

Abstract
The ATR/Chk1 pathway is a critical surveillance network that maintains genomic integrity during DNA replication by stabilizing the replication forks during normal replication to avoid replication stress. One of the many differences between normal cells and cancer cells is the amount of replication stress that occurs during replication. Cancer cells with activated oncogenes generate increased levels of replication stress. This creates an increased dependency on the ATR/Chk1 pathway in cancer cells and opens up an opportunity to preferentially kill cancer cells by inhibiting this pathway. In support of this idea, we have identified a small molecule termed HAMNO ((1Z)-1-[(2-hydroxyanilino)methylidene]naphthalen-2-one), a novel protein interaction inhibitor of replication protein A (RPA), a protein involved in the ATR/Chk1 pathway. HAMNO selectively binds the N-terminal domain of RPA70, effectively inhibiting critical RPA protein interactions which rely on this domain. HAMNO inhibits both ATR autophosphorylation and phosphorylation of RPA32 Ser33 by ATR. By itself, HAMNO treatment creates DNA replication stress in cancer cells that are already experiencing replication stress, but not in normal cells, and it acts synergistically with etoposide to kill cancer cells in vitro and slow tumor growth in vivo. Thus, HAMNO illustrates how RPA inhibitors represent candidate therapeutics for cancer treatment, providing disease selectivity in cancer cells by targeting their differential response to replication stress.
Introduction
One of the intrinsic features that distinguish cancer cells from surrounding normal cells is a deregulated cell cycle. Common cancer-associated mutations in genes such as p53, Myc, Rb and EGFR in head and neck squamous cell carcinomas (HNSCC) promote a promiscuous S-phase entry, which leads to higher levels of replication stress (1-6). Replication stress occurs when the replication fork encounters aberrant DNA structures. While these structures may differ greatly, they all lead to the formation of long stretches of ssDNA that are bound with Replication Protein A (RPA), the main eukaryote ssDNA binding protein (7, 8).
The major structural feature of heterotrimeric RPA is the presence of the oligonucleotide/oligosaccharide binding (OB) folds within each of its six DNA binding domains (DBDs). This OB fold structure consists of beta sheets, forming β-barrel structures that wrap around ssDNA, a common feature in ssDNA binding proteins (9, 10). These OB folds are responsible for RPA’s binding to long stretches of ssDNA, making RPA one of the first responders in the replication stress response. RPA has several functions other than stabilizing ssDNA. One of these functions is to recruit and interact with proteins needed for an effective replication stress response, including ATRIP, Rad17, Rad9, and Nbs1 (11-15). This occurs via the OB fold located on the N-terminal of RPA70, DNA Binding Domain F (DBD-F), which unlike the other OB folds on RPA70 is primarily a protein binding domain. Recruitment of ATRIP, Rad17, Rad9, and Nbs1 leads to the loading of TopBP1 to stalled forks, which stimulates the kinase activity of ATR (11, 12, 15). This triggers a complex network of signaling pathways involved in cell cycle checkpoints, DNA repair and apoptosis that mediates the cellular response to replication stress. In the context of cancer therapy, the activation of these signaling pathways can limit the therapeutic efficacy of radiation and chemotherapeutics and enhance the survival of cancer cells (16).
The presence of higher replication stress in cancer cells compared to normal cells provides a therapeutic opportunity to target RPA. Based on observations of a DNA damage response in early stage cancerous lesions, two laboratories proposed a model referred to as the oncogene-induced DNA damage model for cancer development, which posited that deregulation of growth regulation genes induces DNA replication stress and DNA damage (16-18). Their initial analyses provided evidence that loss of heterozygosity in early stage lesions was due to oncogene-induced replication stress preferentially targeting chromosomal fragile sites. More recent studies have supported these initial observations and provided more evidence of the presence of DNA replication stress in human cancers. These studies were able to examine a very large cancer specimen sample size using high-throughput genomic analyses to show that the most common deletions occur at fragile sites in large genes, and these deletions can be attributed to replication stress (19, 20). Combined, these results provide a strong foundation for developing a novel therapeutic approach that exploits the endogenous replication stress intrinsic to cancer cells (21).
Here, we present the identification and development of a small molecule inhibitor that inhibits the protein interaction domain of RPA70 involved in the replication stress response. By targeting the cellular response to replication stress via RPA, we demonstrate that this approach is particularly toxic to cells that harbor cancer-associated mutations, which promote resistance to conventional chemotherapies.
