

Abstract
A minority of inherited prion diseases (IPD) are caused by four to 12 extra octapeptide repeat insertions (OPRI) in the prion protein gene (PRNP). Only four families affected by IPD with 8-OPRI have been reported, one of them was a three-generation Swedish kindred in which four of seven affected subjects had chorea which was initially attributed to Huntington’s disease (HD). Following the exclusion of HD, this phenotype was labeled Huntington disease-like 1 (HDL1). Here, we provide an update on the Swedish 8-OPRI family, describe the clinical features of one of its affected members with video-recordings, compare the four 8-OPRI families and study the effect of PRNP polymorphic codon 129 and gender on phenotype. Surprisingly, the Swedish kindred displayed the longest survival of all of the 8-OPRI families with a mean of 15.1 years from onset of symptoms. Subjects with PRNP polymorphic codon 129M in the mutated allele had significantly earlier age of onset, longer survival and earlier age of death than 129V subjects. Homozygous 129MM had earlier age of onset than 129VV. Females had a significantly earlier age of onset and earlier age of death than males. Up to 50% of variability in age of onset was conferred by the combined effect of PRNP polymorphic codon 129 and gender. An inverse correlation between early age of onset and long survival was found for this mutation.
Keywords: Huntington disease-like 1, Huntington’s disease, PRNP, PRNP polymorphic codon 129, base pair insertions (BPI), inherited prion disease (IPD), octapeptide repeat insertions (OPRI)
Genotype-phenotype analysis in inherited prion disease with 8 octapeptide repeat insertional mutation
Introduction
Inherited prion diseases (IPD) are a group of rare autosomal dominant, neurodegenerative and fatal diseases caused by mutations in the prion protein gene (PRNP). They constitute up to 15% of all prion diseases. The major IPD phenotypes include familial Creutzfeldt-Jakob disease (fCJD), fatal familial insomnia (FFI) and Gertsmann-Sträussler-Scheinker disease (GSS). Most IPD cases are caused by point mutations and premature stop codon mutations, but a minority of mutations are insertions of extra octapetide repeats (OPRI) in the N-terminal region of PRNP. Insertions of four to 12 OPRI, also known as base pair insertions (BPIs), lead to heterogeneous phenotypes.1-4 These phenotypes include progressive and varying degrees of early-onset cognitive decline/dementia; movement disorders, e.g., parkinsonism, chorea; ataxia; pyramidal symptoms; aphasia; seizures; and psychiatric symptoms/behavioral disturbances. The rarity of PRNP mutations with extra OPRI has limited understanding of genotype-phenotype correlations and the polymorphisms modulating disease expression.
Only four families affected by IPD with 8-OPRIs (192 bp) have been reported. Two of them were unrelated French families (M-E and Che families) displaying some features of GSS.5-8 The third family was a Dutch kindred (A family) displaying predominantly hypokinesia and dementia.9 The fourth family was a three generation Swedish kindred in which four of seven affected subjects had chorea, initially attributed to Huntington’s disease (HD). Following the exclusion of HD in this kindred10 and after identifying the PRNP gene mutation11 this phenotype was labeled Huntington disease-like 1 (HDL1).12 HDL1, which can be more accurately referred to as IPD with 8 OPRI, consists of rapidly progressive early-onset cortical dementia, various movement disorders, and psychiatric symptoms.10-12
The neuropathological features of IPD with 8-OPRI include varying degrees of spongiosis, cell loss, and astrocytosis in different areas of the brain,7,9,10 and the presence of prion protein (PrP) plaques in the cerebellum.8,13,14 Brain homogenates from one subject in each of the Che and M-E families were injected into monkeys.7,8 Only in the first case was transmission achieved7
Here we provide an update on the Swedish kindred affected by an IPD with 8-OPRI and describe the clinical features of one of its affected members with video recordings. Based on this update, we review and compare the features of four reported families with this mutation, and analyze the effect of PRNP polymorphic codon 129, a well-known susceptibility factor for prion diseases, on disease phenotype.
Results
Case presentation of one member of a Swedish kindred affected by IPD with 8-OPRI and update on the Swedish family
The subject presented here was born in 1959 and has been described previously as III:3.10 His mother was also affected by HDL1. The subject was adopted at the age of 3 and was in normal health until the age of 29. He then developed insidious personality changes, severe apathy, cognitive decline and bizarre behavior. He often talked incoherently to himself and displayed aggression toward inanimate objects. Three years later he developed dyspraxia, urinary incontinence, short-term memory impairment, and his speech was limited to short answers. Gait difficulties were evident although chorea was not documented. A CT-scan of the brain revealed general atrophy with particularly reduced volume in the temporal lobes. At the age of 34 he was admitted to a psychiatric ward due to his aggressive behavior. He had then developed severe dementia with expressive aphasia, dysphagia and seizures. The subject was treated with tetrabenazine, although documentation of the rationale for this decision is not available. He was also treated with carbamazepine and zuclopentixol. A stooped posture with antecollis, bradykinesia and rigidity were evident, and may have been due to neuroleptic medication (see Video S1). It is possible that this medication may have prevented the appearance of chorea. No 123I-ioflupane SPECT scan was performed but a HMPAO-SPECT scan showed marked reduction of blood flow in both parietotemporal areas. EEG showed generalized slowing with a 2–4 Hz frequency. When his aggressive behavior waned he was transferred to a nursing home. Gradually he became wheelchair-bound and progressed into a state of akinetic mutism with generalized spasticity. In the last stages of disease he had frequent seizures until dying of pneumonia at the age of 48. Genetic test in this subject and in subjects III:1, III:8 and III:10 revealed an insertion of 8 OPRI in the PRNP gene and homozygosity -MM- at codon 129. At the time of the first description of the Swedish family affected by IPD with 8-OPRI10 three of the seven affected subjects were alive; now all subjects are deceased (Table 1).
Table 1. Age of onset and disease duration in the Swedish family affected by IPD with 8-OPRI.
| Nr in pedigree | Gender | Age of onset/ Age of death, years |
Disease duration, years | PRNP polymorphic codon 129 (mutated allele) | PRNP polymorphic codon 129 (both alleles) |
|---|---|---|---|---|---|
| I:2 | M | 41/52 | 11 | M | M- |
| II:2 | F | 28/42 | 14 | M | M- |
| II:6 | F | 29/43 | 14 | M | M- |
| III:1 | F | 23/46 | 23 | M | MM |
| III:3 | M | 29/48 | 19 | M | MM |
| III:8 | F | 29/42 | 12 | M | MM |
| III:10 | M | 29/41 | 12 | M | MM |
III, 3 is the subject reported here; M, inferred polymorphism.
