

Abstract
Three different genes each located on a different chromosome encode the heavy chains of nonmuscle myosin II in humans and mice. This review explores the functional consequences of the presence of three isoforms during embryonic development and beyond. The roles of the various isoforms in cell division, cell-cell adhesion, blood vessel formation and neuronal cell migration are addressed in animal models and at the cellular level. Particular emphasis is placed on the role of nonmuscle myosin II during cardiac and brain development, and during closure of the neural tube and body wall. Questions addressed include the consequences on organ development, of lowering or ablating a particular isoform as well as the effect of substituting one isoform for another, all in vivo. Finally the roles of the three isoforms in human diseases such as cancer as well as in syndromes affecting a variety of organs in humans are reviewed.
Keywords: Cancer, MYH9-RD, Pentalogy of Cantrell, Congenital Diaphragmatic Hernia, Double Outlet of Right Ventricle, Neuronal Migration, Cell Adhesion, Body Wall Closure, Cytokinesis, Karyokinesis, Placental Vascular Formation
Introduction
In metazoans, the in vivo functions of the contractile protein nonmuscle myosin II (NM II) commence at the moment of conception and remain operative until the death of the organism. Thus two of the genes encoding the NM II heavy chains (NMHCs), Myh9 and Myh10 in humans and mice, are transcribed in the early embryo including the embryonic stem cells.1 The third gene, Myh14 is not expressed at the protein level until embryonic day (E)11.5 in mice.2 Although the 230 kDa heavy chains contain the domains required for the various functions attributed to NM II, the two tightly but non-covalently bound light chains are also required for stabilization (17 kDa, essential light chain) and regulation (20 kDa, regulatory light chain) of the NM II enzymatic activity. The two different light chains are the product of a number of homologous genes but to date there is no definitive evidence that each of the heavy chain isoforms binds to a unique set of light chains. Based on the three different NM II heavy chains, we refer to the isoforms of NM II as NM II-A (Myh9), NM II-B (Myh10), and NM II-C (Myh14). Although all three isoforms are ubiquitously expressed, differences in their protein expression patterns are obvious in developing mouse embryos at E11.5 and later (Fig. 1; for details see refs. 3 and 4).

Figure 1. Expression of NM II in Mouse Embryos. Sections of paraformaldehyde-fixed mouse embryos were stained with antibodies to NMHC II-A (upper row), II-B (middle row), and II-C (lowest row). All three NM IIs are widely distributed throughout the E11.5 embryo (left column). In E11.5 mouse brain (second column), NM II-A is enriched in vasculature, NM II-B is the major isoform detected in neurons, and NM II-C stains intensively in developing pituitary (white arrow). In developing mouse inner ear at E16.5 (third column), NM II-A mostly stains the vasculature, II-B is detected in both mesenchymal and epithelial cells, and II-C is particularly enriched in the developing sensory cells of the cochlea. In the E16.5 mouse intestines (fourth column), both II-A and II-C are intensely stained in the epithelial cells, but II-C is particularly concentrated at the apical border of these cells; II-B appears more intense in the surrounding serosal cells.2
One of the most striking aspects of the myosin molecule (a homodimer of two heavy chains and 2 pairs of light chains) is the marked structural and functional differences between the amino-terminal and carboxyl terminal parts of the molecule (Fig. 2). The former is globular in shape and contains both a MgATP binding domain and a separate but nearby domain which binds actin. NM II is characterized by an actin-activated MgATPase activity which is critical for the conversion of chemical energy to mechanical force and movement. This ATPase activity is required to slide actin-filaments past myosin and to exert tension on actin-filaments. Each of the NM II hexamers has a distinct enzymatic activity and kinetic properties whether measured as heavy meromyosin (HMM, the amino-terminal fragment of NM II) or the full-length molecule.5 In contrast, the carboxyl terminal region is a double-stranded, α-helical coiled-coil extending approximately 1100 amino acid residues and terminating in a nonhelical tail region of 33 (NM II-A), 43 (II-B), or 46 (II-C) amino acids depending on the NMHC isoform. The helical rod, the non-helical tail and the 20kDa light chain contain sites for reversible phosphorylation that regulate the structure and enzymatic activity of NM II.3

Figure 2. Schematic Diagrams of the Myosin Molecule, Filament and Insertions (not drawn to scale). (A) Diagram of a myosin molecule showing the globular head region, the α-helical coiled-coil rod and the short non-helical tail (NHT). The subfragment-1 (S-1), rod and heavy meromyosin (HMM) proteolytic domains are also indicated. (B) An example of a bipolar filament, which is formed by interaction among the rod domains. The in vitro bipolar filament contains an average of 29, 30 and 14 molecules respectively for NM II-A, II-B and II-C.5 Note the significantly smaller size of the II-C filament. (C) Exclusion or inclusion of two alternative insertions at the myosin head increases the total isoforms of NM II-B and II-C to 4 each.
The amino acid sequences in the globular head of NMHC II-B and II-C can be varied by including or excluding two different exons by alternative splicing of pre-mRNA. This has been demonstrated and studied at the protein level for both NM II-B and II-C,6-9 but has only been reported at the level of mRNA for NM II-A.10 The locations for the inserts are identical for NM II-B and II-C, one being in loop 1, near the ATP binding region (Fig. 2) referred to as the B1 (10 amino acids in humans and mice) and C1 (8 a.a.) inserts and the second in loop 2 near the actin binding region, the B2 (21 a.a.) and C2 (33 a.a.in humans and 41 a.a. in mice) inserts. Thus the total number of transcripts identified for NM II-B and II-C is four each (NM II-B0, B1, B2, and B1B2, etc.). Three of the insertions (B1, B2, and C2) are only detected in the nervous system, while the C1 insert is expressed in a large variety of tissues and cells. Although the presence of the B1 and C1 insertion results in an increase in the actin-activated MgATPase activity above that of the non-inserted NM II-B0 and II-C0 isoforms respectively, the MgATPase activity, similar to other NM II isoforms, requires regulatory light chain phosphorylation.8,9,11 This is not the case for the B2 and C2 insertions. Characterization of the actin-activated MgATPase activity of these inserted isoforms has been studied using NM II HMM.6,7 Similar to other NM II HMMs, NM II-B2 has a low actin-activated MgATPase activity when the 20 kDa light chains are unphosphorylated but unlike all NM II HMMs this activity does not substantially increase following phosphorylation. This has recently been verified using baculovirus expressed full-length NM II-B2.5 In contrast, NM II-C2 HMM although unphosphorylated, has a relatively high actin-activated MgATPase activity which is not further activated by 20kDa light chain phosphorylation.6
In an in vivo murine study to uncover the function of the two NM II-B insertions Ma et al. show that deletion of the B1 insertion results in abnormal migration of facial neurons during development. However this abnormality also requires a reduction in NM II-B expression. Deletion of the B2 insertion results in abnormalities in cerebellar development, particularly with respect to Purkinje cell localization and maturation. This is consistent with enriched expression of the B2-inserted NM II-B in mouse cerebellar Purkinje cells. The B2 exon-deleted mice show impaired motor coordination and rotorod performance, particularly at a higher speed (40 rpm). These findings are independent of the amount of NM II-B.12
Having briefly introduced the various isoforms of NM II, the purpose of this review is to highlight what we have learned about the function of NM II and its role in mammalian development with an emphasis on studies performed in vivo. In particular we will emphasize studies from humans and mice. Figure 3 highlights two mouse models carrying a homologous mutation in the motor domain of either NM II-A (R702C) or NM II-B (R709C). Unlike Drosophila which has a single heavy chain of NM II (zipper),13 humans and mice contain three different isoforms raising questions about the ability of one isoform to substitute for another, and if so the consequences both in vivo and in cells. In addition, we explore the information gained from this substitution. In the last section we summarize some of the recent work in an area of growing importance, the role and function of NM IIs in human disease.

Figure 3. (A) Localization of the Mutant Amino Acid. Model of the NMHC II-A motor domain (gray) shows the position (red spheres) of amino acid R702 (R709 in II-B). Green spheres in the panel show ATP at the nucleotide binding site. (B) Enzymatic properties of control and mutant NM II HMMs and mouse phenotypes for R702C NM II-A or R709C NM II-B. IVM, in vitro motility; NSM, no significant movement.
NM II and Vertebrate Development
NM II isoform independent roles in cell-cell adhesion
NM II-A and visceral endoderm cell-cell adhesion
NM II is involved in almost every aspect of vertebrate development. Expression of NM II is essential for the formation and the normal function of the visceral endoderm during early mouse embryonic development. Among the three NM II isoforms, NM II-A is the sole isoform detected in the developing visceral endoderm in mice.14 Mice ablated for NM II-A fail to form E-cadherin mediated cell-cell adhesions between visceral endoderm cells. Instead of forming a normal polarized columnar layer, NM II-A ablated visceral endoderm is disorganized at E6.5 and fails to support gastrulation in NM II-A knockout embryos.14 The development of a functional visceral endoderm does not depend on specific properties of NM II-A since NM II-B15 or II-C1 (unpublished observation, this lab) is also capable of supporting functional visceral endoderm formation in mice. Mice expressing NM II-B or II-C in place of NM II-A were generated by genetically ablating and replacing NMHC II-A with cDNA encoding NMHC II-B (or NMHC II- C1) under control of the endogenous NMHC II-A promoter. These mice develop a normal visceral endoderm and survive beyond organogenesis. E-cadherin localization is restored to the normal cell-cell adhesion complex together with NM II-B (or NM II-C1) in these mice. The function of NM II-A in maintaining cell-cell adhesion in the visceral endoderm does not require full NM II-A motor activity, since R702C NM II-A with a compromised MgATPase activity16 (Fig. 3).can also support functional visceral endoderm formation in mice, despite causing other severe abnormalities such as defects in placental formation later in mouse development 17
NM II-B and spinal neuroepithelial cell-cell adhesion
Similar to the visceral endoderm, expression of NM II is also required to maintain the integrity of the neuroepithelial cell adhesion complex in the cells lining the spinal canal. Unlike the visceral endoderm cells, neuroepithelial cells are enriched in NM II-B.18 Ablation of NM II-B in mice results in disruption of neuroepithelial cells lining the spinal canal, leading to the outgrowth of the neuroepithelial cells to block the spinal canal, and resulting in the development of hydrocephalus in NM II-B knockout mice.18,19 Either NM II-A or a motor impaired NM II-B can substitute for wild type NM II-B in maintaining the integrity of neuroepithelial cell-cell adhesion. Mice expressing NM II-A in place of NM II-B and under control of the endogenous NM II-B promoter show no defects in spinal neuroepithelial cell-cell adhesion and no hydrocephalus.20 Similarly, mice expressing only R709C NM II-B which fails to propel actin-filaments in an in vitro motility assay (Fig. 3),8 also show no defects in neuroepithelial cell-cell adhesion and no evidence of hydrocephalus.18 However hypomorphic mice expressing only ~25% wild type level of R709C NM II-B fail to prevent the disruption of the neuroepithelial cell-cell adhesion complex and develop hydrocephalus.18 Thus it is the quantity of NM II, but not the NM II MgATPase and motor activity that is important to maintain the integrity of cell-cell adhesion.
