL-6 autocrine loop. Inhibition of the constitutive STAT3 pathway induces the cells into apoptosis.4
We first evaluated the effect of AR-42 on the expression of p-STAT3 and gp130 in U266 cells. Figure 3a shows that treatment of U266 cells with low concentrations of AR-42 for 24 hr leads to a significant reduction in gp130 expression as well as tyrosine-phosphorylated STAT3 although total STAT3 was unaffected. While AR-42 reduced p-STAT3 as early as 1–5 hr after drug exposure, gp130 was not significantly reduced at these early time points (Fig. 3b). On the other hand, AR-42 caused only a marginal decrease in p-MEK levels and no significant change in Akt and p-Akt expression (Fig. 3a). Next, we examined whether AR-42 could inhibit IL-6-induced STAT3 phosphorylation in H929 cell lines. H929 cells were pretreated with AR-42 for 24 hr and then stimulated with IL-6 for 15 min. As shown in Figure 3c, IL-6 induced STAT3 phosphorylation was reduced by AR-42. Importantly, exogenous IL-6 did not protect MM cells from AR-42-induced cytotoxicity despite its stimulatory activity on cell growth in H929 and U266 cells not exposed to drug (Fig. 3d; results for U266 not shown). The results indicate that gp130/STAT3 pathway is likely an important target of AR-42 in MM cells.
Figure 3.
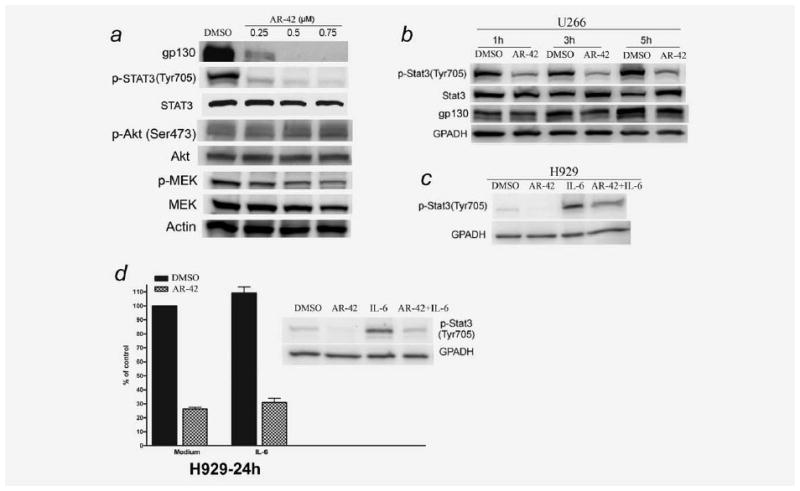
AR-42 downregulates gp130 and reduces constitutive and inducible p-STAT3 in MM cells. (a) U266 cells were treated for 24 hr with or without AR-42 and analyzed for the indicated protein by immunoblotting. (b) AR-42 reduced p-STAT3 as early as 1–5 hr in U266 cells, although gp130 was minimally altered at these early time points. U266 cells were cultured in 10% FBS RPMI 1640 media with DMSO (0.05%) or AR-42 (0.75 μmol/l). (c) AR-42 reduces IL-6-induced phosphorylation of STAT3 in H929. H929 cells were cultured in serum-free RPMI 1640 media for 22 hr followed by culture in serum-free RPMI 1640 media with 0.05% DMSO (control) or AR-42 (0.75 μmol/l) for 24 hr. Cells were then cultured with or without 10 ng/ml recombinant human IL-6 for 15 min and lysates made. (d) Exogenous IL-6 does not protect MM cells from the inhibitory effect of AR-42. H929 cells were treated with AR-42 (0.75 μmol/l) in the presence or absence of recombinant human IL-6 (10 ng/ml) for 24 hr and then analyzed for proliferation by MTS assay (results represent mean of three independent experiments). AR-42 also reduced IL-6 induced p-STAT3 expression in H929 as shown in Western blot.
AR-42 downregulates Bcl-xL expression, and overexpression of Bcl-xL partially protects against AR-42-mediated cytotoxicity of MM cells
The apoptosis inhibitor, Bcl-xL, is one of the key downstream targets of STAT3 in MM cells, considered important for myeloma cell survival and drug resistance.4 As shown in Figure 4a, AR-42 treatment strongly downregulated Bcl-xL expression in U266 cells. Furthermore, overexpression of Bcl-xL using lentivirus gene transduction partially protected against AR-42-induced cell death (Figs. 4b and 4c), indicating that reduction in Bcl-xL may contribute an important role in AR-42-induced apoptosis in MM cells.