Materials and Methods
Reagents
HAMNO (1Z)-1-[(2-hydroxyanilino)methylidene]naphthalen-2-one), also known as NSC111847, was acquired from the Developmental Therapeutics Program of the National Cancer Institute. The primary antibody for RPA32 was purchased from Santa Cruz. The primary antibodies for ATR and ATR-PT1989 were purchased from Genetex. Primary antibodies for RPA32-PS4S8, RPA32-PS33 and GAPDH were purchased from Bethyl Laboratories. The Alexa Fluor 680 and Alexa Fluor 488 secondary antibodies were purchased from Invitrogen. The IRDye700-labeled polyT 30mer oligonucleotide and complementary polyA 30mer oligonucleotides were purchased from IDT.
Docking simulations
The Molegro Virtual Docker Program (CLC bio) was used to dock HAMNO to the DBD-F in-silico. The HAMNO structure used for docking was created using Open Babel. The model of DBD-F chosen for docking was a modified version of the original crystal structure (PDB:2B29) optimized for ligand binding (22, 23). The entire protein structure was used for modeling under default conditions, resulting in five simulating dockings of high predictive affinity. Docking data and results are included in supplementary.
Immunofluorescence and western blot
For immunofluorescence studies, UMSCC38 cells were grown overnight prior to drug treatment, fixed and permeabilized with 100% ice-cold methanol for 10 min, washed and blocked 10% goat serum and 1% BSA in PBS for 30 min at room temperature and the primary antibodies to PS139-H2AX were applied in blocking solution for 1 h at room temperature. An Alexa Fluor 488 conjugated secondary antibody was then incubated in blocking solution for 1 h at room temperature. Cells were mounted in PermaFluor (Fisher) supplemented with 0.5 μg/mL 4’,6-diamidino-2-phenylindole (DAPI) (Roche). Images were captured digitally with a Zeiss Axiovert 200 M microscope, and scored using Mathematica software (Wolfram) as follows. DAPI staining was used to define the region of interest (ROI). H2AX signal within the ROI was determined and background intensity calculated from outside the ROI for each nuclei. 200 cells were assayed per condition. Statistical analysis was then performed with Prism software (Graphpad). For western blot, whole cell lysates were separated by SDS–PAGE, blotted onto nitrocellulose membranes, and probed with primary antibodies to RPA32, followed by Alexa Fluor 680-conjugated anti-rabbit. Images were obtained with an Odyssey Imager (LI-COR).
Cells and clonogenic assays
The squamous cell carcinoma cell lines UMSCC38 and UMSCC11B (kindly obtained from Dr. Thomas G. Carey, University of Michigan, Ann Arbor, MI) were propagated in DMEM with 10% fetal bovine serum (FBS). The immortalized primary oral keratinocyte cell line, OKF4 (obtained from Dr. James G. Rheinwald, Harvard Institutes of Medicine, Boston, MA) was propagated in fortified KBM-2 media (Lonza) with 10% fetal bovine serum (Hyclone). For clonogenic assays, cells were trypsinized and diluted in media to 1000 cells/mL, then dispersed into 60 mm dishes (3 mL) overnight. After addition of HAMNO, cells were grown for 9 d, then fixed in PBS containing 6% glutaraldehyde for 30 min, and then dyed in 0.5% crystal violet for 30 min and rinsed. Colonies containing over 50 cells were counted. For studies requiring etoposide, HAMNO was added one h before addition of 2 μM etoposide. After 2 h of etoposide exposure, media was removed and rinsed with PBS, before adding back media containing HAMNO. Data were analyzed using an unpaired 2-tailed Student t test to determine statistical significance.
Protein purification and Electrophoretic mobility shift assays (EMSA)
RPA was purified using a published protocol as described (24). DBD-F fused to maltose binding protein was generated and purified as described (22). Quality of both proteins were assessed by SDS-PAGE, followed by coomassie staining (22). For ssDNA binding studies, 7 nM RPA was added to 10 nM labeled polyT 30mer in EMSA buffer (10 mM Tris, pH 7.5, 10 mM KCl, 10% glycerol) for 10 min at 25 °C. Samples were run on 1% agarose gels in 40 mM Tris-Acetate buffer, pH 7.5, and then scanned on an infrared scanner. For DNA unwinding assays, 14 nM RPA was added to 10 nM PAGE purified annealed polyA:polyT 30mer oligonucleotides.