Looking into detailed structures of NM II in cell-cell adhesion in the inner ear, Ebrahim et al. analyzed the normal developing mouse organ of Corti which plays an important role in hearing and found that NM II forms a sarcomeric-like belt surrounding the epithelial apical junctions.21 NM II-C is the major NM II isoform located in the apical junction of the organ of Corti in wild type mice. Of note, when NM II-C expression is ablated, both NM II-A and NM II-B will also incorporate into the structure apparently to compensate for the loss of NM II-C. This is one of the rare cases where compensation by the other isoforms has been observed and this is consistent with the findings that mice ablated for NM II-C show no obvious hearing impairment (unpublished observation, this lab). NM II mediated contractility of the belt regulates the geometry of the epithelial cells. This sarcomeric-like structure is also seen in intestinal epithelial cells, and may be ubiquitous in all types of epithelial cells. This requirement for NM II to maintain cell-cell adhesion is also well documented in epithelial cells in culture.22-25
NM II isoform specific functions
NM II-A and mouse placental blood vessel formation
The development of the placenta in mice requires the unique properties of NM II-A.15 Ablation of either NM II-B or II-C, or even both II-B and II-C together shows no obvious effects on placenta formation in mice. Mice ablated for NM II-A die before placenta development. Mice expressing NM II-B in place of NM II-A under control of the endogenous NM II-A promoter survive to E11.5, and show major defects in placental development manifested by a compact and underdeveloped labyrinthine layer lacking fetal blood vessels. Moreover homozygous mice expressing mutant R702C NM II-A also show similar defects, although they are less severe than those shown by mice expressing NM II-B in place of NM II-A.17 In vitro analyses show that mutant R702C HMM II-A displays a 75% reduction in MgATPase activity and a 50% reduction in in vitro motility of actin-filaments compared with wild type HMM II-A.16 Therefore normal placental blood vessel formation requires both the proper levels of NM II-A expression as well as the characteristic NM II-A MgATPase activity. One function of NM II-A in placental development can be attributed to its role in trophoblast-lineage cells.26 Mice specifically ablated for NM II-A in the mouse trophoblast-lineage cells demonstrate placental defects similar to mice in which NM II-B replaces II-A.
NM II-B and neuronal cell migration
A second example of a unique requirement for a specific NM II isoform is pontine and facial neuron migration during mouse brain development, which specifically requires NM II-B. Both the facial and pontine neurons migrate long distances via tangential migration to reach their final destination. Ablation of NM II-B in mice results in a failure of both the facial and pontine neurons to reach their final destination.27 Impaired facial and pontine neuron migration is also observed in homozygous mice expressing motor-impaired R709C NM II-B.18 The requirement for NM II-B motor activity for neuronal migration is consistent with the finding that blebbistatin inhibition of NM II activity blocks neuronal migration (nuclear translocation).28 Studies from mice ablated for both mDia 1 and 3 show that the tangential migration of cortical interneurons is critically mediated by Rho signaling via mDia and Rho kinase which regulate actin-filament dynamics and NM II activity, respectively.29 This signaling appears not to be required for radial neuronal migration. However the in vivo migration of facial and pontine neurons appears not to depend simply on the enzymatic activities of NM II. NM II-A, which shows increased enzymatic properties (MgATPase and in vitro motility) compared with NM II-B, fails to rescue the migration defects of facial and pontine neurons in mice when it replaces NM II-B.18 The unique requirement for a specific NM II isoform (i.e., NM II-B) during neuronal migration is most likely due to the specific kinetic properties of the actin-activated MgATPase activity and duty ratio (the fraction of time that myosin remains strongly bound to actin during the total ATPase cycle time30) which vary significantly among the isoforms thus making it impossible for one isoform to substitute the other.4 Due to its unique load-dependent mechanosensitivity, NM II-B is functionally adapted for sustained tension generation.31-33 Other possibilities include the differences in cellular distribution and different binding partners. This is in sharp contrast to the role of NM IIs in maintaining cell-cell adhesions such as in the visceral endoderm and spinal neuroepithelium, where the crosslinking activity of NM II in binding actin-filaments is more important, and where NM II-A, NM II-B or motor-impaired NM II-A and II-B are interchangeable. Figure 4 summarizes NM II isoform specific and non-specific functions during mouse development.
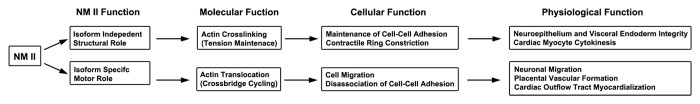
Figure 4. Summary of NM II Isoform Specific and Non-specific Functions in Mouse Development.
NM II-B and heart development
Cardiac myocyte development
The heart is one of the earliest organs of the embryos that forms and functions during development. All three isoforms of NM II are expressed in human and mouse hearts. NM II-B is the major isoform detected in mature cardiac myocytes in mice together with a limited amount of NM II-C. NM II-A is absent in mature cardiac myocytes although transient expression of NM II-A is detected in cardiac myocytes during early outflow tract development.34 The level of NM II-B expression in mouse cardiac myocytes decreases significantly after birth correlating in time with myocyte bi-nucleation and consistent with its role in cytokinesis. Neonatal cardiac myocytes exit the cell cycle, are mostly binucleated and no longer divide. Both NM II-A and II-B are detected in epicardial and endocardial cells as well as in cardiac mesenchymal cells. Expression of NM II-B, but not II-A and II-C, is essential for cardiac myocyte development in mice.35,36 Cardiac myocytes in NM II-B ablated mice fail in cytokinesis which results in an early exit of the cardiac myocytes from mitotic cell cycling. The reduced proliferation of cardiac myocytes contributes to the development of hypoplastic hearts in NM II-B ablated mice which die at E14.5. Interestingly mice expressing a motor impaired R709C NM II-B show no evidence for cytokinesis defects in cardiac myocytes.37 During steady-state cross-bridge cycling, R709C NM II-B spends a very long period strongly bound to actin-filaments due to its extremely high affinity for ADP. R709C NM II-B will rarely detach from actin-filaments thus preventing it from moving actin-filaments during cytokinesis.37 Studies with COS-7 cells reveal that R709C NM II-B can only complete one attachment and detachment cycle during cytokinesis. Moreover prolonged actin attachment favors the simultaneous attachment of both NM II-B heads to actin and consequently increases the resistance of the leading head to actin translocation. In vitro motility assays with R709C NM II-B HMM show no detectible movement of actin-filaments (Fig. 3). Therefore instead of translocating actin-filaments like striated muscle myosin II does during muscle contraction, R709C NM II-B constricts the contractile ring by exerting tension during cytokinesis. These results support the idea that it is the structural (crosslinking actin-filaments) rather than the motor (translocating actin-filaments) properties of NM II which are important for cytokinesis. In addition to cardiac myocyte cytokinesis, NM II-B is also involved in proper alignment of cardiac myocytes in mouse hearts.38 Instead of the normal spiral and parallel alignment of cardiac myocytes in wild type mouse hearts, cardiac myocytes align randomly in NM II-B ablated mouse hearts which impairs the mechanical unity of the heart and its pump function (Fig. 5). The hypoplastic myocardium and impaired pump function of the heart contribute to heart failure in the NM II-B ablated mouse embryos.
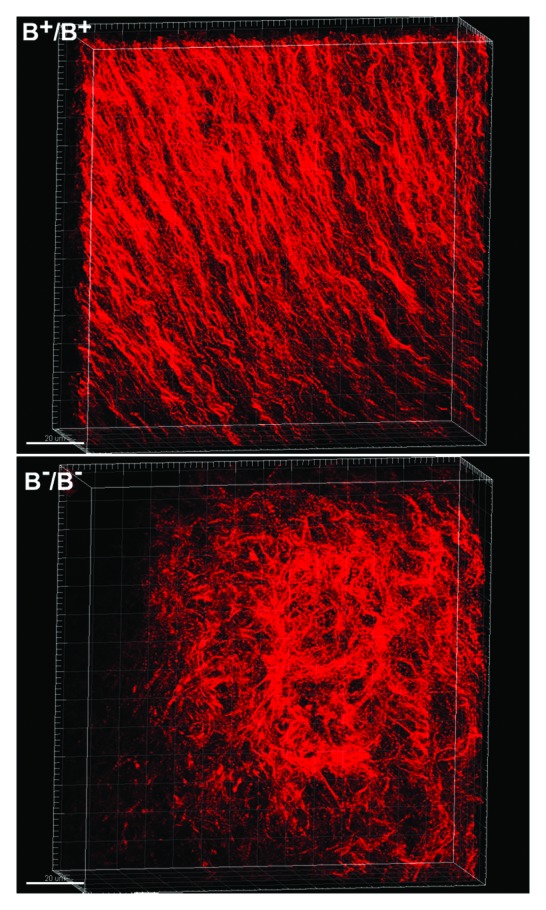
Figure 5. Disrupted Cardiomyocyte Alignment in NM II-B Ablated Mouse Hearts. E13.5 wild type (B+/B+) and NM II-B knockout (B-/B-) mouse hearts stained (wholemount) for actin-filaments with Alexa Fluor® 594 phalloidin. 3D stack of confocal microscope images of the ventricular myocardium shows that B+/B+ cardiomyocytes align in a regular spiral pattern (upper panel), while B-/B- cardiomyocytes are distributed randomly with no obvious pattern (lower panel).