Figure 4.
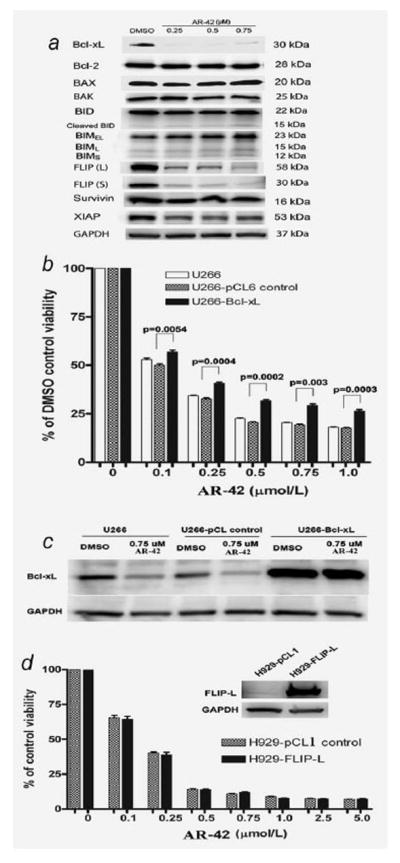
AR-42 downregulates Bcl-xL and c-FLIP. (a) Expression of Bcl-2 family proteins, c-FLIP, IAP family proteins, survivin and XIAP in U266 cells following 24-hr exposure to AR-42 (Western blot representative of three independent experiments). (b, c) Overexpression of Bcl-xL partially protected against AR-42-mediated cytotoxicity in U266 cells. (b) Viability of U266 cells transduced with Bcl-xL and empty pCL6IEGwo vector after treatment with AR-42 for 72 hr (data is representative of three independent experiments). (c) Expression of Bcl-xL in stably transduced U266 cells confirming overexpression. U266 control, U266 cell lines transduced with empty pCL6IEGwo vector; U266-Bcl-xL: U266 transduced cells with overexpression of Bcl-xL. (d) Overexpression of c-FLIP (L) fails to protect against AR-42-induced cell death in H929 cells. Top, overexpression of c-FLIP (L) in stably transduced H929 cells. Bottom, viability of H929 cells transduced with c-FLIP (L) and empty pCL1 vector after treatment with AR-42 for 48 hr (results are the mean of three independent experiments). H929-pCL1, H929 cells transduced with empty pCL1IEG vector; H929-FLIP-L: H929 transduced cells with overexpression of c-FLIP (L).
AR-42 downregulates c-FLIP expression and only minor effects on other anti- or proapoptotic proteins
We also evaluated the relative expression levels of other pro-and antiapoptotic proteins, which have been shown to be important for the survival of MM cell and their resistance to chemotherapeutic agents.30 As shown in a representative experiment in Figure 4a, Bcl-2, Bax, BAK, BID and BIM showed no significant change in expression in MM cells following treatment with AR-42. Similarly, there was minimal if any significant change in the expression of proteins of the inhibitor of apoptosis (IAP) family, including survivin and the X-linked inhibitor of apoptosis (XIAP) following exposure to AR-42 (Fig. 4a).
As AR-42 induced-apoptosis was associated with caspase 8 activation, we investigated the expression of cellular FLICE-like inhibitory protein (c-FLIP) following drug exposure. c-FLIP is an inhibitor of death receptor-mediated apoptosis and exists as a family of alternatively spliced variants, including long (c-FLIP (L)) and short (c-FLIP (S)) splice variants in human cells.31 Upregulation of c-FLIP in MM cells by bone marrow stroma has been reported to mediate resistance to Apo2 ligand/TRAIL induced apoptosis.32 As shown in Figure 4a, AR-42 treatment resulted in a significant reduction of c-FLIP (L) and c-FLIP (S) in U266 (Fig. 4a) and H929 cells (data not shown). Despite this change, however, overexpression of c-FLIP (L) using lentivirus gene transfer failed to protect against drug-induced cell death (Fig. 4d), indicating that reduction in c-FLIP is not likely a dominant mechanism in AR-42-induced apoptosis in MM cells.