Flow cytometry
Cell cycle assessment and γ-H2AX staining were monitored in UMSCC38 and OKF4 cells after 2 h incubation with HAMNO and fixed in 70% ethanol overnight. Cells were washed with PBS and incubated overnight in PBS containing 1% BSA, 10% goat serum and PS139-H2AX antibodies (Millipore), washed and incubated in goat anti-mouse Alexa Fluor 647 antibody for 30 min at RT. Cells were incubated in 50 μg/mL propidium iodide and 100 μg/mL RNase A for 30 min, and 10,000 cells per sample were analyzed on a BD FACSarray (BD Biosciences) using 532 and 635 nm excitations and collecting fluorescent emissions with filters at 585/42 nm and 661/16 nm (yellow and red parameters, respectively). BD FACSarray and WinList™ (Verity House) software were used for data collection and analysis, respectively.
Xenograph tumor model
Athymic nude mice were purchased from NIH and housed at the animal facility at the UNMC College of Dentistry. UMSCC38 and UMSCC11B cells were implanted into 6-week-old female mice by a single subcutaneous injection of tumor cells (2 – 6 × 105 cells in 100 mL of sterile PBS). The growth rates of tumors were determined by daily monitoring of tumor volume with vernier calipers [tumor volume = 1/2(length × width2)]. Once the tumor size reached 50 mm3, etoposide (10 mg/kg mouse) and HAMNO (2 mg/kg) were administered intraperitoneally every day for 3 days. Tumor size was monitored daily and the volume of the tumor was compared among all experimental groups. At least three mice were used per group. Data were analyzed using an unpaired 2-tailed Student t test to determine the statistical significance.
Results
HAMNO is selective for DBD-F
HAMNO (Fig. 1A) was first identified as a RPA DBD-F inhibitor in a high-throughput screen that determined the ability of a small molecule to dissociate a Rad9-GST fusion protein from a RPA-ssDNA complex, an interaction that requires DBD-F (25). Binding of HAMNO to DBD-F was further investigated through in silico methods (Fig. 1B). These studies utilized a crystal structure of DBD-F (23) that was earlier optimized for binding to the in vitro DBD-F inhibitor, fumaropimaric acid (FPA) (22). The site of highest predicted affinity was to a position immediately adjacent to R43 on DBD-F (Fig. 1B: right panel), where the compound would predictively act to hinder protein-protein interaction, as this residue is essential for DBD-F-protein binding (11).
Figure 1.

Structure/Activity of HAMNO. (A) Chemical structure of HAMNO. (B) In-silico docking of HAMNO with DBD-F. Left panel: Docking HAMNO on the entire DBD-F structure results in the most favorable docking site residing in the basic cleft of DBD-F. Areas of positive electrostatic potential are in blue, negative in red. Area within the white outlined square is enlarged on the right panel. Right panel: HAMNO is predicted to bind immediately adjacent to the essential R43 residue. (C) HAMNO binds to DBD-F. Left panel: HAMNO has no effect on the mobility of a single-stranded polyT-30mer. Right panel: Addition of HAMNO to DBD-F complex with polyT 30mer results in the appearance of a band of increased mobility (denoted by a ‘*’), a result of HAMNO interacting with the DBD-F/DNA complex. Concentration of DBD-F and DNA is 5 μM and 100 nM, respectively. Concentration of HAMNO is 50,100,150,200 μM. (D) HAMNO prevents DBD-F dependent unwinding of DNA by RPA (left panel), but does not prevent RPA ssDNA binding (right panel). Concentrations of drug are 200,100,50,25,12.5 μM.
To confirm an interaction of HAMNO with DBD-F in vitro, we took advantage of the ability of DBD-F to weakly bind a labeled oligonucleotide, and looked for changes in mobility and intensity of the complex in the presence of HAMNO using a EMSA (Fig. 1C) (22). In the absence of DBD-F, HAMNO does not bind labeled oligonucleotide (Fig. 1C: left panel). In the presence of both DNA and DBD-F, the addition of HAMNO results in the formation of a band between the free ssDNA and protein-bound ssDNA bands. (Fig. 1C: right panel; denoted by an asterisk). As HAMNO is uncharged at neutral pH, it is unlikely that binding of the compound to the protein-DNA complex alone would result in this newly formed band detected on the gel. Rather, this stimulation of DBD-F binding to DNA by HAMNO suggests a direct interaction of the small molecule with DBD-F, resulting in a conformational shift that alters DNA binding.