In adult cardiac myocytes NM II-B is enriched in the intercalated discs which glue the myocytes together to build a functional unit. Together with other intercalated disc associated proteins such as mXinα,39 NM II-B is important for maintaining the integrity of the intercalated discs. Mice conditionally ablated for NM II-B in cardiac myocytes using αMHC-Cre mice survive to adulthood, but progressively developed cardiomyopathy between 6 to 10 mo of age associated with abnormally widened intercalated discs.40 NM II-C is also detected in the intercalated discs in adult cardiac myocytes, however, expression of NM II-C appears not to be essential. Mice ablated for NM II-C alone are healthy and show no obvious abnormalities. However when NM II-C is ablated together with NM II-B, the doubly ablated cardiac myocytes show a major defect in karyokinesis with abnormal chromatid segregation and mitotic spindle formation in embryonic mouse hearts.36 The role of NM II in regulating karyokinesis may relate to its crosstalk with microtubules. NM II is detected in mitotic spindles together with microtubules in cultured cells. Ablation of NM II or blebbistatin inhibition of NM II activity enhances microtubule polymerization and stabilizes microtubules. Conversely, inhibition of microtubules enhances the formation of the acto-NM II cytoskeleton.41 Thus in addition to cytokinesis, NM II is also involved in karyokinesis during cardiomyocyte cell mitosis. Whether this remains true for other types of cells requires further investigation.
Outflow tract formation
The function of NM II-B is not limited to cardiac myocyte development in hearts. Mice ablated for NM II-B or expressing R709C NM II-B consistently develop cardiac structural defects such as abnormal alignment of the aorta with the right ventricle (DORV) and a ventricular septal defect (VSD).38 Both abnormalities are frequently observed in patients with congenital heart disease. Outflow tract (OFT) myocardialization, where the OFT cardiac myocytes migrate into cardiac cushions and replace the cushion mesenchymal cells, plays an important role in proper positioning of the aorta with respect to the left ventricle.42 Ablation of NM II-B or impairment of NM II-B motor activity by the R709C mutation in NMHC II-B, prevents the OFT cardiac myocytes from migrating into the cushion thereby causing mislocalization of the aorta to the right ventricle in mice.43 The failure of myocardialization appears to be due to the inability of the OFT cardiac myocytes to dissociate cell-cell adhesions when NM II-B is absent or its activity is impaired. Myocardialization is also unique to NM II-B and cannot be effected by NM II-A. Expression of NM II-A in place of NM II-B in mice fails to prevent the aorta from being misplaced to the right ventricle. Consistent with the above was the previous report that noncanonical Wnt planar cell polarity (PCP) mediated RhoA/ROCK1 signaling is involved in OFT myocardialization in mice.44 NM II-B most likely serves as a downstream target of this signaling which regulates myocardialization. Thus NM II functions both in maintaining and dissociating the cell adhesion complex. Similar results were observed from studies using cultured cells.45 These results support the idea that NM II utilizes its structural (crosslinking) property to maintain cell-cell adhesions, while utilizing its dynamic motor property to dissociate cell-cell adhesions.
NM II and neural development
Neural tube closure
Evidence of apical actomyosin accumulation at the hinge area of the folding chick neural tube implicates NM II in neural tube closure.46 The finding that blebbistatin inhibition of NM II results in a failure of neural tube closure in chick embryos supports the requirement for NM II in this process.47 This is further supported by the finding that Rho/Rho kinase mediated NM II activation is required for normal neural tube formation in chick embryos. In Xenopus, morpholino knockdown of NM II-B results in delayed and defective neural tube closure associated with impaired apical actin accumulation and neural convergent extension.48 One key morphogenic signal, Wnt/PCP mediated Rho signaling, has been implicated in regulating NM II activity during neural tube closure. Studies from Nishimura et al. showed that PCP signaling activates NM II via Dishevelled, DAAM1, and PDZ-RhoGEF leading to upregulation of Rho kinase.49 Rho mediated NM II contraction promotes apical constriction and convergence of the neuroepithelium. Instead of directly driving cell shape changes in morphogenesis, NM II mediated periodic apical contraction and cortical tension trigger apical constriction via a tissue clutch which connects the cell-cell contact zone to the myosin motor.50 In addition to PCP signaling, the actin-binding protein Shroom is also involved in regulating neural tube closure by apical recruitment of Rho kinase and actomyosin filaments.51,52 The importance of Rho/NM II in regulating neural tube closure in mammals is still not clear. Mice ablated for Rho kinase develop defects in eye lid and ventral body wall closure, but not neural tube closure.53 Mice ablated for NM II-B also show no defects in neural tube closure.38 It is possible that other kinases such as myosin light chain kinase may compensate for the loss of Rho kinase by activating NM II in these mice. In both mice and humans, all three isoforms of NM II are expressed in neuroepithelial cells, and in mice loss of one isoform can be compensated for by genetically substituting one of the other two isoforms.15 In addition, other cytoskeletal networks may also function to drive neural tube closure in mammals.54
Neuronal cell migration
The function of NM II in neuronal migration is well documented. In cooperation with the microtubules, NM II plays an important role in propelling soma movement during neuron migration.28 Cerebellar granule neurons (CGN) in mice lacking NM II-B activity migrate significantly more slowly than those in wild type mice.27 Blebbistatin treatment stops nuclear migration in cultured neurons. Based on in vitro studies with Medial Ganglionic Eminence (MGE) interneurons in cortical slice preparations and subventricular zone (SVZ) precursors migrating in a 3D matrigel matrix, it has been proposed that NM II functions by breaking adhesions at the cell rear or by causing contractions that squeeze the nucleus forward.55 This is consistent with NM II localization at the rear of migrating nuclei.28 In contrast, studies from CGNs suggest that NM II pulls the centrosome and soma forward, which is supported by NM II capping at the leading area of the migrating nuclei.56,57 The discrepancy between the two hypotheses may reflect the nature of different types of neuronal migration, since the MGE interneurons and SVZ precursors migrate using the glial independent tangential mode while CGNs migrate using the glial dependent radial mode. Both the glial dependent (CGN) and tangential (facial and pontine neurons) migrations are impaired in mice lacking NM II-B function.27 Ablation of NM II-C36 or conditional ablation of NM II-A in neuronal cells (unpublished observation, this lab) shows no effects on neuronal migration in mice.
In addition to neural migration, NM II is also required for interkinetic nuclear translocation of the neural progenitor cells during neurogenesis. Blebbistatin inhibition of NM II activity impairs apical to basal nuclear translocation of cortical neural progenitors from mouse brain in hemisphere rotation (HERO) culture.58 Using high-resolution live-cell imaging of chick neural tubes in slice culture, Das et al. demonstrate that newborn neurons at the ventricular surface detach from the ventricle by apical abscission of the apical membrane, a process that also requires NM II function.59 This same mechanism is also found in mice. Abscission of the apical membrane results in loss of apical cell polarity of the newborn neurons and facilitates neuronal migration and exit from the cell cycle. Thus the function of NM II in neuronal migration is multifaceted, and a detailed mechanism is still far from clear.
NM II and body wall closure
Drosophila dorsal wall closure is one of the best studied developmental processes during morphogenesis. Expression of NM II is essential for dorsal wall closure in the developing Drosophila embryo. Ablation of the sole Drosophila NM II heavy chain gene, zipper, results in a failure of dorsal wall closure.60 During Drosophila dorsal wall closure, NM II together with actin accumulates at the leading edge of the migrating lateral ectodermal-sheets forming an actomyosin cable, the “purse string,” which contracts to produce tension.61 Additionally, the underlying squamous epithelial amnioserosa cells, surrounded by the lateral ectodermal leading cells, contract at the apical surface to produce the pulsed force which pulls the ectodermal sheets dorsally in a ratchet-like mechanism.62,63 These two NM II dependent mechanical events cooperate to close the dorsal wall in the developing Drosophila, where the purse-string helps to effect the transient displacement of the epidermal sheets pulled by the pulsed constriction of the amnioserosa cells.63
Ventral body wall closure of the developing mouse embryo is thought to be equivalent to Drosophila dorsal body wall closure. Mice ablated for Rho kinase, one of the major kinases which activate NM II, develop an upper umbilical hernia which suggests the involvement of NM II in normal mouse ventral body wall closure.53 In contrast to Drosophila, as noted above, mice express three isoforms of NM II. The requirement for normal NM II function in mouse ventral body wall closure is demonstrated by studies in a knockin mouse model expressing the motor-impaired R709C NM II-B. R709C mice with either one allele mutated or both alleles mutated show defects in ventral body wall closure.43 The severity of the defect depends on the amount of mutant NM II-B expressed. Homozygous mice expressing wild type levels of R709C NM II-B show defects in both abdominal and thoracic walls, whereas heterozygous mice expressing 50% mutant NM II-B develop defects limited to the abdominal wall. Homozygous, hypomorphic mice expressing ~25% mutant NM II-B show no defect in body wall closure. R709C NM II-B HMM displays extremely high affinity for actin-filaments which prevents it from translocating actin-filaments in an in vitro motility assay.37 However loss of motor activity from R709C NM II-B does not directly contribute to the failure in body wall closure, since knockout mice expressing no NM II-B have no defects in body wall closure. In addition, mice ablated for both NM II-B and II-C also undergo normal ventral body closure, suggesting expression of NM II-A alone is sufficient for normal ventral body wall closure in mice. One possibility is that expression of R709C NM II-B interferes with the normal function of NM II-A or with another cytoskeletal protein which prevents ventral body wall closure in R709C NM II-B mice. Consistent with a role for NM II-A in body wall closure is the finding that conditional knockout of NM II-A expression in eyelid front epidermal cells by injecting epithelial cell-specific (K17-Cre) lentivirus in vivo in mouse embryos causes defects in eye closure. In studying eyelid closure in mice, Heller et al. utilized multiple techniques and found that NM II-A and α5/β1 integrin/fibronectin mediated migration and intercalation of the eyelid front cells generate tension to pull the eyelid epithelial sheets forward.64 It will be of great interest to determine whether a similar mechanism also governs NM II mediated ventral body wall closure in mice.