Comparison between 8-OPRI families
Age of onset
The mean age of onset (SD) for the Swedish kindred was 29.7 y (±5.4 y) (Fig. 1; Table 2). ANOVA revealed a significant difference between the families, post hoc comparisons using the Tukey HSD test revealed significantly (P = 0.026) earlier mean age of onset (SD) in the Swedish family which was 29.7 y (±5.4 y), when compared with the Che (43.3 ± 6.8 y), and A (43.3 ± 12.4 y) families, but not with the M-E family (31.8 ± 7.0 y) (P = 0.95).
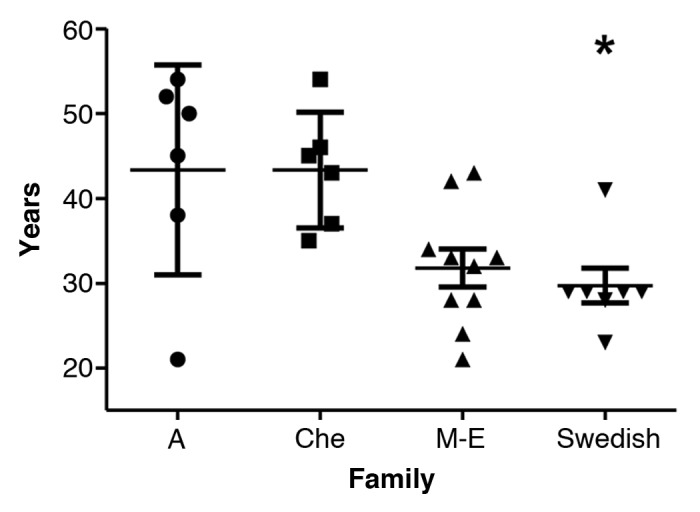
Figure 1. Age of onset with mean and SD in the four 8-OPRI families. *P = 0.026 only when compared with the A and Che families.
Table 2. Comparison of four 8 OPRI families.
| A family | Che family | M-E family | Swedish family | All the families | |
|---|---|---|---|---|---|
| n (females; males) | 6 (2;4) | 6 (1;5) | 11 (7; 4) | 7 (4;3) | 30 (14; 16) |
| PRNP polymorphic codon 129 in mutated allele (n) | 129V6 | 129V6 | 129M11 | 129M7 | 129V,12 129M18 |
| PRNP polymorphic codon 129 in both alleles (nr of homozygous and heterozygous) | 2 pol 129VV 1 pol 129 VM | NA | 4 pol 129MM | 4 pol 129 MM | 1 pol 129 VM 2 pol 129VV 8 pol 129 MM |
|
Age of onset, mean in years; SD (range; missing values) |
43.3; 12.4 (21–54; 0) | 43.3; 6.8 (35–54; 0) |
31.8; 7.0 (21–43; 1) | 29.7; 5.4 (23–41; 0) |
36.1; 6.6 (21-54; 1) |
|
Disease duration, mean in years; SD (range; number for which information is available; missing values; still alive at the time of publication) |
3.6; 2.6 (0.4–6a; 4; 0; 2) |
3.3; 4.9 (0.3–13; 6; 0; 0) |
3.4; 2.3 (1.3–7b; 6; 1; 4) |
15.1; 4.3 (11–23; 7; 0; 0) |
6.9; 6.6 (0.3–23; 23; 1; 6) |
|
Age of death, mean in years; SD (range; n) |
53.6; 3.9 (50.4–59; 4) |
46.6; 8.7 (35.3–56; 6) |
40.6; 4.8 (35-47;7) |
44.9; 4.0 (41–52; 7) |
45.5; 6.9 (35-59; 24) |
a Two subjects were still alive at the time of report publication more than 1 and two years after disease onset. bOne subject was still alive more than 12 y after symptom onset. Disease duration is the longest of all the reported IPD families with 8-OPRI (P < 0.001).
Age of disease onset at less than 30 y was significantly associated with longer disease duration across the four families (log-rank test P < 0.001) as seen in Figure 2A and B.
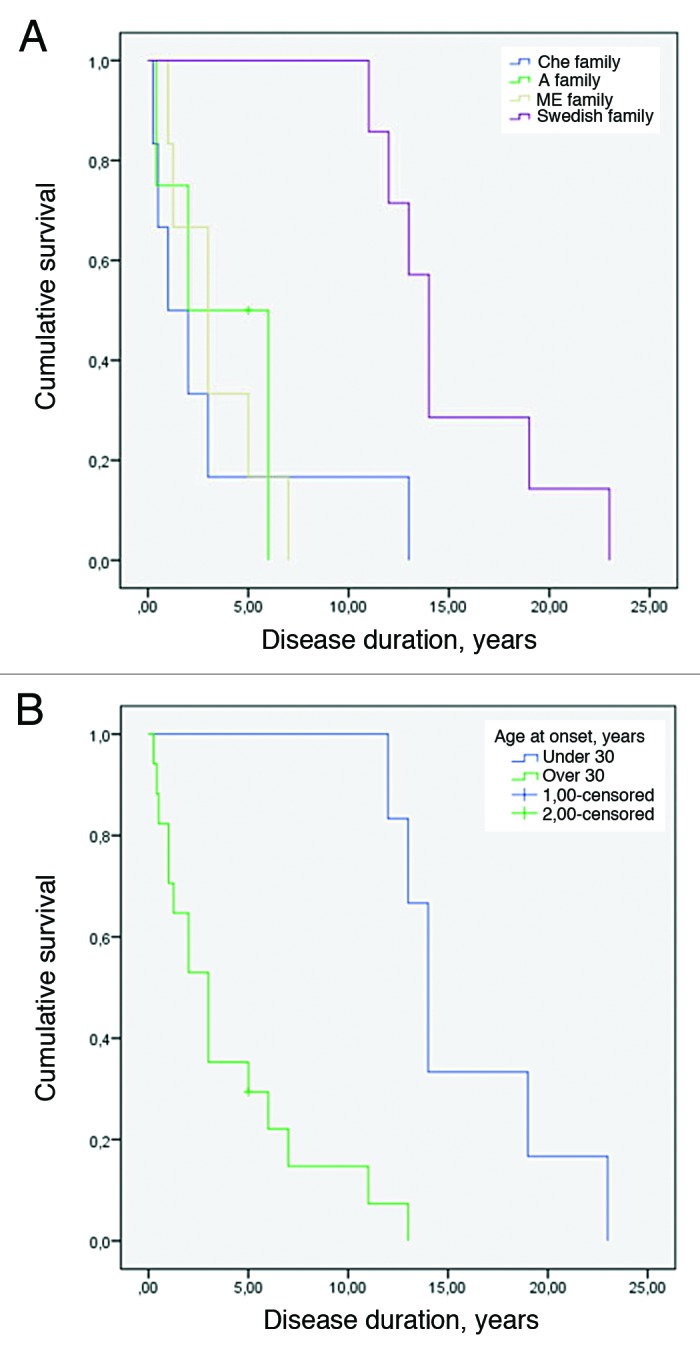
Figure 2. (A) Kaplan-Meier survival curve for the 4 8-OPRI families. The Swedish family had the longest survival (log-rank test with P value = 0.004). (B) Kaplan-Meier survival curve when early age of onset is set as less than 30 y (log-rank test with P value < 0.001).