AR-42 downregulates cyclin D1, increases p21 and p16 expression and induces G1 and G2 cell cycle arrest in MM cells
As the cell cycle promoter cyclin D1 is another key downstream factor of STAT3,5 we investigated the effect of AR-42 on its expression. As shown in Figures 5a and 5b, a reduction in cyclin D1 was observed as early as 5 hr after treatment but was completed reduced by 0.25–0.75 μmol/l of AR-42 after 24 hr, although cyclins A and B1 were unaffected. The promoter of p21 gene is regulated by histone acetylation status and induction of p21 is a hallmark of HDAC inhibitors.33 As increased expression of p21 results in inhibition of proliferation, we examined the effect of AR-42 on its expression and that of p16, another cell cycle inhibitor that has been shown to be transcriptionally silenced in MM.34 Expression of both p21 and p16 proteins was induced by AR-42 as early as 5 hr after treatment but was quite profound at 24 hr (Figs. 5a and 5b). Consistent with the above findings, cell cycle analysis data showed that AR-42 induces G1 and G2 cell cycle arrest in MM cells (Fig. 5c).
Figure 5.
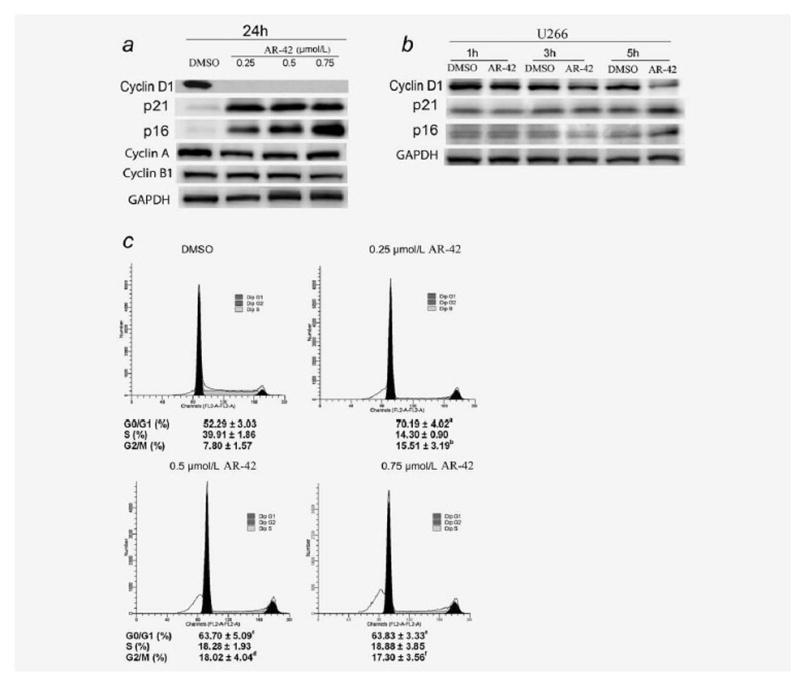
Effect of AR-42 on cell cycle proteins and cell cycle progression. (a) AR-42 induces expression of p16 and p21 and reduces expression of cyclin D1. U266 cells were treated with 0.05% DMSO or AR-42 at the shown concentrations for 24 hr. Lysates were then prepared immediately and analyzed by Western blot for cyclin D1, p21, p16, cyclin A and cyclin B. (b) Early change in cyclin D1, p16 and p21 with 0.05% DMSO or 0.75 μmol/l AR-42 in U266 cells. As noted, a reduction in cyclin D1 and induction of p16 and p21 are noted as early as 5 hr after treatment with AR-42. (c) Cell cycle analysis in AR-42-treated U266 cells showing arrest in G1 and G2 phases. U266 cells were incubated with 0.05% DMSO or with 0.25, 0.5 or 0.75 μmol/l AR-42 for 24 hr. The percentage of cells in each phase of the cell cycle is presented as mean ± SD from three independent experiments. Following treatment with AR-42 for 24 hr, there was a significant increase in the percentage of cells in G0/G1 or G2/M relative to DMSO control (p values: ap = 0.0035; bp = 0.0198; cp = 0.0289; dp = 0.0151; ep = 0.0113; fp = 0.0134; Student t-tests).