NM II and Disease
Mutations and deletions in each of the isoforms of NM II have been associated with a variety of diseases and in some cases mouse models have been generated that mimic the human defects. In this section we review some of the more prominent examples of the abnormalities that have been associated with each of the isoforms. We also address the role of loss or mutation of each isoform with respect to the generation of the phenotype in mice and humans.
NM II and cell division
All three isoforms appear to play a role in cytokinesis, the importance of which depends on the amount of the isoform and the type of cell being studied. Cos7 cells which are devoid of NM II-A contain a high percentage of NM II-B (86%) and significantly less NM II-C (15%).36 Removing NM II-B by siRNA results in a defect in cytokinesis (multinucleation) which can be rescued by introducing any of the three isoforms back into the cells. Removing NM II-C from these cells has no effect on cytokinesis, suggesting that it is the amount of NM II and not the isoform that matters.65 This is in contrast to some cancer cell lines described below.
As noted above, a role for NM II-B in cardiac myocyte cytokinesis has been documented and is supported by the finding of binucleation by E14 in cardiac myocytes of NM II-B knockout, and hypomorphic R709C NM II-B mice.35 Moreover eliminating the hypomorphism in vivo is sufficient to rescue binucleation.37 Ablating NM II-A in early mouse embryos results in abnormal, multi-lobed nuclei in the epithelial cells that constitute the visceral endoderm, a defect that can be rescued by genetically introducing NM II-B or II-C using homologous recombination to ablate and replace NM II-A.14,15 Interestingly, eliminating NM II-B and II-C in the heart by generating a II-C and II-B combined knockout mouse not only affects myocyte cytokinesis but karyokinesis too36 predisposing these cells to a more malignant phenotype.66
NM II-A and cancer
Although NM IIs have been associated with alterations in cell division and indirectly with deleterious alterations in cells, more direct evidence for a role for NM II in cancer has recently been published.67 Schramek et al. provide evidence that NM II-A acts as a tumor suppressor for squamous cell cancer. An in vivo tissue-specific RNAi screen was used in which epithelial cell-specific delivery of lentiviral short hairpin RNA (shRNA) was performed in utero using pregnant mice. Knocking down NMHC II-A induces metastatic squamous cell carcinoma (SCC) in the skin or head and neck in keratin 14-Cre Tgfbr2 conditional knockout mice with a median latency of 3–7 mo. Moreover ablation of NM II-A in non-Tgfbr2 compromised mice results in SCC of the skin after I year. The authors also provide evidence that in human and mouse keratinocyte cell lines NM II-A appears to play a role in regulating posttranscriptional stabilization of p53. Of note, p53 activation appears to depend on NM II-A MgATPase activity since it was abolished by the NM II chemical inhibitor blebbistatin, and NM II-A appears to be necessary for both p53 stability and nuclear retention. How this correlates with the onset of the tumor phenotype is yet to be determined.
With respect to humans, the authors also provide evidence that low expression of NM II-A correlated with poor survival in patients with head and neck SCC and found that MYH9 was mutated in 5% of tumors, with most mutations clustering in the MgATPase domain. One as yet unanswered question, is whether patients with MYH9-related diseases (MYH9-RD),68 who also harbor mutations in both the MgATPase domain and elsewhere throughout the NMHC II-A molecule are more prone to develop skin cancer (see below).
In addition to the studies above, there are a number of studies suggesting that various NM II isoforms play a role in cell differentiation, proliferation and metastatic behavior in cell lines. Of note is a study in mice in which NM II-A was ablated in the epidermis by crossing to keratin 5-Cre mice, which results in epidermal hyperproliferation and disruption of hair follicle development in the newborn mice.26 In a recent article Shin et al.69 present data indicating that NM II-A and II-B act to determine the cell fate of hematopoietic stem cells by regulating cell division. Interestingly, NM II-B is associated with cell self-renewal through asymmetric cell division during which it is downregulated in the differentiated cell but markedly polarized in the stem cell. In contrast, NM II-A which is associated with cell differentiation is activated by dephosphorylation of Ser1943 on a stiff, endosteum-like matrix.
Two proteins associated with the cancer phenotype that interact with NM IIs, the Ca2+-binding protein S100A4, and the tumor suppressor Lgl (Lethal[2] giant larvae), have recently been covered in a review published in this Journal.70 The former has been shown to disassemble filaments of NM II-A and to mediate the cellular location of pseudopodial protrusions during chemotaxis, thereby playing a role in tumor metastatic spread.71 In a recent article, Dahan et al.72 showed that mammalian Lgl1, which is known to interact with NM II-A and inhibit its assembly into filaments, can be phosphorylated by atypical protein kinase Cζ which prevents Lgl1 interaction with NM II-A. Lgl1 can form two distinct complexes in cells, Lgl1-Par6α-aPKCζ and Lgl1-NM II-A. Formation of both complexes is prevented when Lgl1 is phosphorylated. However the connection between the interaction with NM II-A and tumor suppression in humans has yet to be established.
The syndrome, MYH9-related disease (MYH9-RD)
Although loss of NM II-A in the skin of mice results in SCC under certain circumstances, mutations in NM II-A, including missense, nonsense, deletions and duplications, , result in a syndrome originally called by a number of different names in humans but now referred to as MYH9-related disease, MYH9-RD.68 The defects in humans include macrothrombocytopenia (abnormally large and decreased numbers of platelets), aggregates of NM II-A in neutrophils, cataracts, deafness, and glomerulosclerosis. Although the prolonged bleeding times seen in humans are not usually severe enough to cause a problem, the glomerulosclerosis (originally referred to as Epstein’s disease) may be lethal. Two different laboratories have generated a mouse model of MYH9-RD.17,73 Zhang et al. report on 3 different mutations which duplicate the most frequent mutations found in humans: one in the head region of NMHC II-A, R702C, and two in the rod region, D1424N and E1841K. Suzuki et al. report on the effects of the R702C mutation. Although there are some differences between the results obtained, both studies agree that the mouse models do mimic the human phenotypic changes including large platelets, hearing loss, and severe kidney pathology. Of note is that Zhang et al. used a cDNA cassette encoding GFP-labeled human R702C NMHC II-A to ablate the endogenous mouse II-A. This replacement with GFP-labeled NMHC II-A permitted real-time imaging of proplatelet formation. Both labs report a decrease in proplatelet formation accompanied by an increase in platelet bud size. The two mutations in the rod region reported in Zhang et al. (D1424N and E1841K) were generated by directly mutating codons in the corresponding exons of the endogenous gene. Although homozygous R702C mice died between E10.5–11.5, the cause is attributed to a failure in placenta development and appears to be independent of MYH-RD. On the other hand homozygous D1424N and E1841K mice are viable and show a more severe phenotype than the heterozygous mutant mice. Since the mutant NM IIs are expressed at wild type levels as detected by immunoblot analysis, these findings rule out both haploinsufficiency and interference of the mutant myosin with the normal II-A (dominant negative) as mechanisms for the abnormalities found. One possibility with respect to a causative mechanism is an abnormality in filament formation or some similar defect that would be independent of the location of the mutation, but this still requires further work. Of particular note, Zhang et al. also show that conditional ablation of NM II-A exclusively in kidney podocytes results in glomerulosclerosis similar to that found in the mouse models of MYH9-RD.
A potential difference between MYH9-RD in humans and mice is that a number of authors (see Pecci et al.68,74 and references therein) report that in humans the defects in the globular head region of the molecule result in more severe clinical phenotypes than those in the tail domain and that this is particularly true for the glomerulosclerosis. This is in contrast to what is found in the mouse models where the two mutations in the coiled coil region result in somewhat more severe defects than the mutation in the globular head.17 However with respect to humans the conclusion reached by Savoia et al. is still relevant: “Since a definitive prediction of clinical course is not possible from our present state of knowledge it is necessary to regularly follow-up MYH9-RD patients for the development of nephritis, hearing impairment, and cataract formation.”75
In an in vitro study, Chen et al. confirmed that fewer proplatelets are formed in human patients with MYH9-RD compared with controls. Using traction force microscopy the authors demonstrated an elevated level of contractile force in adherent, mutated megakaryocytes. Interestingly the NM II inhibitor blebbistatin as well as the Rho kinase inhibitor Y27632 rescued the proplatelet formation defect and normalized the ultra-structural properties of the MYH-RD megakaryocytes.76 This is consistent with previous studies showing that ablation of NM II-A or treatment with blebbistatin increases proplatelet production in cultured megakaryocytes.77-79 Because the mutations studied in the MYH9-RD patients are in both the NM II head and rod, the mechanism may be through stress fiber stabilization or through an effect on the folded-unfolded myosin molecule equilibrium.
An indication of the ubiquitous location and function of NM II-A in disease processes is illustrated in a recent publication showing that NM II-A plays an important role in contributing to the efficiency of viral infection causing Severe Fever with Thrombocytopenia Syndrome (SFTS).80 Most patients infected with the virus reside in China, Japan, and South Korea and the clinical signs include fever, thrombocytopenia, and leukopenia accompanied by diarrhea and abdominal pain. NM II-A appears to play a critical role in the cellular entry of the SFTS virus into cells by binding to Gn, the envelope glycoprotein. siRNA knockdown of NM II-A but not II-B or II-C reduces SFTS viral infection, and antibodies to NM II-A block infection by the SFTS virus but not by other control viruses. Thus NM II-A appears to play a significant role in a wide variety of human diseases.