We then wanted to confirm that HAMNO is selective for inhibiting only DBD-F, and not DBDs A-E which are important in RPA binding to ssDNA. This selectivity would ensure that HAMNO would affect protein recruitment more adversely than ssDNA binding. To determine this, we took advantage of a DNA unwinding activity by RPA that is dependent on the DBD-F, but does not require DBD-F for stable binding of the resultant ssDNA that is formed (22, 26). To do this, we utilized EMSAs with full length RPA using ssDNA and dsDNA probes. HAMNO did not affect RPA binding to ssDNA at concentrations up to 200 μM, but prevented dsDNA unwinding and subsequent ssDNA binding at 100 μM (Fig. 1D). This inhibition of dsDNA unwinding by HAMNO is nearly equivalent to that by our previously identified in vitro inhibitor, FPA, whose subsequent dissociation constant for DBD-F was determined to be 9.0 μM (22). Together these data show a preference of HAMNO for selectively inhibiting DBD-F at micromolar levels, an ability that would predictively target the replication stress response in replication-stressed cancer cells over normal cells.
HAMNO induces γ-H2AX staining in a cell-cycle specific manner
DBD-F binds ATRIP, Rad9, Rad17 and Nbs1 thereby recruiting and stabilizing the proteins involved in ATR activation (11, 13, 15, 27, 28). Inhibition of DBD-F- interactions with these proteins would likely short-circuit ATR signaling, leading to increased replication stress that can be monitored by pan-nuclear phosphorylation of H2AX in S-phase (29-31). This type of phosphorylation has been shown to be S-phase specific as previously demonstrated with Chk1 inhibitors (32) and differs from the punctate foci that occur in response to double-strand breaks. We evaluated whether HAMNO induces replication stress as assessed through increases in pan-nuclear γ-H2AX staining (Fig. 2A). After UMSCC38 cells were exposed to HAMNO, increased pan-nuclear γ-H2AX staining occurred in a dose dependent manner (Fig. 2A,B). When H2AX phosphorylation was assessed via western blot, cancer derived UMSCC38 cells, as well as another cancer cell line, UMSCC11B, had prominent γ-H2AX staining, particularly after incubation with 20 μM HAMNO. In contrast, the telomerase-immortalized keratinocyte cell line, OKF4, did not show enhanced γ-H2AX staining at this concentration, suggesting that HAMNO is more effective in inducing H2AX phosphorylation in cancer cell lines that are potentiated for oncogene-induced stress. To further validate that pan-nuclear γ-H2AX represents replication stress and the differences in replication stress between HNSCC cells and OKF4 cells is S-phase specific we employed flow cytometry. To measure cell cycle specific γ-H2AX staining we assayed cells in the absence of HAMNO as a negative control which determined the threshold between γ-H2AX positive and γ-H2AX negative cells for each cell cycle phase (Fig. 2D). Both UMSCC38 and OKF4 cells presented increased γ-H2AX staining after addition of HAMNO, with the greatest increase in signal occurring in S-phase (Fig. 2D). In further comparison of the two cell lines, the ratio of γ-H2AX positive to γ-H2AX negative cells in S-phase was six- to eight-fold greater in UMSCC38 cells than OKF4 cells at 20 μM and 50 μM, respectively (Fig. 2E). These data suggest that HAMNO selectively increased γ-H2AX staining in S-phase, indicative of increased replicative stress. These data also indicate that cells predicted to have high levels of oncogene-induced stress, such as mutant p53 squamous cell carcinoma cells, are selectively potentiated for increased replication stress by HAMNO.
Figure 2.

Cell-cycle dependent phosphorylation of H2AX generated by HAMNO. (A) Immunofluorescent detection of γ-H2AX in UMSCC38 cells after 2 h exposure HAMNO. γ-H2AX and DAPI signal are labeled green and blue, respectively. (B). Whisker plot generated from results shown in ‘A’. Ends or whiskers refer to the minimum and maximum of γ-H2AX signal. (C) Western blot of HAMNO induced γ-H2AX staining in two carcinoma cell lines (UMSCC38,UMSCC11B) and the immortalized oral keratinocyte cell line, OKF4. (D) Cell cycle distribution of γ-H2AX induction in UMSCC38 and OKF4 cells after a 2 h treatment of HAMNO. Vertical lines trisect each scatter plot into G1,S and G2/M groups, respectively. (E) Quantitated results of experiment shown in ‘D’ represented as a ratio of γ-H2AX positive to γ-H2AX negative cells for each dose.
HAMNO affects RPA and ATR phosphorylation
Inhibition of DBD-F by HAMNO is expected to inhibit RPA32 phosphorylation directly by inhibiting DBD-F interactions with replication stress response proteins involved in activating ATR (11, 12, 15, 27, 28, 33, 34). We tested this hypothesis under conditions that enhance RPA32 Ser33 phosphorylation (Fig. 3A). Ser33 of RPA32, an ATR substrate, is highly phosphorylated after two hours of treatment with 20 μM of etoposide, which was reduced with the addition of 2 μM HAMNO, and was nearly absent at higher concentrations, demonstrating an in vivo effect of HAMNO as an inhibitor of RPA32 phosphorylation by ATR.