Early age of onset correlated inversely with long disease duration only when data from the Swedish family was added to the calculation (Fig. 3). This Pearson correlation was performed for deceased subjects, age of onset in years vs. disease duration in years, and yielded r = -0.642 (P < 0.001).
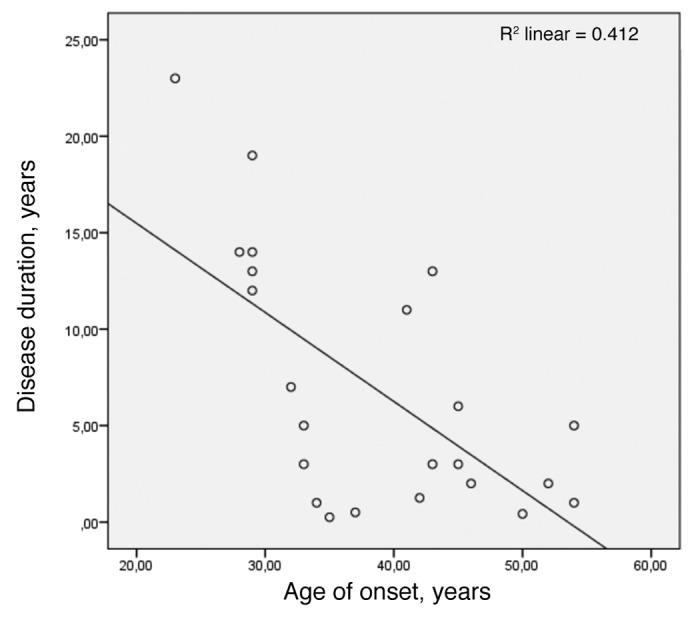
Figure 3. Pearson correlation for early age of onset and long survival (n = 23) with r = -0.642 and P value < 0.001.
Disease duration
The mean disease duration (SD) of the Swedish family was 15.1 y (± 4.3 y) (Fig. 4; Table 2). Survival ranged from 11–23 y in the Swedish family and from 0.3 to 13 y in the other three 8-OPRI families combined. ANOVA revealed a significant difference between the families, post hoc comparisons using the Tukey HSD test revealed that the Swedish kindred had the longest survival of all the four 8-OPRI families (P = 0.004, Fig. 2A). This long survival occurred despite the fact that 2 of the affected with the longest disease duration (19–23 y) were homozygous at codon 129 -MM- in the PRNP gene. Two more-affected in the Swedish kindred were MM and with survivals of 12 and 13 y respectively (Table 2).
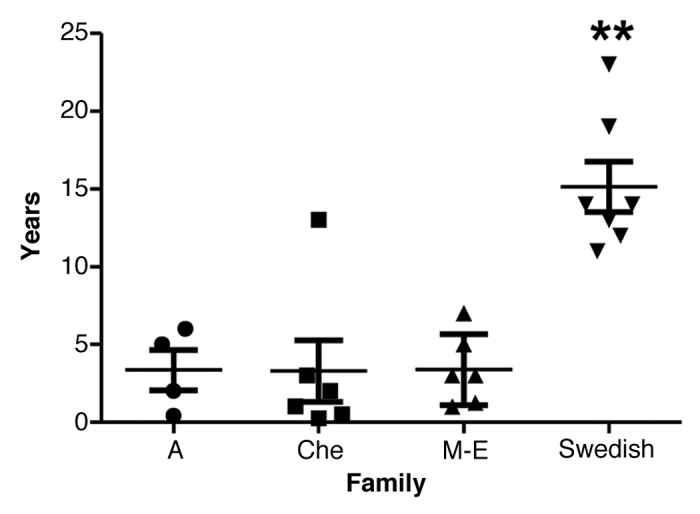
Figure 4. Disease duration with mean and SD in the four 8-OPRI families. ** P = 0.004 when compared with all the three families.
Six subjects (2 in the A family and 4 in M-E family) were still alive at the time of the respective publications and their disease duration was reported as >2 to >12 y.8,9 Update on the other families’ survival was not possible to obtain.
Age of death
The mean age of death for the Swedish family was 44.9 y (±4.0). In the four families combined the mean age of death was 45.5 y (±6.9) (Fig. 5; Table 2). ANOVA revealed significant differences between the families. Post hoc comparisons using the Tukey HSD test revealed that mean age of death was earlier in the M-E family, 40.6 y (±4.8), than in the A family, 53.6 y (±3.9) (P = 0.009). Age of death was not significantly correlated with disease duration.
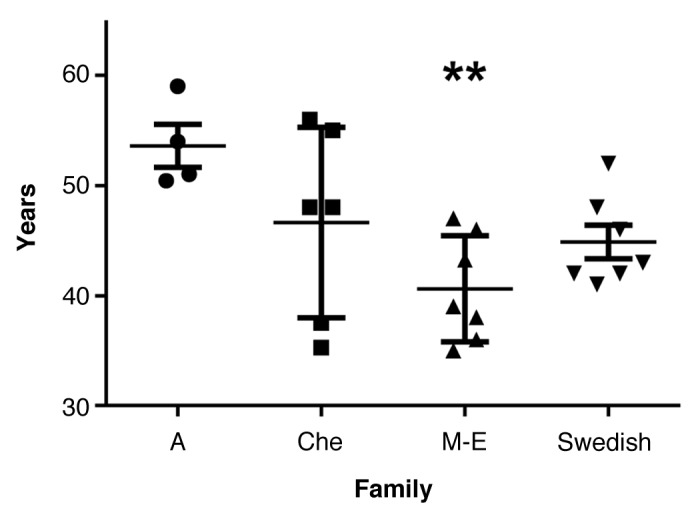
Figure 5. Age of death with mean and SD in the four 8-OPRI families. ** P = 0.009 only when compared with the A family.