NM II-B and disease
In contrast to both NM II-A and II-C, no human diseases have been directly linked to mutations in NM II-B until recently. As noted above mouse models of NM II-B mutation and ablation show defects in the heart and nervous system including the brain. These abnormalities are consistent with the fact that NM II-B is the most prominent isoform of NM II expressed in cardiac myocytes and the nervous system.
Tuzovic et al.81 used whole exomic sequence analysis to search for a possible genetic defect in an 8 y old male who was born after intrauterine growth restriction and who showed evidence of microcephaly, developmental delay, hydrocephalus, cerebral and cerebellar atrophy and a congenital diaphragmatic hernia. Whole exomic sequencing identified a novel heterozygous mutation, E908X in MYH10, the gene encoding NMHC II-B. This mutation is present in the coiled–coil rod region of the myosin molecule and if expressed would be expected to disrupt filament formation. In addition a de novo mutation (T1162M), also in the rod region, was identified in a second proband. This child, who died in infancy was also found to have a congenital diaphragmatic hernia (personal communication, Wendy Chung, Dept. of Pediatrics, Columbia University Medical Center).
In an effort to produce a mouse model of a human NM II-B disease, Ma and Adelstein43 generated mice with a single mutation in the globular head region of NMHC II-B. The selection of an amino acid to mutate (R709C) was governed by searching for a residue that was conserved in all 3 isoforms, as well as across species (Fig. 3). Moreover, mutations in homologous amino acids are known to cause human syndromes in the case of NM II-A (MYH9-RD) and NM II-C (deafness). The resultant homozygous mice died by E14.5 with a phenotype that resembles the human syndrome Pentalogy of Cantrell (POC).82,83 POC is a rare syndrome that includes: 1-a deficiency of the anterior diaphragm, 2-a midline supraumbilical abdominal wall defect, resulting in the heart presentation outside of the thoracic wall, 3-a defect in the diaphragmatic pericardium, 4- intracardiac abnormalities including ventricular septal defect (VSD) and double outlet of the right ventricle (DORV), and 5-a defect in the lower sternum. The mice generated phenocopy all of the above defects including a VSD, DORV, and ectopia cordis.43 Similar to the human mutations in NMHC II-B81 the mice also suffer from a congenital diaphragmatic hernia. In an effort to identify a possible genetic cause for POC in humans and in collaboration with the University of Washington Center for Mendelian Genomics, we are presently carrying out whole exomic sequencing of 11 families with the diagnosis of POC in order to identify a possible causative gene. Of note, with respect to the mouse model of POC, is the presence of abnormalities due to both loss of function such as the VSD and DORV which are present in NM II-B knockout mice, and gain of function defects such as an abnormal ventral wall closure, diaphragmatic herniation and an omphalocele. These latter defects were not seen in the knockout mice.
NM II-C and disease
NM II-C and deafness
Similar to MYH9, mutations in MYH14, the gene encoding NMHC II-C result in sensorineural hearing impairment. Donaudy et al.84 report 4 different mutations (Fig. 6) in MYH14 (S7X, R726S, and L976F) in large pedigrees, as well as one de novo mutation (G376C). All of these mutations result in an autosomal dominant hearing loss. The authors show that in mice NM II-C is localized to all cells of the scala media wall, with the exception of Reissner’s membrane, with significant expression in the organ of Corti and the stria vascularis. This is in contrast to NM II-A which immunolocalizes to the outer hair cells of the organ of Corti, the spiral ligament, and Reissner’s membrane.85 Choi et al.86 describe an autosomal dominant phenotype of peripheral neuropathy, myopathy, hoarseness and hearing loss in a large Korean family with a mutation in MYH14. If confirmed, this could significantly expand the phenotypic range of the gene.
Figure 6.

NM II Mutation and Disease. MYH9-RD mutation list is updated from a previous review.3 *Deletion of two different nucleotides causes D1925fs; del, deletion; dup, duplication; fs, frame shift. Clinical features other than macrothrombocytopenia and Dohle body inclusion are marked: d, deafness; n, nephritis; c, cataracts. Mutations in MYH14 result in deafness. Mutations in MYH10 are associated with congenital diaphragmatic hernia.
NM II-C and Cancer
In a number of tumor cell lines a late step in cytokinesis, abscission, has been shown to require NM II-C and particularly, a specific alternatively spliced isoform of II-C, NM II-C1. As noted above in the Introduction, alternative splicing of the pre-mRNA introduces an 8 amino acid insert near the MgATP-binding region of NMHC II-C0.2 This spliced isoform appears to be the most widely expressed NM II-C isoform in mice and humans. Jana et al.87 used the A549 human lung tumor cell line to demonstrate that NM II-C1, but not NM II-A, which is also present in these cells in a significant amount, localizes to the midbody during abscission. Moreover decreasing the quantity of NM II-C1 in A549 cells, using NMHC II isoform-specific siRNAs inhibits cell proliferation by 5.5-fold in 120 h, by prolonging cytokinesis from 2 to 9 h. This defect in abscission is rescued by transfecting the cells with a plasmid expressing NM II-C1 but not plasmids expressing NM II-A or NM II-B. The NM II-C0 isoform which lacks the C1 insert is able to effect a partial rescue.
Recently the role of NM II-C1 in abscission in tumor cell lines has been expanded and more completely defined. The tumor suppressor BRCA2 functions in DNA repair and similar to NM II-C1 has also been suggested to regulate cytokinesis. It has been shown that this well-known cancer associated protein recruits regulators of abscission to the midbody.88,89 These studies propose that BRCA2 acts as a scaffold to assemble part of the cytokinesis machinery at the midbody. Of note is the ability of a BRCA2 deletion mutant that lacks the DNA repair function, to rescue cytokinesis defects caused by loss of BRCA2. In a related recent publication Takaoka et al.90 demonstrate that following phosphorylation by the mitotic kinase Polo-like kinase 1 (PLK1), BRCA2 localizes to the Flemming body at the center of the midbody where it interacts with NM II-C1 to form a discrete ring-like structure. The interaction of BRCA2 and associated proteins (which include Rho kinase) with NM II-C1 results in phosphorylation of the 20 kDa myosin light chain and thereby activates NM II MgATPase activity. Inhibiting the interaction between NM II-C1 and BRCA2 decreases NM II-C1 MgATPase activity and results in a defective ring and midbody, and interrupts cytokinesis. Analysis of cancer associated mutations in BRCA2 at the PLK1 binding site suggests that they may contribute to cytokinetic defects by altering BRCA2 localization. The authors suggest that BRCA2 dependent NM II-C1 ring formation is a critical step in midbody formation. A defect in this process could explain how chromosome instability arises in breast cancer.
All three isoforms of NM II have been implicated in disease processes in humans (summarized in Fig. 6).3,75,91-95 To date two of the isoforms, NM II-A and II-C have been implicated in cancer. Although two patients with mutations in NM II-B have been identified, the genetic link between NM II-B and cardiac defects, which has been found in mice has yet to be shown in humans. The wide-spread use of whole exomic sequencing, gene manipulation and new developments in next generation sequencing should prove to be productive tools in these areas in the next few years.
Conclusions
NM II plays numerous and diverse roles during vertebrate development. Table 1 summarizes the phenotypes observed in mice when NM II is genetically ablated or mutated. Here we focus on the three different isoforms of NM II and those functions that appear to be isoform specific and those in which one or both isoforms can replace the endogenous isoform. We conclude that those functions which involve the globular motor domain of NM II are less likely to be replaceable leading to restoration of normal function than those that involve the filament forming domain. The kinetic properties and duty ratio of the individual isoforms are critical functions not easily replaced. Studies of chimeras of NM II isoforms help to pinpoint the necessity for the globular head domain of NM II-A for normal formation of the placenta vasculature. Point mutations in the NM II motor (e.g., R709C) can be used to alter the duty ratio and kinetics of NM II-B thus separating the process of translocation of actin filaments from tension during cytokinesis.
Table 1. Abnormalities in NM II ablated or mutated mice.