Discussion
HDAC have recently been investigated as potential targets in the treatment of MM.10–14 However, while a recent clinical trial of vorinostat in MM has reported only modest efficacy,15 HDACi differ in their spectrum of cellular activity and those currently available clinically, such as valproic acid and vorinostat, suffer from low potency and/or poor oral bioavailability. We, therefore, investigated the effects of a novel phenylbutyrate derived-HDACi, AR-42, in MM cell lines and primary myeloma cells. We show that AR-42 has a significant inhibitory effect on IL-6 receptor signaling, downregulating the signaling transduction subunit gp130 and blocking STAT3 phosphorylation, thereby inducing resistance to IL-6 stimulation. AR-42 also decreases the expression and cyclin D1 and Bcl-xL, two major downstream targets of STAT3, and overexpression of Bcl-xL partly protects against AR-2-induced myeloma cell death. Finally, AR-42 upregulates the cell cycle inhibitors p21 and p16 and inhibits cell cycle progression.
The mechanisms of HDACi-induced cytotoxicity may vary depending on the class of HDAC being inhibited and the downstream targets of HDAC in different cancer cells. Our results in MM show that AR-42-induced apoptosis is associated with cleavage of caspases 8, 9 and 3, and PARP cleavage, suggesting that the drug activates both the extrinsic and intrinsic apoptotic pathways. Further, AR-42-induced apoptosis is in large part dependent on caspase activation. The dependence on caspase activation for induction of apoptosis by AR-42 appears to differ from other HDACi tested in MM. While LBH589 also induced caspase and PARP cleavage, caspase inhibition had only modestly protected against cell death,11 suggesting other mechanisms likely dominate. On the other hand, vorinostat-induced cytotoxicity in MM cells was not associated with activation of caspases 8, 9 and 3, but induced cell death in a calpain-dependent manner.14 This difference between vorinostat and AR-42 was also observed in prostate cancer cells.8 Therefore, the mechanisms of cell death by distinct HDACi are specific.
In MM cells, AR-42 also modulates the expression of the antiapoptotic proteins. Of the Bcl-2 family members, only Bcl-xL expression was significantly reduced, while other family members, including Bcl-2, BAX and BAK remain unaffected. Bcl-xL has been found to play an important role in the regulation of apoptosis by at least two different mechanisms, including the interaction with proapoptotic proteins of the Bcl-2 family (such as BAX) and with non-Bcl-2 family proteins, such as Apaf-1, thereby inhibiting Apaf-1-dependent caspase-9 activation, and by a direct pore-forming effect on the outer membrane of mitochondria.35,36 The changes in Bcl-2 family members with AR-42 differ from those previously reported with vorinostat. While a change in Bcl-xL expression has not been reported with vorinostat, treatment of MM cells with the latter drug resulted in upregulation of the proapoptotic proteins Bim, Bak and Bax,37 although these remained unchanged with AR-42. Further, unlike the effect of vorinostat on MM,14 we did not observe cleavage of Bid (Fig. 4a). Together, these results highlight the differences between the actions of different HDACi and suggest that the effect of AR-42 on Bcl-2 family members in MM is predominantly on Bcl-xL, as also previously reported in prostate cancer cells.8 More importantly, overexpression of Bcl-xL partially protects against AR-42-mediated cytotoxicity suggesting that reduction in Bcl-xL plays an important role in AR-42-induced apoptosis in MM cells. In addition to the effect on Bcl-xL, AR-42 also appeared to reduce the expression of c-FLIP. c-FLIP has been reported to regulate apoptosis by inhibiting caspase 8 activation.38 Moreover, c-FLIP is overexpressed in MM and direct silencing of c-FLIP using siRNA has been shown to inhibit the proliferation of MM cells.39,40 However, although AR-42 downregulated c-FLIP in MM cells and may have accounted for facilitation of caspase 8 cleavage, we could not demonstrate that this is a dominant mechanism of cell death as constitutive expression of c-FLIP failed to provide protection against AR-42-induced apoptosis.