Figure 3.
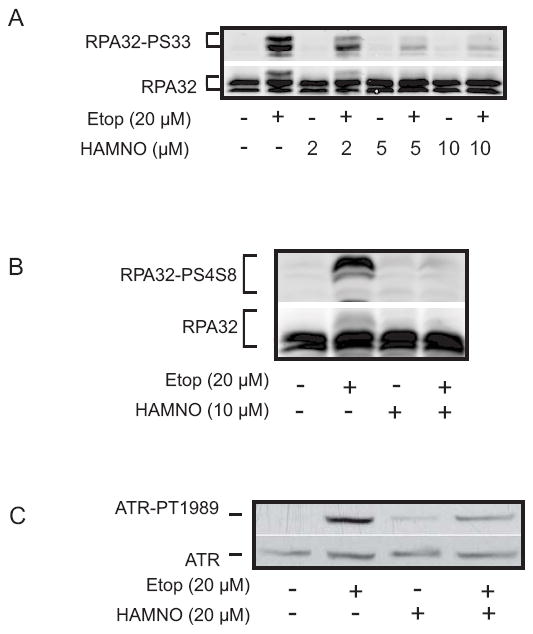
HAMNO alters RPA32 and ATR phosphorylation. (A,B) Etoposide increases RPA32 phosphorylation at S33 (A) and S4/8 (B) in UMSCC38 cells, which is reduced in the presence of increasing concentrations of HAMNO. (C) HAMNO induces ATR autophosphorylation at T1989, but to a lesser extent than etoposide. When HAMNO and etoposide are added simultaneously, ATR is phosphorylated to a lesser extent than etoposide treatment alone, indicating a decrease in ATR signaling.
Ser33 phosphorylation primes further phosphorylation of the N-terminus of RPA32 at other sites, including Ser4 and Ser8 by ATM and DNAPK, a requirement for competent RPA-dependent signaling (35-37). We tested whether inhibition of Ser33 phosphorylation would then inhibit phosphorylation of these downstream sites. As with Ser33, HAMNO also reduced phosphorylation of Ser4 and Ser8 after etoposide treatment (Fig. 3B).
The increase in γ-H2AX staining and decrease in RPA phosphorylation in HAMNO treated cells suggests a deregulation of ATR signaling. To further describe this loss of competent DNA damage response (DDR) signaling, we assessed the autophosphorylation of ATR at T1989, a marker for ATR activity (Fig. 3C) (38, 39). As expected, etoposide strongly induced ATR autophosphorylation, while treatment with HAMNO alone showed a slight increase in T1989 phosphorylation. Interestingly, the addition of etoposide and HAMNO together resulted in a decrease in ATR autophosphorylation as compared to etoposide alone, suggesting that ATR activity has been compromised. Taken together, these data show that HAMNO inhibits RPA32 phosphorylation and decreases ATR autophosphorylation, indicative of HAMNOs ability to affect ATR signaling.
HAMNO enhances etoposide toxicity
The ability of HAMNO to inhibit ATR signaling suggested that the compound may be cytotoxic, and sensitizes cells to DNA damaging agents. HAMNO alone inhibited colony formation in both HNSCC cell lines in the low micromolar range (Fig. 4A). Next, we compared the colony formation of UMSCC38 cells in the presence of increasing concentrations of HAMNO with or without an initial 20 μM/2 h exposure to etoposide (Supplementary Fig. S1). When these two conditions are normalized to the 0 μM HAMNO treatment, HAMNO combined with etoposide significantly inhibited colony formation to a greater degree than HAMNO alone (Fig. 4B). To further examine the potential of HAMNO as an anticancer agent, we tested HAMNO in a mouse xenograft model. In mice, HAMNO slowed the progression of UMSCC11B tumors (Fig.4C). This inhibition was also seen in mice cotreated with sub-lethal etoposide as well as in similarly treated UMSCC38 cells (Fig. 4D,E) These results raise the exciting possibility that a DBD-F inhibitor alone can reduce tumors or can sensitize tumor cells to other chemotherapy agents.
Figure 4.