PRNP polymorphic codon 129
Subjects with PRNP polymorphic codon 129M in the mutated allele had significantly earlier age of onset, longer survival and earlier age of death than 129V subjects (Table 3). Homozygous MM at codon 129 (n = 8) displayed an earlier age of onset than VV homozygous (n = 2) (mean 26.4 y vs. 41.5 y; P < 0.001). Of note, the case with the earliest age of onset, 21 y, was a heterozygous male, 129VM, in the A family.9 Missing information on survival and age of death for the two 129VV subjects precluded further comparisons with 129MM. Age of onset, survival and age of death did not significantly differ between 129M females and 129M males but 129V females (n = 3) had earlier mean age of onset (SD) than males (n = 9) which were 32 y (5.5) vs. 47.1 y (2.0), respectively (P = 0.0085). We also compared the 8-OPRI 129MM cases with their counterparts harboring the following mutations: E200K mutation; P102L mutation; and IPD with 6-OPRI. This comparison was done by performing ANOVA (Table 4). E200K causes fCJD and is the most common conventional PRPN mutation. The P102L mutation was included in this analysis since it is a classic example of GSS15 which shares clinical and neuropathological similarities with 8-OPRI associated IPD. IPD with 6-OPRI is the most common IPD with extra OPRI. Age of onset and disease duration were compared after performing Bonferroni correction for multiple testing which yielded a new α of 0.008 (Table 5). Age of onset was significantly earlier in IPD with 6-OPRI and 8-OPRI when compared with subjects with the E200K and P102L mutations (P < 0.001 in all four comparisons). GSS caused by P102L had earlier age of onset than E200K (P = 0.005) while IPD with 8-OPRI also had earlier age of onset than 6-OPRI, however this difference was not statistically significant (P = 0.02). Subjects affected by IPD with 6-OPRI had significantly longer survival than E200K and P102L subjects (P < 0.001 in both cases). In turn, IPD with 8-OPRI subjects had a longer survival than E200K subjects (P = 0.008), this difference was border-line significant. The longer survival among 8-OPRI subjects when compared with P102L and 6-OPRI subjects was not statistically significant.
Table 3. Differences based on PRNP polymorphic codon 129 in the mutated allele.
| 129M | 129V | Mean difference in years |
P value and 95% CI | |
|---|---|---|---|---|
| Age of onset mean in years, (n) | 30.9 (17) | 43.3 (12) | 12.3 | 0.0002*; -18.41 to -6.371 |
| Disease duration mean in years (n) | 9.7 (13) | 3.3 (10) | 6.4 | 0.0170*; 1.262 to 11.53 |
| Age of death mean in years (n) | 42.7 (14) | 49.4 (10) | 6.7 | 0.0154*; -12.00 to -1.408 |
significant value based on the Student t test at P < 0.05
Table 4. ANOVA for age of onset and disease duration for homozygous 129MM in three IPD: E200K, 6-OPRI and 8-OPRI.
| E200K | P102L | 6-OPRI | 8-OPRI | P value | |
|---|---|---|---|---|---|
|
Age of onset Number of cases Mean in years (SD) |
17 58 (12.8) |
22 46 (12.4) |
30 31.4 (5.7) |
8 26.4 (3.2) |
P < 0.001 (F3,73 statistics = 35.2) |
|
Disease duration Number of cases Mean in years (SD) |
17 0.6 (0.3)* |
15 4.5 (2.0)* |
19 11.4 (4.6) * |
4 16.8 (5.2) |
P < 0.001 (F3,51 statistics = 49.6) |
Information obtained from Mead et al.18 other than that denoted by * which was provided by personal communication also with Professor Simon Mead.
Table 5. Comparison of homozygous 129MM in three IPD- E200K, IPD with 6-OPRI, and IPD with 8-OPRI.
| Homozygous 129MM | Compared PRNP mutation types | Mean difference, years (95% CI) | t statistics | P value |
|---|---|---|---|---|
| Age of onset | E200K and P102L | 12 (3.5, 20.5)e | 2.9 | 0.005* |
| E200K and 6-OPRI | 26.6 (18.6, 34.6)u | 8.1 | < 0.001* | |
| E200K and 8-OPRI | 31.6 (20.3,42.9)u | 9.6 | < 0.001* | |
| P102L and 6-OPRI | 14.6 (7.2, 21.9)u | 5.1 | < 0.001* | |
| P102L and 8-OPRI | 19.6 (8.7 30.5)u | 6.8 | < 0.001* | |
| 6-OPRI and 8-OPRI | 5.0 (-5.4, 15.5)e | 2.4 | 0.02 | |
| Disease duration | E200K and P102L | -3.9 (-7.1, -0.8)u | -7.5 | < 0.001* |
| E200K and 6-OPRI | -10.8 (-13.8, -7.9)u | -10.2 | < 0.001* | |
| E200K and 8-OPRI | -16.2 (-21.1, -11.3)u | -6.2 | 0.008 | |
| P102L and 6-OPRI | -6.9 (-9.9, -3.8)u | -5.8 | < 0.001* | |
| P102L and 8-OPRI | -12.2 (-17.2, -7.3)u | -4.6 | 0.02 | |
| 6-OPRI and 8-OPRI | -5.4 (- 10.2, -0.5)e | -2.1 | 0.05 |
e Equal variance, uunequal variance. *Significant P value, new α = 0.008 after Bonferroni correction.
Gender
The mean age of onset was 9.5 y earlier among females than males (P < 0.001). The mean age of death was 7.8 y earlier among females (P < 0.001). Disease duration did not significantly differ between males and females (Table 6).
Table 6. Gender differences in IPD with8-OPRI.
| Males | Females | Mean difference in years |
P value and 95% CI | |
|---|---|---|---|---|
| Age of onset mean in years (n) | 40.4(16) | 30.7 (13) | 9.8 | 0.0057*; 3.094 to 16.40 |
| Disease duration mean in years (n) | 6.1 (14) | 8.3 (9) | 2.3 | 0.4351; -8.158 to 3.643 |
| Age of death mean in years (n) | 48.8(14) | 41.0 (10) | 7.8 | 0.0037*; 2.824 to 12.80 |
significant value based on the Student t test at P < 0.05
Regression models
Multiple linear regression revealed that both PRNP polymorphic codon 129 (P = 0.002) and gender (P = 0.037) combined conferred up to 50% of variability in age of onset (Table 7). In one model, PRNP polymorphic codon 129 was found to confer 25.3% of variability in survival (P = 0.024) after correcting for gender (Table 8). Additional regression models revealed that age of onset conferred 48% of variability in survival (P = 0.010) and 54.6% of variability in age of death (P = 0.026) after correcting for PRNP polymorphic codon 129 and gender (Tables 9 and 10).
Table 7. Regression model for age of onset (dependent variable) in IPD with 8-OPRI.
| Independent variables | Coefficient (β) | 95% CI | P value |
|---|---|---|---|
| PRNP polymorphic codon 129 | 0.52 | 4.26–16.24 | 0.002* |
| Gender | 0.33 | 0.414–12.28 | 0.0037* |
R2 = 50%
Table 8. Regression model for disease duration (dependent variable) in IPD with 8-OPRI,
| Independent variables | Coefficient (β) | 95% CI | P value |
|---|---|---|---|
| PRNP polymorphic codon 129 | - 0.56 | -13.36- -1.06 | 0.024* |
| Gender | 0.12 | -4.67–7.82 | 0.605 |
R2 = 25.3%.