Interference with progression through the cell cycle is an important mechanism observed with HDACi. Both vorinostat and LBH589 have been shown to induce G1 cell cycle arrest in MM cells and other cancer cell types.11,14 Similar to these HDACi, AR-42 also induced G1 arrest, although an additional effect was also seen on the G2 phase of the cell cycle. AR-42 induced significant expression of the cyclin dependent kinase (CDK) inhibitors p16 and p21. The induction of p16 expression has not been previously reported for other HDACi in MM, although has been observed in other cancer cell types with valproic acid.41,42 On the other hand, induction of p21 expression has previously been reported in MM cells treated with almost all other HDACi.43,44 p16, inhibits G1 CDKs specifically, while p21, inhibits CDK in all phases of the cell cycle45 and may explain the effect of AR-42 on inhibiting progression also through G2. Both p16 and p21 block the formation of cyclin-CDK complexes, allowing Rb to become activated and halting the cell cycle. In addition to inducing p16 and p21, AR-42 also downregulates cyclin D1 in MM cells contributing to arrest in the G1 phase, an effect common to other HDACi.
IL-6 plays an important role in the growth and survival of MM cells,23,46 and the signal transduction subunit gp130 is the central player in receptor complexes formed by IL-6-type cytokines.29 Our results indicate that AR-42 exerts an important inhibitory effect on the IL-6 signal transduction pathway by downregulating the expression of gp130 and inhibiting constitutive and inducible STAT3 phosphorylation. While the mechanism by which gp130 is down regulated remains uncertain, the relatively late time course at which this effect is observed (24 hr) likely indicates that this occurs at the transcription level rather than degradation of the receptor subunit.47,48 Furthermore, inhibition of STAT3 phosphorylation may further contribute to this effect on gp130 as previously reported.47 However, minimal if any changes were observed in the expression of p-Akt or p-MEK, suggesting that the effect of the drug is mainly through the gp130/STAT3 pathway. Another HDACi, atiprimod, has also been found to have a negative effect on STAT3 phosphorylation in MM cells.49 STAT3 directly and indirectly upregulates the expression of genes that are required for uncontrolled proliferation and survival of tumor cells, including Bcl-xL and cyclin D1.50 Indeed, inhibition of STAT3 leads to a decrease in Bcl-xL expression and promotes the induction of apoptosis in U266.4 In our study, the reduction of Bcl-xL and cyclin D1 induced by AR-42 may be, at least in part, through an inhibitory effect on the gp130/STAT3 pathway. Importantly, AR-42 overcomes the growth and survival promoting effect of IL-6.
In conclusion, our results show that the novel orally bioavailable HDACi, AR-42, has potent activity against MM cells with overlapping, but unique, mechanisms of action compared to other HDACi previously reported. Its ability to target mainly the gp130/STAT3 cellular pathway and downstream antiapoptotic and cell cycle proteins at nanomolar concentrations that are likely achievable in vivo,9 suggest its viability as part of the therapeutic armamentarium for MM. Our results provide preclinical rationale for clinical development of AR-42 for MM.
Acknowledgments
Dr. Ching-Shih Chen is the inventor of AR-42 and receives royalty payments from Arno Therapeutics, Inc., Parsippany, NJ.
Abbreviations
- CDK
cycle dependent kinase
- c-FLIP
cellular FLICE-like inhibtor protein
- HDAC
histone deacetylase
- HDACi
HDAC inhibitor
- IAP
inhibitor of apoptosis
- IL-6
interleukin-6
- MM
multiple myeloma
- pH3
phosphorylated histone 3
- PI3K
phosphatidylinositol 3-kinase
- X-IAP
X-linked inhibitor of apoptosis
References
- 1.Barlogie B, Shaughnessy J, Tricot G, Jacobson J, Zangari M, Anaissie E, Walker R, Crowley J. Treatment of multiple myeloma. Blood. 2004;103:20–32. doi: 10.1182/blood-2003-04-1045. [DOI] [PubMed] [Google Scholar]