Additive and synergistic effects of HAMNO with etoposide. (A) HAMNO inhibits colony formation of both UMSCC38 (square) and UMSCC11B cells (triangle). (B) Suppression of colony formation in UMSCC38 cells with HAMNO (triangle) and HAMNO with an initial two hour treatment of 2 μM etoposide (circle). Each data set was normalized to a no HAMNO control.*p<.02,**p<.01 . C. HAMNO reduces the rate of progression of UMSCC11B tumors in mice. Mice received intraperitoneal injections at days 0, 1, 2 with 1mg/kg HAMNO (light gray line) or vehicle (DMSO) in PBS (dark gray line). (D,E) Assessment of tumor size in mice injected with (D) UMSCC11B cells, or (E) UMSCC38 cells. Mice received intraperitoneal injections at days 0, 1, 2 with 10 mg/kg etoposide (dark gray line) or 10 mg/kg etoposide plus 1mg/kg HAMNO (light gray line).
Discussion
The inhibition of the ATR-Chk1 pathway has gained prominence as a potential therapeutic intervention for cancer. This strategy has the potential to strike at the disease in a multitude of ways. First, ATR inhibition alone increases replication stress, even in the absence of exogenous DNA damage, which is amplified in cells with active oncogenes or mutated tumor suppressors, resulting in cell death (30, 32). Second, ATR inhibition restrains replication and G2/M checkpoints, prematurely pushing cells along the cell cycle and increasing genomic instability (30). Third, ATR inhibition may act with synthetic lethality with cancers cells that have an incomplete or incompetent repertoire of DNA repair mechanisms (40). Therefore, the inhibition of ATR and Chk1 will have powerful consequences in the cell.
One major disadvantage of ATR-Chk1 inhibition is the off target effects that are not related to the replication stress response or the DNA damage response (DDR), as both kinases phosphorylate hundreds of proteins, many of which are not DDR related (41, 42). Chk1 expression is essential for mouse embryonic cell survival, indicating that ATR and Chk1 inhibition would negatively affect normal cells(43). ATR and Chk1 null mice are embryonic lethal, further implicating that the inhibition of ATR-Chk1 directly may have unwanted consequences in the organism (44, 45). Therefore, a downstream ATR substrate that specifically modulates the replication stress response, such as RPA, may be a more appropriate target for cancer treatment. Inhibition of RPA phosphorylation by HAMNO allows the replication stress response-specific aspects of ATR signaling to be inhibited, allowing other aspects of ATR signaling to remain unimpeded.
Prior to the discovery of RPA-protein inhibitors, inhibitors of RPA-ssDNA interaction have been characterized and have been shown to be cytotoxic and act with synergy with cisplatin, displaying the potential for RPA as a drug target (46, 47). The discovery of several other DBD-F binding proteins further increased the potential of such an inhibition (11, 14). Our pursuit of DBD-F inhibitors through HTS resulted in the first identified in vitro inhibitor, FPA (25). FPA inhibited the DBD-F with high affinity and selectivity, but had a preponderance of high negative charge that restricted the compound from crossing cell membranes (22, 25). Fesik, et al. also utilized both HTS and a fragment-based NMR spectroscopy method to identify in vitro inhibitors of RPA with affinities similar to FPA (48, 49). HAMNO, with its neutral charge and hydrophobic ring structures allow passage of the compound through the cell membrane, where it can bind to DBD-F, an improvement over FPA.
The ability of HAMNO to work effectively with etoposide attests to the effective strategy of inducing replication stress and reducing the replication stress response to increase cell death selectively in cancer cells that have constitutive DNA replication stress. This approach would be beneficial in the clinic, as the therapeutic efficacy would increase with the addition of a RPA inhibitor and reduce unwanted side effects. HAMNO also has the potential to be used as a stand-alone agent due to its ability to selectively increase cytotoxicity in cancer cells that already have oncogene-induced replicative stress.
This is the first in vivo study showing the potential of a RPA DBD-F inhibitor as a cancer chemotherapeutic agent. The ability of HAMNO to induce cytotoxicity alone and kill cells synergistically with etoposide suggests that HAMNO could be used alone or in combination with other chemotherapeutic agents. HAMNO is tolerated in mice at doses that affect tumor growth, attesting to the clinical potential of this compound. The structure of HAMNO allows for the addition and substitution of many possible moieties that could result in increased affinity to the DBD-F. We are currently testing several HAMNO derivatives for increased DBD-F inhibition.
Supplementary Material
Footnotes
The authors have no conflict of interest to disclose.