Table 9. Regression models for disease duration in IPD with 8-OPRI.
| Independent variables | Coefficient (β) | 95% CI | P value |
|---|---|---|---|
| PRNP polymorphic codon 129 | -0.1 | -8.11–5.52 | 0.69 |
| Gender | 0.32 | -1.45–9.87 | 0.14 |
| Age of onset | -0.76 | -0.94- -0.15 | 0.01* |
R2 = 48%
Table 10. Regression models for age of death in IPD with 8-OPRI.
| Independent variables | Coefficient (β) | 95% CI | P value |
|---|---|---|---|
| PRNP polymorphic codon 129 | 0.09 | 8.11–5.52 | 0.69 |
| Gender | 0.30 | 1.45–9.97 | 0.14 |
| Age of onset | 0.59 | 0.06–0.85 | 0.026* |
R2 = 54.6%
Clinical features in IPD with 8-OPRI
Disease onset was studied in the four families affected by IPD with 8-OPRI. Information on symptom onset was available in 22/30 cases. Psychiatric symptoms/behavioral disturbances alone at disease onset were present in 27% of cases while a movement disorder alone was reported in 22% of cases and cognitive decline alone in 9% of cases. A combination of psychiatric symptoms and cognitive decline together at disease onset was reported in 18%, while a movement disorder and psychiatric symptoms at onset were reported in 14%. Psychiatric, symptoms, movement disorders and cognitive decline together were reported in 14% of cases.
During the course of the disease, psychiatric features commonly included personality changes and a wide variety of symptoms such as manias, perseveration, aggressive behavior, emotional lability and in some cases psychotic symptoms, anxiety and depression. Aggressive behavior was reported in 30% and psychotic symptoms in 13% of all cases during the course of disease. Authors reported “unsteadiness, gait difficulties and coordination problems” as the presenting symptom which suggests ataxia. Ataxia was present in 50% of cases during in the course of disease, followed by parkinsonism (43%), myoclonus (20%) and rarely chorea (13%). Chorea was described in 4 subjects from the Swedish family only. It was not possible to discriminate whether parkinsonism was caused by medication or was part of the natural history of the disease. The presence of ataxia and other movement disorders together were reported in 25% of cases. Other neurological features included dysarthria, apraxia, aphasia, frontal release signs, seizures, urine incontinence and pyramidal abnormalities. Progressive cognitive decline eventually leading to dementia was present at some point in the course of disease in all of the 22 cases and appears to be a universal feature. Information on this feature was not reported in the remaining 8 subjects.
Imaging modalities included mostly CT-scans and MRI of the brain. These displayed varying degrees of cerebral and/or cerebellar atrophy, in one case hyperintensities of the basal ganglia were described.9 Only two 8-OPRI cases were studied with SPECT, one is described in the present study. EEG was performed on 13/30: 5 displayed slow activity, di and/or triphasic waves was reported in 4 subjects, 2 displayed “encephalopathic findings,” 1 had pseudo-rhythmic appearance and 1 had irregular paroxysmal discharges. Determination of 14–3-3 levels in the CSF was not performed in the Swedish family, nor were they reported to have been performed in the other three families.
Of note, early prodromal symptoms, originating in childhood, were reported in the M-E family. One subject displayed aggressiveness at the age of 13 as well as early learning disabilities and clumsiness. Two more individuals from the same family also displayed early learning disabilities.8 We did not find evidence of premorbid features in the Swedish family or in the other two 8-OPRI families. Anticipation is suggested in the A and M-E families only.
Cause of death was reported in 10 cases: 3 died of pneumonia, 3 died of hyperthermia, 2 of cachexia, 1 of tuberculosis and 1 of myocardial infarction. All the three subjects who died of hyperthermia belonged to the M-E family.8
Discussion and Conclusion
We provide an update on the Swedish family affected by IPD with 8 OPRI and a video case displaying the major phenotype features. There are only two 8-OPRI cases where functional imaging- HMPAO-SPECT scan- has been reported. An HMPAO-SPECT in a subject from the A family had reduced cortical perfusion, and her MRI showed diffuse cortical and cerebellar atrophy.9 The same modality in the case we describe here displayed a marked bilateral reduction of blood flow in atrophied parietotemporal areas. This abnormality resembles what is typically seen in Alzheimer disease.16,17
The Swedish kindred affected by IPD with 8-OPRI displayed the longest survival of all four described families. Another striking finding was a significant inverse correlation between early age of onset and long disease duration, a pattern that has been described in a large British 6-OPRI kindred18 and in sporadic prion diseases.19 This inverse correlation in IPD with 6 and 8 OPRI can be contrasted with what is seen in HD and kuru where a direct correlation between early age of onset and shorter survival has been described.20,21 Similar to IPD with 6-OPRI, age of onset was not correlated with age of death.18
Both PRNP polymorphic codon 129 and gender combined conferred up to 50% of variability in age of onset but did not have a significant effect in the variability of survival or age of death. The effect of PRNP polymorphic codon 129 resembles again what was found for IPD with 6-OPRI.18 Furthermore, PRNP polymorphic codon 129 was found to confer 25.3% of variability in survival after correcting for gender. Age of onset, on the other hand, conferred 48% and 54.6% of the variability of survival and age of death, respectively, after correcting for PRNP polymorphic codon 129 and gender.
129M subjects had significantly earlier age of onset, longer survival and earlier age of death than 129V. In contrast to what Goldfarb reported for IPD with extra OPRIS, we found that 129V subjects had a shorter survival than 129M subjects.22 Homozygous129MM in this small sample size had an earlier age of onset than 129VV. Again, the scarcity of heterozygous cases precludes further comparisons with homozygous cases, but we found a remarkable modulation of PRNP polymorphic codon129 on age on onset and disease duration.
Age of onset among 129MM 8-OPRI subjects was significantly earlier than their counterparts affected by fCJD caused by the E200K mutation, GSS caused by P102L mutation and IPD with 6-OPRI. These findings provide strong counterevidence to previous reports in which larger OPRI were associated with shorter survival.23 Survival was also longer among 8-OPRI 129MM subjects than for fCJD subjects harboring the E200K mutation. Survival was not significantly longer among 8-OPRI 129MM subjects when compared with 6-OPRI and E200K counterparts. However, this difference is in our opinion clinically important (5.4 respectively 12.2 y), particularly when considering the possible future use of neuroprotective treatments.