- 2.Baumann P, Mandl-Weber S, Volkl A, Adam C, Bumeder I, Oduncu F, Schmidmaier R. Dihydroorotate dehydrogenase inhibitor A771726 (leflunomide) induces apoptosis and diminishes proliferation of multiple myeloma cells. Mol Cancer Ther. 2009;8:366–75. doi: 10.1158/1535-7163.MCT-08-0664. [DOI] [PubMed] [Google Scholar]
- 3.Hideshima T, Richardson P, Anderson KC. Novel therapeutic approaches for multiple myeloma. Immunol Rev. 2003;194:164–76. doi: 10.1034/j.1600-065x.2003.00053.x. [DOI] [PubMed] [Google Scholar]
- 4.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, Dalton WS, Jove R. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10:105–15. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 5.Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin Cancer Res. 2002;8:945–54. [PubMed] [Google Scholar]
- 6.Villar-Garea A, Esteller M. Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int J Cancer. 2004;112:171–8. doi: 10.1002/ijc.20372. [DOI] [PubMed] [Google Scholar]
- 7.Marsoni S, Damia G, Camboni G. A work in progress: the clinical development of histone deacetylase inhibitor. Epigenetics. 2008;3:164–71. doi: 10.4161/epi.3.3.6253. [DOI] [PubMed] [Google Scholar]
- 8.Kulp SK, Chen CS, Wang DS, Chen CY, Chen CS. Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin Cancer Res. 2006;12:5199–206. doi: 10.1158/1078-0432.CCR-06-0429. [DOI] [PubMed] [Google Scholar]
- 9.Lavelle D, Chen YH, Hankewych M, DeSimone J. Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am J Hematol. 2001;68:170–8. doi: 10.1002/ajh.1174. [DOI] [PubMed] [Google Scholar]
- 10.Kaiser M, Zavrski I, Sterz J, Jakob C, Fleissner C, Kloetzel PM, Sezer O, Heider U. The effects of the histone deacetylase inhibitor valproic acid on cell cycle, growth suppression and apoptosis in multiple myeloma. Haematologica. 2006;91:248–51. [PubMed] [Google Scholar]
- 11.Maiso P, Carvajal-Vergara X, Ocio EM, Lopez-Perez R, Mateo G, Gutierrez N, Atadja P, Pandiella A, San Miguel JF. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–9. doi: 10.1158/0008-5472.CAN-05-4186. [DOI] [PubMed] [Google Scholar]
- 12.Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, Mitsiades N, Shringarpure R, LeBlanc R, Chauhan D, Munshi N, Schlossman R, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–22. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 13.Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, Akiyama M, Chauhan D, Munshi N, Gu X, Bailey C, Joseph M, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci USA. 2004;101:540–5. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitsiades N, Mitsiades CS, Richardson PG, McMullan C, Poulaki V, Fanourakis G, Schlossman R, Chauhan D, Munshi NC, Hideshima T, Richon VM, Marks PA, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–62. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 15.Richardson P, Mitsiades C, Colson K, Reilly E, McBride L, Chiao J, Sun L, Ricker J, Rizvi S, Oerth C, Atkins B, Fearen I, et al. Phase I trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) in patients with advanced multiple myeloma. Leuk Lymphoma. 2008;49:502–7. doi: 10.1080/10428190701817258. [DOI] [PubMed] [Google Scholar]
- 16.Lu Q, Wang DS, Chen CS, Hu YD, Chen CS. Structure-based optimization of phenylbutyrate-derived histone deacetylase inhibitors. J Med Chem. 2005;48:5530–5. doi: 10.1021/jm0503749. [DOI] [PubMed] [Google Scholar]
- 17.Catley L, Weisberg E, Tai YT, Atadja P, Remiszewski S, Hideshima T, Mitsiades N, Shringarpure R, LeBlanc R, Chauhan D, Munshi NC, Schlossman R, et al. NVP-LAQ824 is a potent novel histone deacetylase inhibitor with significant activity against multiple myeloma. Blood. 2003;102:2615–22. doi: 10.1182/blood-2003-01-0233. [DOI] [PubMed] [Google Scholar]