References
- 1.Gleich LL, Salamone FN. Molecular genetics of head and neck cancer. Cancer control : journal of the Moffitt Cancer Center. 2002;9:369–78. doi: 10.1177/107327480200900502. [DOI] [PubMed] [Google Scholar]
- 2.Rabinowits G, Haddad RI. Overcoming resistance to EGFR inhibitor in head and neck cancer: a review of the literature. Oral Oncol. 2012;48:1085–9. doi: 10.1016/j.oraloncology.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 3.Bosco EE, Mayhew CN, Hennigan RF, Sage J, Jacks T, Knudsen ES. RB signaling prevents replication-dependent DNA double-strand breaks following genotoxic insult. Nucleic acids research. 2004;32:25–34. doi: 10.1093/nar/gkg919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montana MF, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol. 2011;18:1331–5. doi: 10.1038/nsmb.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–97. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 6.Huang TH, Huo L, Wang YN, Xia W, Wei Y, Chang SS, et al. Epidermal Growth Factor Receptor Potentiates MCM7-Mediated DNA Replication through Tyrosine Phosphorylation of Lyn Kinase in Human Cancers. Cancer cell. 2013;23:796–810. doi: 10.1016/j.ccr.2013.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nature reviews Molecular cell biology. 2010;11:208–19. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 8.Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, et al. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer research. 2010;70:9693–702. doi: 10.1158/0008-5472.CAN-10-2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Murzin AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. The EMBO journal. 1993;12:861–7. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philipova D, Mullen JR, Maniar HS, Lu J, Gu C, Brill SJ. A hierarchy of SSB protomers in replication protein A. Genes Dev. 1996;10:2222–33. doi: 10.1101/gad.10.17.2222. [DOI] [PubMed] [Google Scholar]
- 11.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins, including RAD9, to regulate ATR signaling. Molecular and cellular biology. 2008;28:7345–53. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zou L, Elledge SJ. Science. Vol. 300. New York, NY: 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes; pp. 1542–8. [DOI] [PubMed] [Google Scholar]
- 13.Majka J, Binz SK, Wold MS, Burgers PM. Replication protein A directs loading of the DNA damage checkpoint clamp to 5’-DNA junctions. The Journal of biological chemistry. 2006;281:27855–61. doi: 10.1074/jbc.M605176200. [DOI] [PubMed] [Google Scholar]
- 14.Oakley GG, Tillison K, Opiyo SA, Glanzer JG, Horn JM, Patrick SM. Physical interaction between replication protein A (RPA) and MRN: involvement of RPA2 phosphorylation and the N-terminus of RPA1. Biochemistry. 2009;48:7473–81. doi: 10.1021/bi900694p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shiotani B, Nguyen HD, Hakansson P, Marechal A, Tse A, Tahara H, et al. Two Distinct Modes of ATR Activation Orchestrated by Rad17 and Nbs1. Cell reports. 2013;3:1651–62. doi: 10.1016/j.celrep.2013.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 17.Gorgoulis VG, Vassiliou LV, Karakaidos P, Zacharatos P, Kotsinas A, Liloglou T, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–13. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 18.Halazonetis TD, Gorgoulis VG, Bartek J. Science. Vol. 319. New York, NY: 2008. An oncogene-induced DNA damage model for cancer development; pp. 1352–5. [DOI] [PubMed] [Google Scholar]
- 19.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bignell GR, Greenman CD, Davies H, Butler AP, Edkins S, Andrews JM, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–8. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Molecular oncology. 2011;5:368–73. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glanzer JG, Carnes KA, Soto P, Liu S, Parkhurst LJ, Oakley GG. A small molecule directly inhibits the p53 transactivation domain from binding to replication protein A. Nucleic acids research. 2013;41:2047–59. doi: 10.1093/nar/gks1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bochkareva E, Kaustov L, Ayed A, Yi GS, Lu Y, Pineda-Lucena A, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15412–7. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Henricksen LA, Umbricht CB, Wold MS. Recombinant replication protein A: expression, complex formation, and functional characterization. J Biol Chem. 1994;269:11121–32. [PubMed] [Google Scholar]
- 25.Glanzer JG, Liu S, Oakley GG. Small molecule inhibitor of the RPA70 N-terminal protein interaction domain discovered using in silico and in vitro methods. Bioorganic & medicinal chemistry. 2011;19:2589–95. doi: 10.1016/j.bmc.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lao Y, Lee CG, Wold MS. Replication protein A interactions with DNA. 2. Characterization of double-stranded DNA-binding/helix-destabilization activities and the role of the zinc-finger domain in DNA interactions. Biochemistry. 1999;38:3974–84. doi: 10.1021/bi982371m. [DOI] [PubMed] [Google Scholar]