Females had earlier age of onset and age of death than males (mean difference 9.8 y and 7.8 y, respectively). This difference was more striking than in vCJD (which had a mean difference of 9.5 and 2 y, respectively).24 The role of gender in prion diseases has otherwise yielded contradicting results, for instance females had longer survival in sporadic prion disease19 but not in IPD with 8-OPRI as shown here. Similarly to IPD with 8-OPRIs, individuals affected by vCJD display both younger age of onset and longer disease duration when compared with sCJD.25 There is, however, an important survival difference with 8-OPRI associated IPD; the median survival time for vCJD is only 1.1 y.19
Both age of onset and survival for IPD with 8-OPRIs reported here differ substantially from those estimated by Kovács, being 39 y and 3 y, respectively, in a comprehensive review in which data from the Swedish family was not included.23 In his analysis, Kovács included the other three 8-OPRI families (Che, A, and M-E).7-9 Our observations should be qualified by the complex effect of PRNP polymorphic codon 129 and the scarcity of available data on heterozygotes.
It is unknown how PRNP mutations with additional OPRI arise, the result of an unequal recombination phenomenon has been proposed.6,18 The tandem repeat region, represented by R1-R2-R2-R3-R4, spans between codons 51 and 91 in the PRNP gene. R1 encodes a nonapeptide while R2-R4 encode octapetides.6,26 Additional OPRI in the reported 8-OPRI families display two silent nucleotide substitutions designated as R2a and R3g. The insertion sites of additional OPRI are the same only for the M-E and Che families, their order differ in the Swedish and A family (Table 11). Despite these differences, the amino acid sequence of R2-R4 OPRI in all four families will remain the same (PHGGGWGQ). Thus, the only difference in terms of protein structure is still the PRNP polymorphic codon 129 as previously observed by Laplanche when he compared the M-E family with the Che and A families.8
Table 11. Structure of the OPRI region in a normal case and in the four 8-OPRI families.
| Family | OPRI region | Codon 129 in the PRNP gene |
|---|---|---|
| Wild-type | R1-R2-R2-R3-R4 | M or V |
| Che | R1-R2-R2-R3-R2-R2-R2-R2-R2-R2-R2-R2a-R4 | V |
| M-E | R1-R2-R2-R3-R2-R2-R2-R2a-R2-R2-R2-R3-R4 | M |
| A | R1-R2-R2-R3g-R3-R2-R2-R2-R2-R2-R2-R3-R4 | V |
| Swedish | R1-R2-R2-R3g-R2-R2-R2-R3g-R3g-R2-R2-R3-R4 | M |
Also unknown is the mechanism of disease in IPD with additional OPRI. The tandem OPRI sequences were initially believed to lack a role in PrP misfolding since mice expressing prion proteins devoid of four and five OPRI were able to sustain disease upon transmission in a model of scrapie.27,28 However, incubation time for the transgene animals was longer and the neuropathology milder than in wild-type animals.28 There is though evidence supporting a role of additional OPRI in the pathogenesis of IPD. For instance, Leliveld suggested that the wild-type OPRI sequences act as reversible copper-dependent switch of prion conformation,29 this switch function was suggested to become irreversible with additional OPRI.29 Additional OPRI were also found to increase the rate of protease-resistant prion formation30 and to reinforce PrP misfolding.31
Psychiatric symptoms alone, movement disorders alone, and a combination of psychiatric symptoms with cognitive decline, are the most common features of disease onset in IPD with 8 OPRI. Also similar to IPD with 6 OPRI is the universal feature of progressive cognitive decline leading to dementia. It is otherwise difficult to associate 129M and 129V with the other main features of this mutation, neurological and psychiatric, since different authors emphasized various aspects of this heterogeneous disorder. Laplanche, for instance, emphasized the psychiatric features of the M-E family but the same family’s phenotype was labeled as GSS in the original description.5,8 This is not a coincidence since ataxia is the most common movement disorder in IPD with 8-OPRI. Similarity with GSS was also mentioned by Goldfarb7; van Gool et al. emphasized the cognitive deficits and Xiang the features resembling Huntington’s disease.9,10 Atrophy of the caudate nucleus was seen in 3/4 of the choreic subjects in the Swedish family,10 this abnormality likely contributed to uphold the suspicion of HD.
OMIM includes HDL1 as a prion disease phenotype of its own,12 however given the wide expressivity of IPD with 8-OPRI and the wide intrafamilial variation, the HDL1 term may be inappropriate and misleading. For instance, ataxia but not chorea was the most common movement disorder described for this mutation. Second, chorea was not a universal trait in the Swedish kindred, but was described in 4/7 individuals. The subject from the Swedish family we describe here exhibited rather parkinsonian features but was not clearly documented as having chorea. Whether this parkinsonism was induced or aggravated by neuroleptics remains unclear. Finally, chorea is not an uncommon feature in other OPRI-associated prion diseases or in sporadic prion diseases. This is illustrated by the fact that some individuals from the large English 6-OPRI kindred were diagnosed with clinical HD.18,32 Other disadvantages of using the HDL term have been summarized by Walker.33 Using the term IPD followed by the mutation34 or OPRI-associated prion disease or prion encephalopathy with OPRIs would appear to be more suitable terms.13
A premorbid personality disorder has been described for some IPD cases.18,34 The striking presence of early learning disabilities in three subjects from the M-E kindred, aggressiveness and prolonged psychiatric symptoms seem to support this observation.8 We did not find evidence of premorbid features in the other IPD families with 8-OPRI. Another interesting finding in the M-E family is the fact that three affected subjects died of hyperthermia. A neuropathological study in one of these individuals displayed marked spongiosis in the thalamus.8 This abnormality and the suggestion of dysautonomia resemble the features of FFI.35
The differential diagnosis of OPRI-associated IPD is broad and includes sporadic and acquired prion diseases as well as familial and sporadic disorders with early-onset dementia like HD, spinocerebellar ataxias caused by CAG expansions (poly-Q SCA), dentatorubropallidoluysian atrophy (DRPLA), MAPT-related disorders and familial Alzheimer disease.2,18 The MAPT-related disorders encompass frontotemporal dementia with or without parkinsonism linked to chromosome 17 (FTDP-17), progressive supranuclear palsy (PSP) and corticobasal degeneration.36
Despite our findings, questions remain regarding genotype-phenotype associations for IPD with 8-OPRI. For instance, it is still unclear what determines the striking gender differences we found as well as the wide phenotype heterogeneity documented so far and a premorbid personality disorder in some cases. Other disease-modifying factors for IPD with 8-OPRI, such as polymorphisms other than PRPN polymorphic codon 129, remain to be studied as well. Whether differences in symptomatic treatment and/or care may have influenced the remarkable survival differences in the four 8-OPRI families is also unknown. Two other unclear aspects of disease are the progression rate and the suggestion of anticipation. Progression rate cannot be assessed in the published work on this mutation. Anticipation is suggested in the A and M-E families but the sample size in every generation is again too small to make firm conclusions. Anticipation has otherwise been suggested for fCJD caused by the E200K mutation.37 The small size of this sample with missing information on genotype and survival for some subjects constitute a weakness in our study.