| Mouse Models | Lethality | Placenta | Heart | Brain | Others |
|---|---|---|---|---|---|
| A-/A- (ref. 14) | E6.5 | Disorganized visceral endoderm | |||
| AK5/AK5 (ref. 26) (Keratin 5-Cre) |
E9.5 to E13.5 | Reduced labyrinth vascularization | Hyperproliferative epidermis | ||
| APod/APod (ref. 96) (Podocin-Cre) |
Adult | Focal segmental glomerulosclerosis | |||
| APf4/APf4 (ref. 79) (Platelet Factor 4-Cre) |
Adult | Macrothrombocytopenia | |||
| B-/B- (refs. 19, 27, 35, 38) | E14.5 | No placenta defects | Reduced compact myocardium, Defective cardiac myocyte cytokinesis, VSD, DORV | Hydrocephalus, Defects in neuronal migration, Disrupted neuroepithelial cell-cell adhesion | |
| BαMHC/BαMHC (ref. 40) (αMHC-Cre) |
Adult | Cardiomyopathy, Disruption of intercalated discs | |||
| BNest/BNest (ref. 40) (Nestin-Cre) |
P14 | Hydrocephalus, Defects in neuronal migration, Disrupted neuroepithelial cell-cell adhesion | |||
| BDdx4/BDdx4 (ref. 97) (Germ Cell Ddx4-Cre) |
Adult | Male germ cell cytokinesis defects | |||
| C-/C- (ref. 36) | Adult | No obvious defects | |||
| B-C-/B-C- (ref. 36) | E14.5 | Reduced compact myocardium, Defective cardiac myocyte karyokinesis, VSD, DORV | Hydrocephalus, Defects in neuronal migration, Disrupted neuroepithelial cell-cell adhesion | ||
| Ba*/Ba* (ref. 20) (NM II-A replacing for II-B) |
E16.5 | Defective cardiac myocyte cytokinesis, VSD, DORV, (milder than B-/B- hearts) | Defects in neuronal migration, No hydrocephalus | ||
| Ab*/Ab* (ref. 15) (NM II-B replacing for II-A) |
E9.5 to E10.5 | Reduced labyrinth vascularization | Normal visceral endoderm | ||
| Aba/Aba (ref. 15) (NM II-B head/II-A rod chimera replacing for II-A) |
E9.5 to E10.5 | Reduced labyrinth vascularization | Normal visceral endoderm | ||
| Aab/Aab (ref. 15) (NM II-A-head/II-B-rod chimera replacing for II-A) |
E10.5 to E11.5 | Reduced labyrinth vascularization | Normal visceral endoderm | ||
| A+/AR709C (refs. 17 and 73) | Adult | Macrothrombocytopenia, Cataract, Focal segmental glomerulosclerosis | |||
| AR709C/AR709C (ref. 17) | E10.5 to E11.5 | Reduced labyrinth vascularization | Normal visceral endoderm | ||
| A+/AD1424N (ref. 17) AD1424N/AD1424N (ref. 17) | Adult | Macrothrombocytopenia, Cataract, Focal segmental glomerulosclerosis | |||
| A+/AE1841K (ref. 17) AE1841K/AE1841K (ref. 17) |
Adult | Macrothrombocytopenia, Cataract, Focal segmental glomerulosclerosis | |||
| B+/BR709C (ref. 43) | Adult | Diaphragmatic and umbilical hernia | |||
| BR709C/BR709C (ref. 43) | E14.5 to E16.5 | Reduced compact myocardium,ASD, VSD, DORV, No defects in cytokinesis | Defects in neuronal migration, No hydrocephalus | Major midline fusion defects: cleft palate, split lower sternum, large omphalocele |
The role of NM II mutations and ablation in human disease is an emerging and exciting area of research, particularly when coupled with the use of RNAi screens and next generation genomic sequencing. At least two of the isoforms, NM II-A and NM II-C1 have been implicated as possible tumor suppressors. Mouse models of the NM II-A mutations already described in humans should make it possible to study the pathogenesis of the various abnormalities associated with MYH9-RD at the cellular level and coupled with stem cell technology to pursue models for therapeutic purposes. Clearly we are standing on the threshold of a new and productive age of research into the function of NM IIs in development and disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Mary Anne Conti for her many significant contributions to this manuscript and Dr. Sarah M. Heissler (Laboratory of Molecular Physiology, NMLBI) for the model of NMII-A motor domain in Figure 3. Dr. Sachiyo Kawamoto, Dr. Elena Grant, and members of the Laboratory of Molecular Cardiology also provided critical comments on the manuscript. This research was supported by the Division of Intramural Research, NHLBI.
Glossary
Abbreviations:
- ASD
Atrial Septal Defect
- CGN
Cerebellar Granule Neuron
- DORV
Double Outlet of Right Ventricle
- E
Embryonic Day
- HMM
Heavy Meromyosin
- MGE
Medial Ganglionic Eminence
- MYH9-RD
MYH9-Related Disease
- NM II
Nonmuscle Myosin II
- NMHC
Nonmuscle Myosin Heavy Chain
- OFT
Outflow Tract
- POC
Pentalogy of Cantrell
- SCC
Squamous Cell Carcinoma
- SFTS
Severe Fever with Thrombocytopenia Syndrome
- SVZ
Subventricular Zone
- VSD
Ventricular Septal Defect
References
- 1.Walker A, Su H, Conti MA, Harb N, Adelstein RS, Sato N. Non-muscle myosin II regulates survival threshold of pluripotent stem cells. Nat Commun. 2010;1:71. doi: 10.1038/ncomms1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Golomb E, Ma X, Jana SS, Preston YA, Kawamoto S, Shoham NG, Goldin E, Conti MA, Sellers JR, Adelstein RS. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J Biol Chem. 2004;279:2800–8. doi: 10.1074/jbc.M309981200. [DOI] [PubMed] [Google Scholar]
- 3.Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–90. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Heissler SM, Manstein DJ. Nonmuscle myosin-2: mix and match. Cell Mol Life Sci. 2013;70:1–21. doi: 10.1007/s00018-012-1002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Billington N, Wang A, Mao J, Adelstein RS, Sellers JR. Characterization of three full-length human nonmuscle myosin II paralogs. J Biol Chem. 2013;288:33398–410. doi: 10.1074/jbc.M113.499848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jana SS, Kim KY, Mao J, Kawamoto S, Sellers JR, Adelstein RS. An alternatively spliced isoform of non-muscle myosin II-C is not regulated by myosin light chain phosphorylation. J Biol Chem. 2009;284:11563–71. doi: 10.1074/jbc.M806574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim KY, Kawamoto S, Bao J, Sellers JR, Adelstein RS. The B2 alternatively spliced isoform of nonmuscle myosin II-B lacks actin-activated MgATPase activity and in vitro motility. Biochem Biophys Res Commun. 2008;369:124–34. doi: 10.1016/j.bbrc.2007.11.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim KY, Kovács M, Kawamoto S, Sellers JR, Adelstein RS. Disease-associated mutations and alternative splicing alter the enzymatic and motile activity of nonmuscle myosins II-B and II-C. J Biol Chem. 2005;280:22769–75. doi: 10.1074/jbc.M503488200. [DOI] [PubMed] [Google Scholar]
- 9.Pato MD, Sellers JR, Preston YA, Harvey EV, Adelstein RS. Baculovirus expression of chicken nonmuscle heavy meromyosin II-B. Characterization of alternatively spliced isoforms. J Biol Chem. 1996;271:2689–95. doi: 10.1074/jbc.271.5.2689. [DOI] [PubMed] [Google Scholar]
- 10.Li Y, Lalwani AK, Mhatre AN. Alternative splice variants of MYH9. DNA Cell Biol. 2008;27:117–25. doi: 10.1089/dna.2007.0661. [DOI] [PubMed] [Google Scholar]
- 11.Heissler SM, Manstein DJ. Comparative kinetic and functional characterization of the motor domains of human nonmuscle myosin-2C isoforms. J Biol Chem. 2011;286:21191–202. doi: 10.1074/jbc.M110.212290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma X, Kawamoto S, Uribe J, Adelstein RS. Function of the neuron-specific alternatively spliced isoforms of nonmuscle myosin II-B during mouse brain development. Mol Biol Cell. 2006;17:2138–49. doi: 10.1091/mbc.E05-10-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansfield SG, al-Shirawi DY, Ketchum AS, Newbern EC, Kiehart DP. Molecular organization and alternative splicing in zipper, the gene that encodes the Drosophila non-muscle myosin II heavy chain. J Mol Biol. 1996;255:98–109. doi: 10.1006/jmbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 14.Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J Biol Chem. 2004;279:41263–6. doi: 10.1074/jbc.C400352200. [DOI] [PubMed] [Google Scholar]
- 15.Wang A, Ma X, Conti MA, Liu C, Kawamoto S, Adelstein RS. Nonmuscle myosin II isoform and domain specificity during early mouse development. Proc Natl Acad Sci U S A. 2010;107:14645–50. doi: 10.1073/pnas.1004023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu A, Wang F, Sellers JR. Mutations in human nonmuscle myosin IIA found in patients with May-Hegglin anomaly and Fechtner syndrome result in impaired enzymatic function. J Biol Chem. 2002;277:46512–7. doi: 10.1074/jbc.M208506200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Conti MA, Malide D, Dong F, Wang A, Shmist YA, Liu C, Zerfas P, Daniels MP, Chan CC, et al. Mouse models of MYH9-related disease: mutations in nonmuscle myosin II-A. Blood. 2012;119:238–50. doi: 10.1182/blood-2011-06-358853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X, Bao J, Adelstein RS. Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II-B-ablated and mutated mice. Mol Biol Cell. 2007;18:2305–12. doi: 10.1091/mbc.E07-01-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tullio AN, Bridgman PC, Tresser NJ, Chan CC, Conti MA, Adelstein RS, Hara Y. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J Comp Neurol. 2001;433:62–74. doi: 10.1002/cne.1125. [DOI] [PubMed] [Google Scholar]
- 20.Bao J, Ma X, Liu C, Adelstein RS. Replacement of nonmuscle myosin II-B with II-A rescues brain but not cardiac defects in mice. J Biol Chem. 2007;282:22102–11. doi: 10.1074/jbc.M702731200. [DOI] [PubMed] [Google Scholar]