- 18.Shao Y, Gao Z, Marks PA, Jiang X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2004;101:18030–5. doi: 10.1073/pnas.0408345102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai D, Wang Y, Ottmann OG, Barth PJ, Neubauer A, Burchert A. FLT3-ITD-, but not BCR/ABL-transformed cells require concurrent Akt/mTor blockage to undergo apoptosis after histone deacetylase inhibitor treatment. Blood. 2006;107:2094–7. doi: 10.1182/blood-2005-08-3317. [DOI] [PubMed] [Google Scholar]
- 20.Amin HM, Saeed S, Alkan S. Histone deacetylase inhibitors induce caspase-dependent apoptosis and downregulation of daxx in acute promyelocytic leukaemia with t(15;17) Br J Haematol. 2001;115:287–97. doi: 10.1046/j.1365-2141.2001.03123.x. [DOI] [PubMed] [Google Scholar]
- 21.Doi S, Soda H, Oka M, Tsurutani J, Kitazaki T, Nakamura Y, Fukuda M, Yamada Y, Kamihira S, Kohno S. The histone deacetylase inhibitor FR901228 induces caspase-dependent apoptosis via the mitochondrial pathway in small cell lung cancer cells. Mol Cancer Ther. 2004;3:1397–402. [PubMed] [Google Scholar]
- 22.Lucas DM, Davis ME, Parthun MR, Mone AP, Kitada S, Cunningham KD, Flax EL, Wickham J, Reed JC, Byrd JC, Grever MR. The histone deacetylase inhibitor MS-275 induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia cells. Leukemia. 2004;18:1207–14. doi: 10.1038/sj.leu.2403388. [DOI] [PubMed] [Google Scholar]
- 23.Klein B, Zhang XG, Lu ZY, Bataille R. Interleukin-6 in human multiple myeloma. Blood. 1995;85:863–72. [PubMed] [Google Scholar]
- 24.Hitzler JK, Martinez-Valdez H, Bergsagel DB, Minden MD, Messner HA. Role of interleukin-6 in the proliferation of human multiple myeloma cell lines OCI-My 1 to 7 established from patients with advanced stage of the disease. Blood. 1991;78:1996–2004. [PubMed] [Google Scholar]
- 25.Muto A, Hori M, Sasaki Y, Saitoh A, Yasuda I, Maekawa T, Uchida T, Asakura K, Nakazato T, Kaneda T, Kizaki M, Ikeda Y, et al. Emodin has a cytotoxic activity against human multiple myeloma as a Janus-activated kinase 2 inhibitor. Mol Cancer Ther. 2007;6:987–94. doi: 10.1158/1535-7163.MCT-06-0605. [DOI] [PubMed] [Google Scholar]
- 26.Heinrich PC, Behrmann I, Muller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem J. 1998;334(Pt 2):297–314. doi: 10.1042/bj3340297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ogata A, Chauhan D, Teoh G, Treon SP, Urashima M, Schlossman RL, Anderson KC. IL-6 triggers cell growth via the Ras-dependent mitogen-activated protein kinase cascade. J Immunol. 1997;159:2212–21. [PubMed] [Google Scholar]
- 28.Hsu JH, Shi Y, Hu L, Fisher M, Franke TF, Lichtenstein A. Role of the AKT kinase in expansion of multiple myeloma clones: effects on cytokine-dependent proliferative and survival responses. Oncogene. 2002;21:1391–400. doi: 10.1038/sj.onc.1205194. [DOI] [PubMed] [Google Scholar]
- 29.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen Q, Ray S, Hussein MA, Srkalovic G, Almasan A. Role of Apo2L/TRAIL and Bcl-2-family proteins in apoptosis of multiple myeloma. Leuk Lymphoma. 2003;44:1209–14. doi: 10.1080/1042819031000068052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kataoka T, Schroter M, Hahne M, Schneider P, Irmler M, Thome M, Froelich CJ, Tschopp J. FLIP prevents apoptosis induced by death receptors but not by perforin/granzyme B, chemotherapeutic drugs, and gamma irradiation. J Immunol. 1998;161:3936–42. [PubMed] [Google Scholar]
- 32.Perez LE, Parquet N, Shain K, Nimmanapalli R, Alsina M, Anasetti C, Dalton W. Bone marrow stroma confers resistance to Apo2 ligand/TRAIL in multiple myeloma in part by regulating c-FLIP. J Immunol. 2008;180:1545–55. doi: 10.4049/jimmunol.180.3.1545. [DOI] [PubMed] [Google Scholar]
- 33.Golay J, Cuppini L, Leoni F, Mico C, Barbui V, Domenghini M, Lombardi L, Neri A, Barbui AM, Salvi A, Pozzi P, Porro G, et al. The histone deacetylase inhibitor ITF2357 has anti-leukemic activity in vitro and in vivo and inhibits IL-6 and VEGF production by stromal cells. Leukemia. 2007;21:1892–900. doi: 10.1038/sj.leu.2404860. [DOI] [PubMed] [Google Scholar]