Knowledge regarding the spectrum of OPRI-associated prion diseases continues to broaden. The most recently described mutation in this subgroup was an insertion of 12 OPRI causing a FTD-like disorder.3 To further clarify genotype-phenotype associations for IPD with extra OPRI as well as rate of disease progression additional thorough phenotype characterizations using validated tools such as the recently published scale for prion diseases,38 functional imaging studies, EEG and biochemical analysis of neurodegenerative markers in the CSF and plasma would be needed. We therefore encourage other researchers and clinicians to report cases of IPDs with extra OPRI.
In conclusion, we found an inverse correlation between early age of onset and long survival for IPD with 8-OPRI. PRNP polymorphic codon 129 and gender have a complex effect on this disease’s phenotype. OPRI-associated prion diseases should be considered in the differential diagnosis of individuals with a family history of early-onset progressive dementia with movement disorders and psychiatric symptoms.
Methods
The study was approved by the Ethical Review Board in Stockholm (Etikprövningsnämnden) and written consent was obtained from a next-of-kin.
PCR for the PRNP gene for subjects III:1, III.3, III:8 and III:10 in the Swedish family was performed according to the details reported by Moore et al.12
Statistical analysis and graphs, survival curves, linear regression, ANOVA tests and Person’s correlation, were performed using SPSS package 20. Significance rates were calculated using the two-tailed, unpaired Student’s t test with a value of P < 0.05 accepted as significant. When the Levene’s test for equality of variance was significant (P < 0.05) we have reported the results for unequal variance.
Disease duration in all the four families was defined as the time elapsed between age of onset and age of death; this definition was the same in three of the reported 8-OPRI families.7,9,10 For the M-E family we have used the information provided in the publication and applied our definition of disease duration.8 Updated information on disease duration for 3 subjects of the Swedish family (III:3, III:8, and III:10) was obtained from chart reviews. Genotype information was available in 24/30 individuals from the four reported families affected by IPD with 8 OPRI.7-10 Genotype data on the remaining 6 subjects was missing, 3 belonged to generation I in the A family and 3 to the Swedish family (I:2, II:2, and II:6). Genotype on the mutated allele for the missing 6 subjects was inferred based on the clinical phenotype, course of disease, family history and neuropathological features. Since affected subjects in generation II in the A family were all VV, then those in generation I were inferred to be at least 129V. Similarly, in the Swedish family, all the affected in generation III were MM which implies that generation I and II would have to be 129M in the mutated allele, at least. Phenotype information was also obtained from previous publications7-10 and from chart reviews (for the case description). Percentage ratios are the number of cases displaying a particular clinical feature or a combination of features divided by the total number of cases.
Supplementary Material
Acknowledgments
We thank the kind assistance provided by Professor Simon Mead at the MRC prion unit, UCL Institute of Neurology, Queen Square; London, UK and by Dr Mesfin Tessma at the Department of Learning, Informatics, Management and Ethics (LIME); the Karolinska Institute, Stockholm, Sweden.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14:273–81. doi: 10.1038/sj.ejhg.5201544. [DOI] [PubMed] [Google Scholar]
- 2.Mastrianni JA. The genetics of prion diseases. Genet Med. 2010;12:187–95. doi: 10.1097/GIM.0b013e3181cd7374. [DOI] [PubMed] [Google Scholar]
- 3.Kumar N, Boeve BF, Boot BP, Orr CF, Duffy J, Woodruff BK, Nair AK, Ellison J, Kuntz K, Kantarci K, et al. Clinical characterization of a kindred with a novel 12-octapeptide repeat insertion in the prion protein gene. Arch Neurol. 2011;68:1165–70. doi: 10.1001/archneurol.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd S, Mead S, Collinge J. Genetics of prion disease. Top Curr Chem. 2011;305:1–22. doi: 10.1007/128_2011_157. [DOI] [PubMed] [Google Scholar]
- 5.Foncin JF, Cardot JL, Martinet Y, Arnott G. [Gerstmann-Sträussler-Scheinker disease. Anatomoclinical and genealogical study] Rev Neurol (Paris) 1982;138:123–35. [PubMed] [Google Scholar]
- 6.Goldfarb LG, Brown P, McCombie WR, Goldgaber D, Swergold GD, Wills PR, Cervenakova L, Baron H, Gibbs CJ, Jr., Gajdusek DC. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci U S A. 1991;88:10926–30. doi: 10.1073/pnas.88.23.10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldfarb LG, Brown P, Vrbovská A, Baron H, McCombie WR, Cathala F, Gibbs CJ, Jr., Gajdusek DC. An insert mutation in the chromosome 20 amyloid precursor gene in a Gerstmann-Sträussler-Scheinker family. J Neurol Sci. 1992;111:189–94. doi: 10.1016/0022-510X(92)90067-U. [DOI] [PubMed] [Google Scholar]
- 8.Laplanche JL, Hachimi KH, Durieux I, Thuillet P, Defebvre L, Delasnerie-Lauprêtre N, Peoc’h K, Foncin JF, Destée A. Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain. 1999;122:2375–86. doi: 10.1093/brain/122.12.2375. [DOI] [PubMed] [Google Scholar]
- 9.van Gool WA, Hensels GW, Hoogerwaard EM, Wiezer JH, Wesseling P, Bolhuis PA. Hypokinesia and presenile dementia in a Dutch family with a novel insertion in the prion protein gene. Brain. 1995;118:1565–71. doi: 10.1093/brain/118.6.1565. [DOI] [PubMed] [Google Scholar]
- 10.Xiang F, Almqvist EW, Huq M, Lundin A, Hayden MR, Edström L, Anvret M, Zhang Z. A Huntington disease-like neurodegenerative disorder maps to chromosome 20p. Am J Hum Genet. 1998;63:1431–8. doi: 10.1086/302093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore RC, Xiang F, Monaghan J, Han D, Zhang Z, Edström L, Anvret M, Prusiner SB. Huntington disease phenocopy is a familial prion disease. Am J Hum Genet. 2001;69:1385–8. doi: 10.1086/324414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Online Mendelian Inheritance in Man. OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: # 603218: 02.06.2012. World Wide Web URL:http://omim.org [cited 2013 Sep 29].