- 21.Ebrahim S, Fujita T, Millis BA, Kozin E, Ma X, Kawamoto S, Baird MA, Davidson M, Yonemura S, Hisa Y, et al. NMII forms a contractile transcellular sarcomeric network to regulate apical cell junctions and tissue geometry. Curr Biol. 2013;23:731–6. doi: 10.1016/j.cub.2013.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ivanov AI, Hunt D, Utech M, Nusrat A, Parkos CA. Differential roles for actin polymerization and a myosin II motor in assembly of the epithelial apical junctional complex. Mol Biol Cell. 2005;16:2636–50. doi: 10.1091/mbc.E05-01-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smutny M, Cox HL, Leerberg JM, Kovacs EM, Conti MA, Ferguson C, Hamilton NA, Parton RG, Adelstein RS, Yap AS. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 2010;12:696–702. doi: 10.1038/ncb2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shewan AM, Maddugoda M, Kraemer A, Stehbens SJ, Verma S, Kovacs EM, Yap AS. Myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell-cell contacts. Mol Biol Cell. 2005;16:4531–42. doi: 10.1091/mbc.E05-04-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ivanov AI, Bachar M, Babbin BA, Adelstein RS, Nusrat A, Parkos CA. A unique role for nonmuscle myosin heavy chain IIA in regulation of epithelial apical junctions. PLoS One. 2007;2:e658. doi: 10.1371/journal.pone.0000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crish J, Conti MA, Sakai T, Adelstein RS, Egelhoff TT. Keratin 5-Cre-driven excision of nonmuscle myosin IIA in early embryo trophectoderm leads to placenta defects and embryonic lethality. Dev Biol. 2013;382:136–48. doi: 10.1016/j.ydbio.2013.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma X, Kawamoto S, Hara Y, Adelstein RS. A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol Biol Cell. 2004;15:2568–79. doi: 10.1091/mbc.E03-11-0836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schaar BT, McConnell SK. Cytoskeletal coordination during neuronal migration. Proc Natl Acad Sci U S A. 2005;102:13652–7. doi: 10.1073/pnas.0506008102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinohara R, Thumkeo D, Kamijo H, Kaneko N, Sawamoto K, Watanabe K, Takebayashi H, Kiyonari H, Ishizaki T, Furuyashiki T, et al. A role for mDia, a Rho-regulated actin nucleator, in tangential migration of interneuron precursors. Nat Neurosci. 2012;15:373–80, S1-2. doi: 10.1038/nn.3020. [DOI] [PubMed] [Google Scholar]
- 30.De La Cruz EM, Ostap EM. Relating biochemistry and function in the myosin superfamily. Curr Opin Cell Biol. 2004;16:61–7. doi: 10.1016/j.ceb.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Kovács M, Thirumurugan K, Knight PJ, Sellers JR. Load-dependent mechanism of nonmuscle myosin 2. Proc Natl Acad Sci U S A. 2007;104:9994–9. doi: 10.1073/pnas.0701181104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang F, Kovacs M, Hu A, Limouze J, Harvey EV, Sellers JR. Kinetic mechanism of non-muscle myosin IIB: functional adaptations for tension generation and maintenance. J Biol Chem. 2003;278:27439–48. doi: 10.1074/jbc.M302510200. [DOI] [PubMed] [Google Scholar]
- 33.Rosenfeld SS, Xing J, Chen LQ, Sweeney HL. Myosin IIb is unconventionally conventional. J Biol Chem. 2003;278:27449–55. doi: 10.1074/jbc.M302555200. [DOI] [PubMed] [Google Scholar]
- 34.Ma X, Adelstein RS. In vivo studies on nonmuscle myosin II expression and function in heart development. Front Biosci (Landmark Ed) 2012;17:545–55. doi: 10.2741/3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda K, Kishi H, Ma X, Yu ZX, Adelstein RS. Ablation and mutation of nonmuscle myosin heavy chain II-B results in a defect in cardiac myocyte cytokinesis. Circ Res. 2003;93:330–7. doi: 10.1161/01.RES.0000089256.00309.CB. [DOI] [PubMed] [Google Scholar]
- 36.Ma X, Jana SS, Conti MA, Kawamoto S, Claycomb WC, Adelstein RS. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol Biol Cell. 2010;21:3952–62. doi: 10.1091/mbc.E10-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma X, Kovács M, Conti MA, Wang A, Zhang Y, Sellers JR, Adelstein RS. Nonmuscle myosin II exerts tension but does not translocate actin in vertebrate cytokinesis. Proc Natl Acad Sci U S A. 2012;109:4509–14. doi: 10.1073/pnas.1116268109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, Westphal H, Preston YA, Adelstein RS. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci U S A. 1997;94:12407–12. doi: 10.1073/pnas.94.23.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gustafson-Wagner EA, Sinn HW, Chen YL, Wang DZ, Reiter RS, Lin JL, Yang B, Williamson RA, Chen J, Lin CI, et al. Loss of mXinalpha, an intercalated disk protein, results in cardiac hypertrophy and cardiomyopathy with conduction defects. Am J Physiol Heart Circ Physiol. 2007;293:H2680–92. doi: 10.1152/ajpheart.00806.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma X, Takeda K, Singh A, Yu ZX, Zerfas P, Blount A, Liu C, Towbin JA, Schneider MD, Adelstein RS, et al. Conditional ablation of nonmuscle myosin II-B delineates heart defects in adult mice. Circ Res. 2009;105:1102–9. doi: 10.1161/CIRCRESAHA.109.200303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Even-Ram S, Doyle AD, Conti MA, Matsumoto K, Adelstein RS, Yamada KM. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat Cell Biol. 2007;9:299–309. doi: 10.1038/ncb1540. [DOI] [PubMed] [Google Scholar]
- 42.van den Hoff MJ, Moorman AF, Ruijter JM, Lamers WH, Bennington RW, Markwald RR, Wessels A. Myocardialization of the cardiac outflow tract. Dev Biol. 1999;212:477–90. doi: 10.1006/dbio.1999.9366. [DOI] [PubMed] [Google Scholar]
- 43.Ma X, Adelstein RS. A point mutation in myh10 causes major defects in heart development and body wall closure. Circ Cardiovasc Genet. 2014;7:257–65. doi: 10.1161/CIRCGENETICS.113.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips HM, Murdoch JN, Chaudhry B, Copp AJ, Henderson DJ. Vangl2 acts via RhoA signaling to regulate polarized cell movements during development of the proximal outflow tract. Circ Res. 2005;96:292–9. doi: 10.1161/01.RES.0000154912.08695.88. [DOI] [PubMed] [Google Scholar]
- 45.Ivanov AI, McCall IC, Parkos CA, Nusrat A. Role for actin filament turnover and a myosin II motor in cytoskeleton-driven disassembly of the epithelial apical junctional complex. Mol Biol Cell. 2004;15:2639–51. doi: 10.1091/mbc.E04-02-0163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee HY, Kosciuk MC, Nagele RG, Roisen FJ. Studies on the mechanisms of neurulation in the chick: possible involvement of myosin in elevation of neural folds. J Exp Zool. 1983;225:449–57. doi: 10.1002/jez.1402250313. [DOI] [PubMed] [Google Scholar]
- 47.Kinoshita N, Sasai N, Misaki K, Yonemura S. Apical accumulation of Rho in the neural plate is important for neural plate cell shape change and neural tube formation. Mol Biol Cell. 2008;19:2289–99. doi: 10.1091/mbc.E07-12-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rolo A, Skoglund P, Keller R. Morphogenetic movements driving neural tube closure in Xenopus require myosin IIB. Dev Biol. 2009;327:327–38. doi: 10.1016/j.ydbio.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishimura T, Honda H, Takeichi M. Planar cell polarity links axes of spatial dynamics in neural-tube closure. Cell. 2012;149:1084–97. doi: 10.1016/j.cell.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 50.Roh-Johnson M, Shemer G, Higgins CD, McClellan JH, Werts AD, Tulu US, Gao L, Betzig E, Kiehart DP, Goldstein B. Triggering a cell shape change by exploiting preexisting actomyosin contractions. Science. 2012;335:1232–5. doi: 10.1126/science.1217869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hildebrand JD. Shroom regulates epithelial cell shape via the apical positioning of an actomyosin network. J Cell Sci. 2005;118:5191–203. doi: 10.1242/jcs.02626. [DOI] [PubMed] [Google Scholar]
- 52.Simões SdeM, Mainieri A, Zallen JA. Rho GTPase and Shroom direct planar polarized actomyosin contractility during convergent extension. J Cell Biol. 2014;204:575–89. doi: 10.1083/jcb.201307070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shimizu Y, Thumkeo D, Keel J, Ishizaki T, Oshima H, Oshima M, Noda Y, Matsumura F, Taketo MM, Narumiya S. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol. 2005;168:941–53. doi: 10.1083/jcb.200411179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suzuki M, Morita H, Ueno N. Molecular mechanisms of cell shape changes that contribute to vertebrate neural tube closure. Dev Growth Differ. 2012;54:266–76. doi: 10.1111/j.1440-169X.2012.01346.x. [DOI] [PubMed] [Google Scholar]
- 55.Bellion A, Baudoin JP, Alvarez C, Bornens M, Métin C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J Neurosci. 2005;25:5691–9. doi: 10.1523/JNEUROSCI.1030-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Solecki DJ, Trivedi N, Govek EE, Kerekes RA, Gleason SS, Hatten ME. Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron. 2009;63:63–80. doi: 10.1016/j.neuron.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He M, Zhang ZH, Guan CB, Xia D, Yuan XB. Leading tip drives soma translocation via forward F-actin flow during neuronal migration. J Neurosci. 2010;30:10885–98. doi: 10.1523/JNEUROSCI.0240-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schenk J, Wilsch-Bräuninger M, Calegari F, Huttner WB. Myosin II is required for interkinetic nuclear migration of neural progenitors. Proc Natl Acad Sci U S A. 2009;106:16487–92. doi: 10.1073/pnas.0908928106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Das RM, Storey KG. Apical abscission alters cell polarity and dismantles the primary cilium during neurogenesis. Science. 2014;343:200–4. doi: 10.1126/science.1247521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Young PE, Richman AM, Ketchum AS, Kiehart DP. Morphogenesis in Drosophila requires nonmuscle myosin heavy chain function. Genes Dev. 1993;7:29–41. doi: 10.1101/gad.7.1.29. [DOI] [PubMed] [Google Scholar]
- 61.Kiehart DP, Galbraith CG, Edwards KA, Rickoll WL, Montague RA. Multiple forces contribute to cell sheet morphogenesis for dorsal closure in Drosophila. J Cell Biol. 2000;149:471–90. doi: 10.1083/jcb.149.2.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franke JD, Montague RA, Kiehart DP. Nonmuscle myosin II generates forces that transmit tension and drive contraction in multiple tissues during dorsal closure. Curr Biol. 2005;15:2208–21. doi: 10.1016/j.cub.2005.11.064. [DOI] [PubMed] [Google Scholar]