- 34.Wong IH, Ng MH, Lee JC, Lo KW, Chung YF, Huang DP. Transcriptional silencing of the p16 gene in human myeloma-derived cell lines by hypermethylation. Br J Haematol. 1998;103:168–75. [PubMed] [Google Scholar]
- 35.Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95:4386–91. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vander Heiden MG, Li XX, Gottleib E, Hill RB, Thompson CB, Colombini M. Bcl-xL promotes the open configuration of the voltage-dependent anion channel and metabolite passage through the outer mitochondrial membrane. J Biol Chem. 2001;276:19414–9. doi: 10.1074/jbc.M101590200. [DOI] [PubMed] [Google Scholar]
- 37.Fandy TE, Shankar S, Ross DD, Sausville E, Srivastava RK. Interactive effects of HDAC inhibitors and TRAIL on apoptosis are associated with changes in mitochondrial functions and expressions of cell cycle regulatory genes in multiple myeloma. Neoplasia. 2005;7:646–57. doi: 10.1593/neo.04655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kataoka T. The caspase-8 modulator c-FLIP. Crit Rev Immunol. 2005;25:31–58. doi: 10.1615/critrevimmunol.v25.i1.30. [DOI] [PubMed] [Google Scholar]
- 39.Munshi NC, Hideshima T, Carrasco D, Shammas M, Auclair D, Davies F, Mitsiades N, Mitsiades C, Kim RS, Li C, Rajkumar SV, Fonseca R, et al. Identification of genes modulated in multiple myeloma using genetically identical twin samples. Blood. 2004;103:1799–806. doi: 10.1182/blood-2003-02-0402. [DOI] [PubMed] [Google Scholar]
- 40.Suvannasankha A, Crean CD, Shanmugam R, Farag SS, Abonour R, Boswell HS, Nakshatri H. Antimyeloma effects of a sesquiterpene lactone parthenolide. Clin Cancer Res. 2008;14:1814–22. doi: 10.1158/1078-0432.CCR-07-1359. [DOI] [PubMed] [Google Scholar]
- 41.Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:3697–702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Valentini A, Gravina P, Federici G, Bernardini S. Valproic acid induces apoptosis, p16INK4A upregulation and sensitization to chemotherapy in human melanoma cells. Cancer Biol Ther. 2007;6:185–91. doi: 10.4161/cbt.6.2.3578. [DOI] [PubMed] [Google Scholar]
- 43.Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci USA. 2004;101:1241–6. doi: 10.1073/pnas.0307708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lavelle D, Chen YH, Hankewych M, DeSimone J. Histone deacetylase inhibitors increase p21(WAF1) and induce apoptosis of human myeloma cell lines independent of decreased IL-6 receptor expression. Am J Hematol. 2001;68:170–8. doi: 10.1002/ajh.1174. [DOI] [PubMed] [Google Scholar]
- 45.Deane NG, Parker MA, Beauchamp RD. Cell proliferation: a matter of time and place. Surgery. 2005;138:1–7. doi: 10.1016/j.surg.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 46.Bruno B, Giaccone L, Rotta M, Anderson K, Boccadoro M. Novel targeted drugs for the treatment of multiple myeloma: from bench to bedside. Leukemia. 2005;19:1729–38. doi: 10.1038/sj.leu.2403905. [DOI] [PubMed] [Google Scholar]
- 47.Blanchard F, Wang Y, Kinzie E, Duplomb L, Godard A, Baumann H. Oncostatin M regulates the synthesis and turnover of gp130, leukemia inhibitory factor receptor alpha, and oncostatin M receptor beta by distinct mechanisms. J Biol Chem. 2001;276:47038–45. doi: 10.1074/jbc.M107971200. [DOI] [PubMed] [Google Scholar]
- 48.Graf D, Haselow K, Munks I, Bode JG, Haussinger D. Caspase-mediated cleavage of the signal-transducing IL-6 receptor subunit gp130. Arch Biochem Biophys. 2008;477:330–8. doi: 10.1016/j.abb.2008.06.009. [DOI] [PubMed] [Google Scholar]
- 49.Amit-Vazina M, Shishodia S, Harris D, Van Q, Wang M, Weber D, Alexanian R, Talpaz M, Aggarwal BB, Estrov Z. Atiprimod blocks STAT3 phosphorylation and induces apoptosis in multiple myeloma cells. Br J Cancer. 2005;93:70–80. doi: 10.1038/sj.bjc.6602637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat Rev Cancer. 2004;4:97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]