- 27.Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. ProcNatlAcadSciUSA. 2003;100:13827–32. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D. Function of a conserved checkpoint recruitment domain in ATRIP proteins. Molecular and cellular biology. 2007;27:3367–77. doi: 10.1128/MCB.02238-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, et al. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic acids research. 2012 doi: 10.1093/nar/gks849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–7. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. The Journal of biological chemistry. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 32.Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. MolCell Biol. 2005;25:3553–62. doi: 10.1128/MCB.25.9.3553-3562.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olson E, Nievera CJ, Klimovich V, Fanning E, Wu X. RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. The Journal of biological chemistry. 2006;281:39517–33. doi: 10.1074/jbc.M605121200. [DOI] [PubMed] [Google Scholar]
- 34.Lee J, Dunphy WG. Rad17 plays a central role in establishment of the interaction between TopBP1 and the Rad9-Hus1-Rad1 complex at stalled replication forks. Molecular biology of the cell. 2010;21:926–35. doi: 10.1091/mbc.E09-11-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zernik-Kobak M, Vasunia K, Connelly M, Anderson CW, Dixon K. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. JBiolChem. 1997;272:23896–904. doi: 10.1074/jbc.272.38.23896. [DOI] [PubMed] [Google Scholar]
- 36.Oakley GG, Loberg LI, Yao J, Risinger MA, Yunker RL, Zernik-Kobak M, et al. UV-induced hyperphosphorylation of replication protein a depends on DNA replication and expression of ATM protein. MolBiolCell. 2001;12:1199–213. doi: 10.1091/mbc.12.5.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anantha RW, Vassin VM, Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. The Journal of biological chemistry. 2007;282:35910–23. doi: 10.1074/jbc.M704645200. [DOI] [PubMed] [Google Scholar]
- 38.Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, et al. ATR autophosphorylation as a molecular switch for checkpoint activation. Molecular cell. 2011;43:192–202. doi: 10.1016/j.molcel.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nam EA, Zhao R, Glick GG, Bansbach CE, Friedman DB, Cortez D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. The Journal of biological chemistry. 2011;286:28707–14. doi: 10.1074/jbc.M111.248914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mu JJ, Wang Y, Luo H, Leng M, Zhang J, Yang T, et al. A proteomic analysis of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) substrates identifies the ubiquitin-proteasome system as a regulator for DNA damage checkpoints. The Journal of biological chemistry. 2007;282:17330–4. doi: 10.1074/jbc.C700079200. [DOI] [PubMed] [Google Scholar]
- 41.Blasius M, Forment JV, Thakkar N, Wagner SA, Choudhary C, Jackson SP. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome biology. 2011;12:R78. doi: 10.1186/gb-2011-12-8-r78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dronkert ML, de Wit J, Boeve M, Vasconcelos ML, van Steeg H, Tan TL, et al. Disruption of mouse SNM1 causes increased sensitivity to the DNA interstrand cross-linking agent mitomycin C. Molecular and cellular biology. 2000;20:4553–61. doi: 10.1128/mcb.20.13.4553-4561.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448–59. [PMC free article] [PubMed] [Google Scholar]
- 44.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, et al. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol. 2000;10:479–82. doi: 10.1016/s0960-9822(00)00447-4. [DOI] [PubMed] [Google Scholar]
- 45.Takai H, Tominaga K, Motoyama N, Minamishima YA, Nagahama H, Tsukiyama T, et al. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(-/-) mice. Genes & development. 2000;14:1439–47. [PMC free article] [PubMed] [Google Scholar]
- 46.Anciano Granadillo VJ, Earley JN, Shuck SC, Georgiadis MM, Fitch RW, Turchi JJ. Targeting the OB-Folds of Replication Protein A with Small Molecules. J Nucleic Acids. 2010;2010:304035. doi: 10.4061/2010/304035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shuck SC, Turchi JJ. Targeted inhibition of Replication Protein A reveals cytotoxic activity, synergy with chemotherapeutic DNA-damaging agents, and insight into cellular function. Cancer research. 2010;70:3189–98. doi: 10.1158/0008-5472.CAN-09-3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Souza-Fagundes EM, Frank AO, Feldkamp MD, Dorset DC, Chazin WJ, Rossanese OW, et al. A high-throughput fluorescence polarization anisotropy assay for the 70N domain of replication protein A. Analytical biochemistry. 2012;421:742–9. doi: 10.1016/j.ab.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patrone JD, Kennedy JP, Frank AO, Feldkamp MD, Vangamudi B, Pelz NF, et al. Discovery of Protein-Protein Interaction Inhibitors of Replication Protein A. ACS medicinal chemistry letters. 2013;4:601–5. doi: 10.1021/ml400032y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.