- 13.Vital C, Gray F, Vital A, Parchi P, Capellari S, Petersen RB, Ferrer X, Jarnier D, Julien J, Gambetti P. Prion encephalopathy with insertion of octapeptide repeats: the number of repeats determines the type of cerebellar deposits. Neuropathol Appl Neurobiol. 1998;24:125–30. doi: 10.1046/j.1365-2990.1998.00098.x. [DOI] [PubMed] [Google Scholar]
- 14.Vital C, Gray F, Vital A, Ferrer X, Julien J. Prion disease with octapeptide repeat insertion. Clin Exp Pathol. 1999;47:153–9. [PubMed] [Google Scholar]
- 15.Webb TEF, Poulter M, Beck J, Uphill J, Adamson G, Campbell T, Linehan J, Powell C, Brandner S, Pal S, et al. Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain. 2008;131:2632–46. doi: 10.1093/brain/awn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jobst KA, Smith AD, Barker CS, Wear A, King EM, Smith A, Anslow PA, Molyneux AJ, Shepstone BJ, Soper N, et al. Association of atrophy of the medial temporal lobe with reduced blood flow in the posterior parietotemporal cortex in patients with a clinical and pathological diagnosis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1992;55:190–4. doi: 10.1136/jnnp.55.3.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dougall NJ, Bruggink S, Ebmeier KP. Systematic review of the diagnostic accuracy of 99mTc-HMPAO-SPECT in dementia. Am J Geriatr Psychiatry. 2004;12:554–70. doi: 10.1176/appi.ajgp.12.6.554. [DOI] [PubMed] [Google Scholar]
- 18.Mead S, Poulter M, Beck J, Webb TEF, Campbell TA, Linehan JM, Desbruslais M, Joiner S, Wadsworth JD, King A, et al. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain. 2006;129:2297–317. doi: 10.1093/brain/awl226. [DOI] [PubMed] [Google Scholar]
- 19.Pocchiari M, Puopolo M, Croes EA, Budka H, Gelpi E, Collins S, Lewis V, Sutcliffe T, Guilivi A, Delasnerie-Laupretre N, et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain. 2004;127:2348–59. doi: 10.1093/brain/awh249. [DOI] [PubMed] [Google Scholar]
- 20.Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatry. 1999;66:52–6. doi: 10.1136/jnnp.66.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collinge J, Whitfield J, McKintosh E, Beck J, Mead S, Thomas DJ, Alpers MP. Kuru in the 21st century--an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–74. doi: 10.1016/S0140-6736(06)68930-7. [DOI] [PubMed] [Google Scholar]
- 22.Golfarb LG, Cervenakova L, Brown P, Gajdusek C. Transmissible Subacute Spongiform Encephalopathies : Prion Diseases. Court L, Dodet B, editors. Elsevier Science Ltd; 1996. [Google Scholar]
- 23.Kovács GG, Trabattoni G, Hainfellner JA, Ironside JW, Knight RSG, Budka H. Mutations of the prion protein gene phenotypic spectrum. J Neurol. 2002;249:1567–82. doi: 10.1007/s00415-002-0896-9. [DOI] [PubMed] [Google Scholar]
- 24.Loeuillet C, Boelle P-Y, Lemaire-Vieille C, Baldazza M, Naquet P, Chambon P, Cesbron-Delauw MF, Valleron AJ, Gagnon J, Cesbron JY. Sex effect in mouse and human prion disease. J Infect Dis. 2010;202:648–54. doi: 10.1086/654818. [DOI] [PubMed] [Google Scholar]
- 25.Corato M, Cereda C, Cova E, Ferrarese C, Ceroni M. Young-onset CJD: age and disease phenotype in variant and sporadic forms. Funct Neurol. 2006;21:211–5. [PubMed] [Google Scholar]
- 26.Kretzschmar HA, Stowring LE, Westaway D, Stubblebine WH, Prusiner SB, Dearmond SJ. Molecular cloning of a human prion protein cDNA. DNA. 1986;5:315–24. doi: 10.1089/dna.1986.5.315. [DOI] [PubMed] [Google Scholar]
- 27.Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996;15:1255–64. [PMC free article] [PubMed] [Google Scholar]
- 28.Flechsig E, Shmerling D, Hegyi I, Raeber AJ, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron. 2000;27:399–408. doi: 10.1016/S0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 29.Leliveld SR, Dame RT, Wuite GJL, Stitz L, Korth C. The expanded octarepeat domain selectively binds prions and disrupts homomeric prion protein interactions. J Biol Chem. 2006;281:3268–75. doi: 10.1074/jbc.M510606200. [DOI] [PubMed] [Google Scholar]
- 30.Moore RA, Herzog C, Errett J, Kocisko DA, Arnold KM, Hayes SF, Priola SA. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci. 2006;15:609–19. doi: 10.1110/ps.051822606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leliveld SR, Stitz L, Korth C. Expansion of the octarepeat domain alters the misfolding pathway but not the folding pathway of the prion protein. Biochemistry. 2008;47:6267–78. doi: 10.1021/bi800253c. [DOI] [PubMed] [Google Scholar]
- 32.Wild EJ, Mudanohwo EE, Sweeney MG, Schneider SA, Beck J, Bhatia KP, Rossor MN, Davis MB, Tabrizi SJ. Huntington’s disease phenocopies are clinically and genetically heterogeneous. Mov Disord. 2008;23:716–20. doi: 10.1002/mds.21915. [DOI] [PubMed] [Google Scholar]
- 33.Walker RH. Update on the Non-Huntington’s Disease Choreas with Comments on the Current Nomenclature. Tremor Hyperkinetic Movements New York N. 2012;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Collinge J, Brown J, Hardy J, Mullan M, Rossor MN, Baker H, Crow TJ, Lofthouse R, Poulter M, Ridley R, et al. Inherited prion disease with 144 base pair gene insertion. 2. Clinical and pathological features. Brain. 1992;115:687–710. doi: 10.1093/brain/115.3.687. [DOI] [PubMed] [Google Scholar]
- 35.Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003;2:167–76. doi: 10.1016/S1474-4422(03)00323-5. [DOI] [PubMed] [Google Scholar]
- 36.Van Swieten JC, Rosso SM, Heutink P. MAPT-Related Disorders. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong C-T, Stephens K, editors. GeneReviewsTM [Internet]. Seattle (WA): University of Washington, Seattle; 1993 [cited 2013 Sep 26]. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1505/
- 37.Rosenmann H, Kahana E, Korczyn AD, Kahana I, Chapman J, Gabizon R. Preliminary evidence for anticipation in genetic E200K Creutzfeldt-Jakob disease. Neurology. 1999;53:1328–9. doi: 10.1212/WNL.53.6.1328. [DOI] [PubMed] [Google Scholar]
- 38.Thompson AGB, Lowe J, Fox Z, Lukic A, Porter M-C, Ford L, Gorham M, Gopalakrishnan GS, Rudge P, Walker AS, et al. The Medical Research Council prion disease rating scale: a new outcome measure for prion disease therapeutic trials developed and validated using systematic observational studies. Brain. 2013;136:1116–27. doi: 10.1093/brain/awt048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.