- 63.Solon J, Kaya-Copur A, Colombelli J, Brunner D. Pulsed forces timed by a ratchet-like mechanism drive directed tissue movement during dorsal closure. Cell. 2009;137:1331–42. doi: 10.1016/j.cell.2009.03.050. [DOI] [PubMed] [Google Scholar]
- 64.Heller E, Kumar KV, Grill SW, Fuchs E. Forces generated by cell intercalation tow epidermal sheets in mammalian tissue morphogenesis. Dev Cell. 2014;28:617–32. doi: 10.1016/j.devcel.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bao J, Jana SS, Adelstein RS. Vertebrate nonmuscle myosin II isoforms rescue small interfering RNA-induced defects in COS-7 cell cytokinesis. J Biol Chem. 2005;280:19594–9. doi: 10.1074/jbc.M501573200. [DOI] [PubMed] [Google Scholar]
- 66.Michor F, Iwasa Y, Vogelstein B, Lengauer C, Nowak MA. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol. 2005;15:43–9. doi: 10.1016/j.semcancer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 67.Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, Reva B, Fuchs E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014;343:309–13. doi: 10.1126/science.1248627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pecci A, Klersy C, Gresele P, Lee KJ, De Rocco D, Bozzi V, Russo G, Heller PG, Loffredo G, Ballmaier M, et al. MYH9-related disease: a novel prognostic model to predict the clinical evolution of the disease based on genotype-phenotype correlations. Hum Mutat. 2014;35:236–47. doi: 10.1002/humu.22476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shin JW, Buxboim A, Spinler KR, Swift J, Christian DA, Hunter CA, Léon C, Gachet C, Dingal PC, Ivanovska IL, et al. Contractile forces sustain and polarize hematopoiesis from stem and progenitor cells. Cell Stem Cell. 2014;14:81–93. doi: 10.1016/j.stem.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dulyaninova NG, Bresnick AR. The heavy chain has its day: regulation of myosin-II assembly. Bioarchitecture. 2013;3:77–85. doi: 10.4161/bioa.26133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stein U, Arlt F, Walther W, Smith J, Waldman T, Harris ED, Mertins SD, Heizmann CW, Allard D, Birchmeier W, et al. The metastasis-associated gene S100A4 is a novel target of beta-catenin/T-cell factor signaling in colon cancer. Gastroenterology. 2006;131:1486–500. doi: 10.1053/j.gastro.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 72.Dahan I, Petrov D, Cohen-Kfir E, Ravid S. The tumor suppressor Lgl1 forms discrete complexes with NMII-A and Par6α-aPKCζ that are affected by Lgl1 phosphorylation. J Cell Sci. 2014;127:295–304. doi: 10.1242/jcs.127357. [DOI] [PubMed] [Google Scholar]
- 73.Suzuki N, Kunishima S, Ikejiri M, Maruyama S, Sone M, Takagi A, Ikawa M, Okabe M, Kojima T, Saito H, et al. Establishment of mouse model of MYH9 disorders: heterozygous R702C mutation provokes macrothrombocytopenia with leukocyte inclusion bodies, renal glomerulosclerosis and hearing disability. PLoS One. 2013;8:e71187. doi: 10.1371/journal.pone.0071187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pecci A, Panza E, Pujol-Moix N, Klersy C, Di Bari F, Bozzi V, Gresele P, Lethagen S, Fabris F, Dufour C, et al. Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9-related disease. Hum Mutat. 2008;29:409–17. doi: 10.1002/humu.20661. [DOI] [PubMed] [Google Scholar]
- 75.Savoia A, Germeshausen M, De Rocco D, Henschel B, Kratz CP, Kuhlen M, Rath B, Steuhl KP, Wermes C, Ballmaier M. MYH9-related disease: Report on five German families and description of a novel mutation. Ann Hematol. 2010;89:1057–9. doi: 10.1007/s00277-010-0928-y. [DOI] [PubMed] [Google Scholar]
- 76.Chen Y, Boukour S, Milloud R, Favier R, Saposnik B, Schlegel N, Nurden A, Raslova H, Vainchenker W, Balland M, et al. The abnormal proplatelet formation in MYH9-related macrothrombocytopenia results from an increased actomyosin contractility and is rescued by myosin IIA inhibition. J Thromb Haemost. 2013;11:2163–75. doi: 10.1111/jth.12436. [DOI] [PubMed] [Google Scholar]
- 77.Chang Y, Auradé F, Larbret F, Zhang Y, Le Couedic JP, Momeux L, Larghero J, Bertoglio J, Louache F, Cramer E, et al. Proplatelet formation is regulated by the Rho/ROCK pathway. Blood. 2007;109:4229–36. doi: 10.1182/blood-2006-04-020024. [DOI] [PubMed] [Google Scholar]
- 78.Chen Z, Naveiras O, Balduini A, Mammoto A, Conti MA, Adelstein RS, Ingber D, Daley GQ, Shivdasani RA. The May-Hegglin anomaly gene MYH9 is a negative regulator of platelet biogenesis modulated by the Rho-ROCK pathway. Blood. 2007;110:171–9. doi: 10.1182/blood-2007-02-071589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eckly A, Rinckel JY, Laeuffer P, Cazenave JP, Lanza F, Gachet C, Léon C. Proplatelet formation deficit and megakaryocyte death contribute to thrombocytopenia in Myh9 knockout mice. J Thromb Haemost. 2010;8:2243–51. doi: 10.1111/j.1538-7836.2010.04009.x. [DOI] [PubMed] [Google Scholar]
- 80.Sun Y, Qi Y, Liu C, Gao W, Chen P, Fu L, Peng B, Wang H, Jing Z, Zhong G, et al. Nonmuscle myosin heavy chain IIA is a critical factor contributing to the efficiency of early infection of severe fever with thrombocytopenia syndrome virus. J Virol. 2014;88:237–48. doi: 10.1128/JVI.02141-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tuzovic LY, Zeng W, Li X, Lu H, Gonzalez K, Chung WK. A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice. Rare Dis. 2013;1:e26144-1–e26144-5. doi: 10.4161/rdis.26144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Engum SA. Embryology, sternal clefts, ectopia cordis, and Cantrell’s pentalogy. Semin Pediatr Surg. 2008;17:154–60. doi: 10.1053/j.sempedsurg.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 83.Mallula KK, Sosnowski C, Awad S. Spectrum of Cantrell’s pentalogy: case series from a single tertiary care center and review of the literature. Pediatr Cardiol. 2013;34:1703–10. doi: 10.1007/s00246-013-0706-4. [DOI] [PubMed] [Google Scholar]
- 84.Donaudy F, Snoeckx R, Pfister M, Zenner HP, Blin N, Di Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4) Am J Hum Genet. 2004;74:770–6. doi: 10.1086/383285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lalwani AK, Goldstein JA, Kelley MJ, Luxford W, Castelein CM, Mhatre AN. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am J Hum Genet. 2000;67:1121–8. doi: 10.1086/321212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Choi BO, Kang SH, Hyun YS, Kanwal S, Park SW, Koo H, Kim SB, Choi YC, Yoo JH, Kim JW, et al. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14. Hum Mutat. 2011;32:669–77. doi: 10.1002/humu.21488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jana SS, Kawamoto S, Adelstein RS. A specific isoform of nonmuscle myosin II-C is required for cytokinesis in a tumor cell line. J Biol Chem. 2006;281:24662–70. doi: 10.1074/jbc.M604606200. [DOI] [PubMed] [Google Scholar]
- 88.Daniels MJ, Wang Y, Lee M, Venkitaraman AR. Abnormal cytokinesis in cells deficient in the breast cancer susceptibility protein BRCA2. Science. 2004;306:876–9. doi: 10.1126/science.1102574. [DOI] [PubMed] [Google Scholar]
- 89.Mondal G, Rowley M, Guidugli L, Wu J, Pankratz VS, Couch FJ. BRCA2 localization to the midbody by filamin A regulates cep55 signaling and completion of cytokinesis. Dev Cell. 2012;23:137–52. doi: 10.1016/j.devcel.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takaoka M, Saito H, Takenaka K, Miki Y, Nakanishi A. BRCA2 phosphorylated by PLK1 moves to the midbody to regulate cytokinesis mediated by nonmuscle myosin IIC. Cancer Res. 2014;74:1518–28. doi: 10.1158/0008-5472.CAN-13-0504. [DOI] [PubMed] [Google Scholar]
- 91.de Rocco D, Heller PG, Girotto G, Pastore A, Glembotsky AC, Marta RF, Bozzi V, Pecci A, Molinas FC, Savoia A. MYH9 related disease: a novel missense Ala95Asp mutation of the MYH9 gene. Platelets. 2009;20:598–602. doi: 10.3109/09537100903349620. [DOI] [PubMed] [Google Scholar]
- 92.De Rocco D, Zieger B, Platokouki H, Heller PG, Pastore A, Bottega R, Noris P, Barozzi S, Glembotsky AC, Pergantou H, et al. MYH9-related disease: five novel mutations expanding the spectrum of causative mutations and confirming genotype/phenotype correlations. Eur J Med Genet. 2013;56:7–12. doi: 10.1016/j.ejmg.2012.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pecci A, Panza E, De Rocco D, Pujol-Moix N, Girotto G, Podda L, Paparo C, Bozzi V, Pastore A, Balduini CL, et al. MYH9 related disease: four novel mutations of the tail domain of myosin-9 correlating with a mild clinical phenotype. Eur J Haematol. 2010;84:291–7. doi: 10.1111/j.1600-0609.2009.01398.x. [DOI] [PubMed] [Google Scholar]
- 94.Sun XH, Wang ZY, Yang HY, Cao LJ, Su J, Yu ZQ, Bai X, Ruan CG. Clinical, pathological, and genetic analysis of ten patients with MYH9-related disease. Acta Haematol. 2013;129:106–13. doi: 10.1159/000342123. [DOI] [PubMed] [Google Scholar]
- 95.Vettore S, De Rocco D, Gerber B, Scandellari R, Bianco AM, Balduini CL, Pecci A, Fabris F, Savoia A. A G to C transversion at the last nucleotide of exon 25 of the MYH9 gene results in a missense mutation rather than in a splicing defect. Eur J Med Genet. 2010;53:256–60. doi: 10.1016/j.ejmg.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 96.Léon C, Eckly A, Hechler B, Aleil B, Freund M, Ravanat C, Jourdain M, Nonne C, Weber J, Tiedt R, et al. Megakaryocyte-restricted MYH9 inactivation dramatically affects hemostasis while preserving platelet aggregation and secretion. Blood. 2007;110:3183–91. doi: 10.1182/blood-2007-03-080184. [DOI] [PubMed] [Google Scholar]
- 97.Yang F, Wei Q, Adelstein RS, Wang PJ. Non-muscle myosin IIB is essential for cytokinesis during male meiotic cell divisions. Dev Biol. 2012;369:356–61. doi: 10.1016/j.ydbio.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]